
Abstract
Background
Osteosarcoma is the most common bone tumor that occurs in children.
Methods
To identify co-expression modules and pathways correlated with osteosarcoma and its clinical characteristics, we performed weighted gene co-expression network analysis (WGCNA) on RNA-seq data of osteosarcoma with 52 samples. Then we performed pathway enrichment analysis on genes from significant modules.
Results
A total of 5471 genes were included in WGCNA, and 16 modules were identified. Module-trait analysis identified that a module involved in microtubule bundle formation, drug metabolism-cytochrome P450, and IL-17 signaling pathway was negatively correlated with osteosarcoma and positively correlated with metastasis; a module involved in DNA replication was positively correlated with osteosarcoma; a module involved in cell junction was positively correlated with metastasis; and a module involved in heparin binding negatively correlated with osteosarcoma. Moreover, expression levels in four of the top ten differentially expressed genes were validated in another independent dataset.
Conclusions
Our analysis might provide insight for molecular mechanisms of osteosarcoma.
Similar content being viewed by others

Background
Osteosarcoma is an aggressive cancer in the skeletal system that most commonly occurs in children [1]. The proportion of patients who receive a complete clinical response after standard chemotherapy and multimodal treatment of surgery is about 70–75% [2]. For patients diagnosed with metastases, the overall 5-year survival rate was approximately 30%, and for patients with relapsed osteosarcoma, the rate decreased to 15% [3]. Since the development of chemotherapy, survival in localized high-grade osteosarcoma has improved considerably. However, there is still no worldwide consensus on a standard chemotherapy approach [3]. Therefore, exploring the molecular mechanism involved in disease progression and metastasis and its relationship with drug response is critical for identifying effective drugs that overcome drug resistance of tumor.
During these decades, with the dramatic improvement on sequencing technology, the increasing accumulation of sequencing data provides us a data resource to investigate molecular mechanisms of diseases including critical genes, pathways, and networks based on an analysis of omics data. Weighted gene co-expression network analysis (WGCNA) is a method that is widely used in exploration of co-expression modules correlated with traits or phenotypes based on expression data (e.g., expression microarray, transcriptome data) [4]. And the R software package for WGCNA was developed to perform weighted correlation network analysis, including procedures of network construction, module identification, gene selection, calculations of topological properties, data simulation, and visualization [4].
In this study, we performed weighted gene co-expression network analysis based on data from RNA-seq of osteosarcoma to identify critical co-expression modules and pathways correlated with osteosarcoma and its clinical characteristics, which might provide new insights for exploring the underlying molecular mechanisms of osteosarcoma.
Materials and methods
Expression profiles of osteosarcoma
Expression profiles of osteosarcoma (GSE87624) were obtained from the Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/). This data included RNA-seq profiles of 52 samples including 44 osteosarcoma tissues, 3 normal bone tissues, 1 osteoblast, and 4 osteosarcoma cell lines based on Illumina HiSeq 2000. After quality control, read mapping, and normalization of read counts of the RNA-seq data, the values of fragments per kilobase million (FPKM) in each gene were calculated.
Co-expression module analysis of osteosarcoma
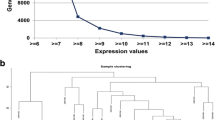
Weighted gene co-expression network analysis (WGCNA) was used to investigate co-expression modules related with osteosarcoma and its clinical characteristics. Genes with the top 25% variance of expression values among samples were included in WGCNA. Then sample clustering was performed to detect whether there were outliers in these samples.
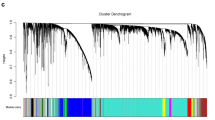
After sample clustering, scale independence and mean connectivity analysis of modules with different power values were performed to determine the soft threshold of module analysis. The power value was set from 1 to 20, and then the values of scale independence and mean connectivity were generated according to these power values. The power value was determined when the scale independence value was 0.9. Then co-expression matrix was calculated under the determined power value, with the minimal module size of 30 and the merge cut height of 0.25. A cluster dendrogram among modules and an eigengene adjacency heatmap between modules were generated.
Module-trait analysis based on clinical characteristics of osteosarcoma
Information on the clinical characteristics of samples with osteosarcoma, including sample type (osteosarcoma/normal), tissue type (osteosarcoma/normal), cell-line type (osteosarcoma/normal), and osteosarcoma type (metastasis/primary/unknown), was collected to identify significant co-expression modules related with the clinical characteristics (as trait). Module-trait relationships were calculated according to the correlation between modules and traits; modules that were significantly correlated with individual traits (P value < 0.05, module size < 500) were identified; and genes in significant modules were then exported for further analysis.
Pathway enrichment analysis of significant co-expression modules
To identify the biological pathways and functions of significant modules, pathway enrichment analysis was conducted by using the R package “clusterProfiler v3.4.4” [5, 6] with genes in significant co-expression modules related with osteosarcoma. Pathways were annotated by information from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database [7] and Gene Ontology (GO) terms [8], and the P value was adjusted by the Benjamin-Hochberg methods [9]. Pathways with a P value < 0.05 were considered as significant pathways.
Validation of differentially expressed genes in independent dataset
To validate whether differentially expressed genes identified in GSE87624 were also differentially expressed in other expression datasets, we investigated the expression levels of the top ten differentially expressed genes in another expression dataset (GSE12865), which included 14 samples (12 osteosarcoma tumor samples and 2 normal human osteoblasts as control). Differentially expressed genes of osteosarcoma were analyzed by limma package in R.
Results
Expression profiles of osteosarcoma
For GSE87624, FPKM values of 21,884 genes from 52 samples were obtained. Then log2-transformed FPKM values were used for further analysis. Information of clinical characteristics of samples included the sample type (osteosarcoma/normal), tissue type (osteosarcoma/normal), cell-line type (osteosarcoma/normal), and osteosarcoma type (metastasis/primary/unknown). Clinical information for osteosarcoma patients is shown in Table 1.
Co-expression module analysis of osteosarcoma
By selecting genes with the threshold of the top 25% variance of expression values, a total of 5471 genes were included in WGCNA. As shown in Fig. 1, by sample clustering, no outliers were observed in 52 samples, thus all samples were included in the analysis.

Sample clustering for detecting outliers
Then the soft threshold was determined by scale independence and mean connectivity analysis of modules with different power values ranging from 1 to 20. As shown in Fig. 2, when the power value was set to 4, the scale independence value achieved 0.9 and lower mean connectivity. Therefore, the co-expression matrix was calculated under the determined power value of 4. As shown in Figs. 3 and 4, a total of 16 modules with different genes were generated and displayed with different colors, including 198 genes in a black module, 837 genes in a blue module, 689 genes in a brown module, 60 genes in a cyan module, 408 genes in a green module, 95 genes in a green-yellow module, 31 in a light cyan module, 131 genes in a magenta module, 50 genes in a midnight blue module, 171 genes in a pink module, 127 genes in a purple module, 302 genes in a red module, 63 genes in a salmon module, 88 genes in a tan module, 1413 genes in a turquoise module, and 428 genes in a yellow module.

Scale independence and mean connectivity analysis

Cluster dendrogram among modules

Eigengene adjacency heatmap between modules
Module-trait analysis based on clinical characteristics of osteosarcoma
Clinical characteristics of samples with osteosarcoma were collected as a trait, and module-trait relationships were calculated according to the correlation between modules and traits. As shown in Fig. 5, for the sample type of osteosarcoma, the most significant modules with negative correlation were purple and green-yellow; for the tissue type of osteosarcoma, the most significant modules with negative correlation were purple and greenyellow and the positively correlated module was brown; and for the osteosarcoma type of metastasis, the most significant modules with positive correlation were purple and yellow.

Module-trait relationships
Pathway enrichment analysis
By pathway enrichment analysis, with the threshold of Benjamin-adjusted P value < 0.05, we obtained significant GO terms and KEGG pathways enriched in significant modules (see Additional file 1). For the purple module (Additional file 1: Table S1), we identified 22 GO terms and 6 KEGG pathways and microtubule bundle formation, IL-17 signaling pathway, and drug metabolism-cytochrome P450 were identified; for the brown module (Additional file 1: Table S2), we identified 58 GO terms and 3 KEGG pathways, which were mainly related with DNA replication and mitotic nuclear division; for the yellow module (Additional file 1: Table S3), we identified 26 GO terms, which were mainly related with the anchored component of membrane and cell junction; and for the green-yellow module (Additional file 1: Table S4), we identified 3 GO terms and 2 KEGG pathways, which were mainly related with regulation of lipolysis in adipocytes and heparin binding.
Validation of differentially expressed genes in independent datasets
With the threshold of Benjamin-adjusted P value < 0.05 and |log2 (fold change)| > 1, we obtained a total of 369 differentially expressed genes of osteosarcoma in GSE87624. The top ten differentially expressed genes were BPIFA1, AGR2, MEPE, MSMB, BPIFB1, BGLAP, SLPI, HBA2, LCN2, and SERPINB3. In an independent dataset GSE12865, we observed that all ten genes had the same trend of expression level as that in GSE87624, in which four genes, MEPE, BPIFB1, HBA2, and SERPINB3, were also identified to have significantly differential expression levels (shown in Table 2.
Discussion
In this study, we performed a weighted gene co-expression network analysis (WGCNA) to investigate co-expression modules related with osteosarcoma and its clinical characteristics. Significant modules were identified to be correlated with osteosarcoma. For the purple module, which was mainly related with microtubule bundle formation, drug metabolism-cytochrome P450, and IL-17 signaling pathway and was identified to be negatively correlated with the trait of osteosarcoma, while being positively correlated with the trait of metastasis in osteosarcoma, previous studies have reported that increase of microtubule destabilization was related with G1/G2 phase cell cycle arrest and apoptosis, and microtubule inhibitors could trigger autophagy and cell death in osteosarcoma cell line [10]. Besides, IL-17A/IL-17RA interaction promoted metastasis of osteosarcoma cells [11]. Moreover, the resistance of osteosarcoma to chemotherapy was related to cytochrome P450 3A4 [12]. Our results might provide supporting evidence for these previous findings.
For the brown module, which was mainly involved in DNA replication and mitotic nuclear division and was observed to be positively correlated with the trait of osteosarcoma tissue, previous studies have revealed that genes involved with DNA replication and DNA damage were associated with radiosensitivity of osteosarcoma [13], as well as drug sensitivity [14]. Our results indicated that genes in the brown module might be related with carcinogenesis of osteosarcoma and might provide insights for exploring drug targets for the treatment of osteosarcoma.
For yellow module, which was mainly related with the anchored component of membrane and cell junction and was observed to be positively correlated with the trait of metastasis in osteosarcoma, it has been reported that pathways related with cell junction were involved in metastasis in various tumors, e.g., lung cancer [15], osteosarcoma [16], and pancreatic cancer [17], which might be due to the molecules in cell junction, and the anchored component of membrane felicitated the migration and invasion tumor cells into other tissues and organs [18]. Our results suggest that genes in the anchored component of membrane and cell junction might also play important roles in the metastasis of osteosarcoma and were worthy of further investigation.
For the green-yellow module, which was mainly related with regulation of lipolysis in adipocytes and heparin binding, it was observed to be negatively correlated with the trait of osteosarcoma. In accordance with our results, there were studies showing that heparin could reduce the osteosarcoma proliferation and growth [19, 20], and heparin binding sites might be potential therapeutic targets for osteosarcoma [21].
In the validation section, among the top ten differentially expressed genes in GSE87624, we validated four genes, MEPE, BPIFB1, HBA2, and SERPINB3, also had significantly differential expression levels in another expression dataset (GSE12865), demonstrating the results identified from the first dataset can be supported by an independent dataset. For MEPE, there were researches revealing its involvement with osteosarcoma [22,23,24]; for BPIFB1, it has been reported that it could inhibit radioresistance in nasopharyngeal carcinoma [25, 26]; for HBA2, previous studies have identified different expression levels in the bone marrow of prostate cancer patients [27]; and for SERPINB3, it has been identified as mediators of Ras-driven inflammation and oncogenesis [28]. The effect of these genes on osteosarcoma was worthy of further investigation.
Conclusions
By WGCNA methods on expression data, we identified significant co-expression modules and pathways correlated with osteosarcoma, as well as metastasis of osteosarcoma, and our analysis might provide insights for the molecular mechanisms of osteosarcoma.
Abbreviations
- GEO:
-
Gene Expression Omnibus
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- WGCNA:
-
Weighted gene co-expression network analysis
References
Luetke A, Meyers PA, Lewis I, Juergens H. Osteosarcoma treatment - where do we stand? A state of the art review. Cancer Treat Rev. 2014;40:523–32.
Stiller CA. International patterns of cancer incidence in adolescents. Cancer Treat Rev. 2007;33:631–45.
Anninga JK, Gelderblom H, Fiocco M, Kroep JR, Taminiau AH, Hogendoorn PC, Egeler RM. Chemotherapeutic adjuvant treatment for osteosarcoma: where do we stand? Eur J Cancer. 2011;47:2431–45.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16:284–7.
Yu G, Wang LG, Yan GR, He QY. DOSE: an R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics. 2015;31:608–9.
Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999;27:29–34.
Gene Ontology Consortium. Going forward. Nucleic Acids Res. 2015;43:D1049–56.
Huang d W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
Vetter NS, Kolb EA, Mills CC, Sampson VB. The microtubule network and cell death are regulated by an miR-34a/Stathmin 1/betaIII-tubulin axis. Mol Cancer Res. 2017;15:953–64.
Wang M, Wang L, Ren T, Xu L, Wen Z. IL-17A/IL-17RA interaction promoted metastasis of osteosarcoma cells. Cancer Biol Ther. 2012;14:155–63.
Mensah-Osman EJ, Thomas DG, Tabb MM, Larios JM, Hughes DP, Giordano TJ, Lizyness ML, Rae JM, Blumberg B, Hollenberg PF, Baker LH. Expression levels and activation of a PXR variant are directly related to drug resistance in osteosarcoma cell lines. Cancer. 2007;109:957–65.
Dai N, Qing Y, Cun Y, Zhong Z, Li C, Zhang S, Shan J, Yang X, Dai X, Cheng Y, et al. miR-513a-5p regulates radiosensitivity of osteosarcoma by targeting human apurinic/apyrimidinic endonuclease. Oncotarget. 2018;9:25414–26.
Li S, Cui Z, Meng X. Knockdown of PARP-1 inhibits proliferation and ERK signals, increasing drug sensitivity in osteosarcoma U2OS cells. Oncol Res. 2016;24:279–86.
Liu MX, Zhou KC, Cao Y. MCRS1 overexpression, which is specifically inhibited by miR-129*, promotes the epithelial-mesenchymal transition and metastasis in non-small cell lung cancer. Mol Cancer. 2014;13:245.
Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, Yu M, Pely A, Engstrom A, Zhu H, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158:1110–22.
Tan X, Tamori Y, Egami H, Ishikawa S, Kurizaki T, Takai E, Hirota M, Ogawa M. Analysis of invasion-metastasis mechanism in pancreatic cancer: involvement of tight junction transmembrane protein occludin and MEK/ERK signal transduction pathway in cancer cell dissociation. Oncol Rep. 2004;11:993–8.
Barbieri I, Pensa S, Pannellini T, Quaglino E, Maritano D, Demaria M, Voster A, Turkson J, Cavallo F, Watson CJ, et al. Constitutively active Stat3 enhances neu-mediated migration and metastasis in mammary tumors via upregulation of Cten. Cancer Res. 2010;70:2558–67.
Ichikawa J, Cole HA, Magnussen RA, Mignemi NA, Butler M, Holt GE, O'Rear L, Yuasa M, Pabla B, Haro H, et al. Thrombin induces osteosarcoma growth, a function inhibited by low molecular weight heparin in vitro and in vivo: procoagulant nature of osteosarcoma. Cancer. 2012;118:2494–506.
Saito M, Ichikawa J, Ando T, Schoenecker JG, Ohba T, Koyama K, Suzuki-Inoue K, Haro H. Platelet-derived TGF-beta induces tissue factor expression via the Smad3 pathway in osteosarcoma cells. J Bone Miner Res. 2018.
Lamoureux F, Picarda G, Garrigue-Antar L, Baud'huin M, Trichet V, Vidal A, Miot-Noirault E, Pitard B, Heymann D, Redini F. Glycosaminoglycans as potential regulators of osteoprotegerin therapeutic activity in osteosarcoma. Cancer Res. 2009;69:526–36.
Siggelkow H, Schmidt E, Hennies B, Hufner M. Evidence of downregulation of matrix extracellular phosphoglycoprotein during terminal differentiation in human osteoblasts. Bone. 2004;35:570–6.
Orimo H, Shimada T. Effects of phosphates on the expression of tissue-nonspecific alkaline phosphatase gene and phosphate-regulating genes in short-term cultures of human osteosarcoma cell lines. Mol Cell Biochem. 2006;282:101–8.
Prideaux M, Wijenayaka AR, Kumarasinghe DD, Ormsby RT, Evdokiou A, Findlay DM, Atkins GJ. SaOS2 osteosarcoma cells as an in vitro model for studying the transition of human osteoblasts to osteocytes. Calcif Tissue Int. 2014;95:183–93.
Wei F, Tang L, He Y, Wu Y, Shi L, Xiong F, Gong Z, Guo C, Li X, Liao Q, et al. BPIFB1 (LPLUNC1) inhibits radioresistance in nasopharyngeal carcinoma by inhibiting VTN expression. Cell Death Dis. 2018;9:432.
Wei F, Wu Y, Tang L, He Y, Shi L, Xiong F, Gong Z, Guo C, Li X, Liao Q, et al. BPIFB1 (LPLUNC1) inhibits migration and invasion of nasopharyngeal carcinoma by interacting with VTN and VIM. Br J Cancer. 2018;118:233–47.
Guzvic M, Braun B, Ganzer R, Burger M, Nerlich M, Winkler S, Werner-Klein M, Czyz ZT, Polzer B, Klein CA. Combined genome and transcriptome analysis of single disseminated cancer cells from bone marrow of prostate cancer patients reveals unexpected transcriptomes. Cancer Res. 2014;74:7383–94.
Catanzaro JM, Sheshadri N, Zong WX. SerpinB3/B4: mediators of Ras-driven inflammation and oncogenesis. Cell Cycle. 2014;13:3155–6.
Acknowledgements
We thank authors providing expression data of clubfoot.
Funding
The present research was supported by the Foundation of Shenzhen Health and Family Planning Commission (grant no. SZFZ 2017081), the National Natural Science Foundation of China (grant no. 81702110) and the Foundation of the Second Hospital of Tianjin Medical University (grant no. 2016ydey05).
Availability of data and materials
All data and material were available in the GEO database.
Author information
Authors and Affiliations
Contributions
JMH contributed to the study concepts; JSW contributed to the study design and data analysis/interpretation; JSW and YGW contributed to the data acquisition, statistical analysis, manuscript preparation, and manuscript definition of intellectual content; YSZ and XDL contributed to the manuscript editing; and SXD, PX, GZZ, and JMH contributed to the manuscript revision/review. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. Significant pathways of the purple module. Table S2. Significant pathways of the brown module. Table S3. Significant pathways of the yellow module. Table S4. Significant pathways of the green-yellow module. (DOCX 27 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, Js., Wang, Yg., Zhong, Ys. et al. Identification of co-expression modules and pathways correlated with osteosarcoma and its metastasis. World J Surg Onc 17, 46 (2019). https://doi.org/10.1186/s12957-019-1587-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12957-019-1587-7