
Abstract
Background
To examine the clinical value of miR-198-5p in lung squamous cell carcinoma (LUSC).
Methods
Gene Expression Omnibus (GEO) microarray datasets were used to explore the miR-198-5p expression and its diagnostic value in LUSC. Real-time reverse transcription quantitative polymerase chain reaction was used to evaluate the expression of miR-198-5p in 23 formalin-fixed, paraffin-embedded (FFPE) LUSC tissues and corresponding non-cancerous tissues. The correlation between miR-198-5p expression and clinic pathological features was assessed. Meanwhile, putative target messenger RNAs of miR-198-5p were identified based on the analysis of differentially expressed genes in the Cancer Genome Atlas (TCGA) and 12 miRNA prediction tools. Subsequently, the putative target genes were sent to Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway analyses.
Results
MiR-198-5p was low expressed in LUSC tissues. The combined standard mean difference (SMD) values of miR-198-5p expression based on GEO datasets were − 0.30 (95% confidence interval (CI) − 0.54, − 0.06) and − 0.39 (95% CI − 0.83, 0.05) using fixed effect model and random effect model, respectively. The sensitivity and specificity were not sufficiently high, as the area under the curve (AUC) was 0.7749 (Q* = 0.7143) based on summarized receiver operating characteristic (SROC) curves constructed using GEO datasets. Based on the in-house RT-qPCR, miR-198-5p expression was 4.3826 ± 1.7660 in LUSC tissues and 4.4522 ± 1.8263 in adjacent normal tissues (P = 0.885). The expression of miR-198-5p was significantly higher in patients with early TNM stages (I-II) than that in cases with advanced TNM stages (III-IV) (5.4400 ± 1.5277 vs 3.5690 ± 1.5228, P = 0.008). Continuous variable-based meta-analysis of GEO and PCR data displayed the SMD values of − 0.26 (95% CI − 0.48, − 0.04) and − 0.34 (95% CI − 0.71, 0.04) based on fixed and random effect models, respectively. As for the diagnostic value of miR-198-5p, the AUC based on the SROC curve using GEO and PCR data was 0.7351 (Q* = 0.6812). In total, 542 genes were identified as the targets of miR-198-5p. The most enriched Gene Ontology terms were epidermis development among biological processes, cell junction among cellular components, and protein dimerization activity among molecule functions. The pathway of non-small cell lung cancer was the most significant pathway identified using Kyoto Encyclopedia of Genes and Genomes analysis.
Conclusion
The expression of miR-198-5p is related to the TNM stage. Thus, miR-198-5p might play an important role via its target genes in LUSC.
Similar content being viewed by others


Background
Lung cancer ranks first among all cancers in terms of incidence, and it is also the most important cause of cancer death all over the world [1]. Non-small cell lung cancer (NSCLC) accounts for around 85% of all lung cancers, with 30% of NSCLC cases being classified as lung squamous cell carcinoma (LUSC) [2,3,4,5,6,7]. Many patients are diagnosed with NSCLC at an advanced phase, which is attributable for the high mortality [8]. Therefore, more effective biomarkers are urgently needed in LUSC.
MicroRNAs (miRNAs) are ∼ 22-nt long endogenous RNAs that play significant roles in various cellular processes [9]. Previous studies have shown that miRNAs can target mRNAs involved in most of the developmental processes and are thus associating with many diseases [10]. Moreover, miRNAs have also been found to be involved in cancer [11]. Studies have found that miR-198-5p plays a vital role in many human cancers, including lung cancer [12]. A previous study [12] investigated the relationship between FGFR1 and miR-198-5p. However, the expression pattern of miR-198-5p in LUSC remains unknown. Additionally, the prospective target genes of miR-198-5p in LUSC have not yet been identified. Therefore, the relationship between miR-198-5p and lung squamous cell carcinoma as well as the underlying mechanism remains unknown.
To evaluate the clinical significance of miR-198-5p in LUSC, we examined miR-198-5p expression in LUSC tissues and carried out additional specific analyses to uncover its clinicopathological role. Furthermore, we performed big data analysis based on the Gene Expression Omnibus (GEO) and the Cancer Genome Atlas (TCGA). Subsequently, bioinformatics examinations were conducted to investigate the probable mechanism of miR-198-5p in LUSC.
Methods
Retrieval of data and publications from TCGA and GEO
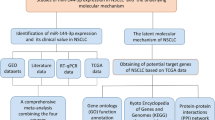
The flowchart representing the main design of our study is shown in Fig. 1. We downloaded miRNA expression data from the Cancer Genome Atlas (TCGA) associated with LUSC. All data were converted to a log2 scale. We also retrieved data from the Gene Expression Omnibus (GEO) and ArrayExpress to assess the expression pattern of miR-198-5p in LUSC and corresponding non-tumor samples. The search terms were as follows: (“lung” OR “pulmonary” OR “respiratory” OR “bronchioles” OR “bronchi” OR “alveoli” OR “pneumocytes” OR “air way” [MeSH]) AND (“cancer” OR “carcinoma” OR “tumor” OR “neoplas” OR “malignan” “squamous cell carcinoma” OR “adenocarcinoma” [MeSH]) OR/AND (“MicroRNA” OR “miRNA” OR “MicroRNA” OR “Small Temporal RNA” OR “noncoding RNA” OR “ncRNA” OR “small RNA” [MeSH]). Datasets with expression levels of miR-198-5p in LUSC and corresponding non-tumor samples were included. Other types of tumor or other miRNA were excluded. The number of samples in the tumor and non-tumor groups was at least three. The expression level of miR-198-5p in the datasets was converted to a log2 scale. The number, mean, and standard deviation of miR-198-5p levels in the tumor and non-tumor groups were calculated. We also searched PubMed, Web of Science, Science Direct, Google Scholar, Ovid, LILACS, Wiley Online Library, EMBASE, Cochrane Central Register of Controlled Trials, Chong Qing VIP, CNKI, Wan Fang, and China Biology Medicine disc; however, no publications regarding miR-198-5p expression in LUSC were found in these databases.

Flowchart representing the main design of our study
RT-qPCR
After the formalin-fixed, paraffin-embedded (FFPE) sections were dewaxed, total RNA was achieved from these sections with the miRNeasy FFPE Kit (QIAGEN) according to manufacturer’s instruction. The concentration of RNA was measured using Nanodrop 2000. MiR-191 (CAACGGAAUCCCAAAAGCAGCU) and miR-103 (AGCAGCAUUGUACAGGGCUAUGA) were used as stably expressed control miRNAs as previously reported [13]. Applied Biosystems 7900 PCR system was used to perform real-time quantitative PCR and detect miR-198-5p expression (GGUCCAGAGGGGAGAUAGGUUC). The relative expression of miR-198-5p was calculated using the formula 2−ΔCq.
Statistical analysis
Paired sample t test and independent sample t test were performed using SPSS 23.0 to determine the association between miR-198-5p expression and various clinicopathological parameters based on real-time RT-qPCR and microarray data. P < 0.05 was regarded as being statistically significant. Receiver operating characteristic (ROC) curves were constructed using SPSS 23.0.
Concerning the meta-analysis based on all accessible data, we used Stata 14 to determine the combined expression value of miR-198-5p in tumor and non-tumor groups and its relationship with both standard mean difference (SMD) and summarized receiver operating characteristic (SROC). The fixed effect model was initially used. The random effect model was used when heterogeneity was detected. The data were considered heterogeneous when I2 > 50%. Subgroup analysis and sensitivity analysis were carried out to find out the source of heterogeneity. We tested publication bias with funnel plots. SPSS 23.0 was employed to explore the diagnostic value of miR-198-5p in LUSC based on GEO data. Then, we performed a diagnostic meta-analysis using MetaDiSc 1.4. Meta-regression and threshold effect analysis were performed to determine the source of heterogeneity. The data from TCGA were not included in the analysis because of missing data.
Differentially expressed mRNAs in LUSC based on TCGA
The expression level of each mRNA transformed into the log2 scale was evaluated using DESeq R package. We obtained 9860 differentially expressed genes in LUSC, including 6092 upregulated and 3768 downregulated genes.
Selection of putative target genes of miR-198-5p
Predictions were conducted in silico with miRWalk 2.0 (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/). Genes that were present in more than 5 of the 12 prediction online tools were selected for further analysis. The selected genes were cross-referenced with the differentially expressed genes in TCGA. The overlapping genes were considered the putative targets of miR-198-5p in LUSC.
Bioinformatics analyses
Gene Ontology (GO) annotation via DAVID (https://david.ncifcrf.gov/) was performed, including biological processes (BP), cellular components (CC), and molecular functions (MF). The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways associated with the putative target genes were also analyzed in DAVID. The results of GO annotation and KEGG pathway analysis were visualized using BiNGO and EnrichmentMap plugins in Cytoscape version 3.5.0. We used STRING (https://string-db.org/) to build interaction maps of the proteins encoded by the putative target genes.
Validation of the putative target genes in the most significant KEGG pathway based on TCGA data
We selected the genes in the most significant KEGG pathway “non-small cell lung cancer” for further analysis. The differences in the expression levels of E2F2, E2F3, TGFA, PRKCG, CDK6, EGF, and CDK4 between LUSC and non-tumor tissues were analyzed based on TCGA data.
Results
GEO data mining to determine the expression and diagnostic value of miR-198-5p
Considering the meta-analysis of miR-198-5p expression-based GEO data, eight datasets were included in our study. The scatter plots based on the GEO datasets are shown in Fig. 2. Forest plots using both fixed effect model (Fig. 3a) and random effect model (Fig. 3b) represented the expression level of miR-198-5p in LUSC. The combined effect sizes were − 0.30 (95% CI − 0.54, − 0.06) and − 0.39 (95% CI − 0.83, 0.05) based on the fixed and random effect models, respectively. Subgroup analysis showed that there was no heterogeneity among the studies from Asia (I2 = 0.0%) (Fig. 3c). The corresponding funnel plot is shown in Fig. 4a (P > 0.05). Sensitivity analysis (Fig. 4b) showed that the dataset GSE40738 might be a source of heterogeneity. The forest plot after the removal of GSE40738 is shown in Fig. 4c. The adjusted combined SMD value was − 0.56 (95% CI − 0.86, − 0.27) with I2 = 33.1%. The receiver operating characteristic curves based on the included datasets from GEO database are shown in Fig. 5. The pooled sensitivity, specificity, positive likelihood ratio, negative likelihood ratio, and diagnostic odds ratio were 0.47 (95% CI 0.40, 0.54), 0.76 (95% CI 0.69, 0.82), 2.19 (95% CI 1.58, 3.05), 0.57 (95% CI 0.38, 0.84), and 4.64 (95% CI 2.04, 10.56), respectively (Fig. 6). The area under the curve (AUC) based on the summarized receiver operating characteristic (SROC) curve was 0.7749 (Q* = 0.7143) (Fig. 7). We did not find a threshold effect of miR-198-5p in the study (P = 0.058). Only study region was determined to be a covariant in the meta-regression, and thus it was likely not a source of heterogeneity (P = 0.0550) (Table 1).

Scatterplots based on the included GEO datasets. a GSE14936. b GSE16025. c GSE19945. d GSE25508. e GSE40738. f GSE47525. g GSE51853. h GSE74190

Continuous variable meta-analysis based on GEO datasets. a Forest plot based on fixed effect model b Forest plot based on random effect model c Subgroup analysis by region

Continuous variable meta-analysis based on GEO datasets. a Funnel plot. b Sensitivity analysis. c Forest plot after the elimination of GSE40738

Receiver operating characteristic (ROC) curves based on GEO datasets. a GSE14936. b GSE16025. c GSE19945. d GSE25508. e GSE40738. f GSE47525. g GSE51853. h GSE74190

Pooled sensitivity, specificity, positive likelihood ratio, negative likelihood ratio, diagnostic score, and odds ratio obtained using MetaDisc 1.4 based on GEO datasets

Summarized receiver operating characteristic (SROC) curve of GEO datasets
Clinical value of miR-198-5p in LUSC assessed using RT-qPCR
Using RT-qPCR, the expression of miR-198-5p in the LUSC tissues was (4.3826 ± 1.7660) compared with that in the non-tumor tissues (4.4522 ± 1.8263, P = 0.885) (Fig. 8a, b). The other clinicopathological features of the LUSC case are shown in Table 2. Notably, the expression level of miR-198-5p in patients with early TNM stage (I-II) was (5.4400 ± 1.5277) compared to (3.5690 ± 1.5228) in patients with advanced TNM stage (III-IV) (P = 0.008) (Fig. 8c, d).

Relationship between miR-198-5p expression and TNM stage in lung squamous cell carcinoma (LUSC) and diagnostic value of miR-198-5p expression. Scatterplots (a LUSC and adjacent non-cancerous tissues and c TNM stage). Receiver operating characteristic (ROC) curves (b LUSC and adjacent non-cancerous tissues and d TNM stage)
Meta-analysis of miR-198-5p expression based on PCR and GEO data
The combined SMD values based on GEO and PCR data were − 0.26 (95% CI − 0.48, − 0.04) (Fig. 9a) and − 0.34 (95% CI − 0.71, 0.04) (Fig. 9b) using the fixed and random effect models, respectively. In subgroup analysis, no heterogeneity among the studies from Asia was observed (I2 = 0.0%) (Fig. 9c). The corresponding funnel plot is shown in Fig. 10a (P > 0.05). Once again, study region and the dataset GSE40738 were identified as sources of heterogeneity (Fig. 10b). After eliminating GSE40738 from the analysis, the SMD changed to − 0.46 (95% CI − 0.72, − 0.20) (Fig. 10c). The pooled sensitivity, specificity, positive likelihood ratio, negative likelihood ratio, and diagnostic odds ratio were 0.52 (95% CI 0.45, 0.58), 0.70 (95% CI 0.63, 0.76), 1.97 (95% CI 1.25, 3.13), 0.56 (95% CI 0.38, 0.82), and 4.23 (95% CI 2.07, 8.64), respectively (Fig. 11). The area under the curve (AUC) based on the summarized receiver operating characteristic (SROC) curve was 0.7351 (Q* = 0.6812) (Fig. 12). We also observed threshold effect of miR-198-5p in this study (P = 0.013). In this case, study region was not accountable for heterogeneity (P = 0.2107) (Table 3).

Continuous variable meta-analysis based on GEO datasets and in-house RT-qPCR results. a: Forest plot based on fixed effect model, (b): forest plot based on random effect model, (c): subgroup analysis by region

Continuous variable meta-analysis based on GEO datasets and in-house RT-qPCR results. a Funnel plot. b Sensitivity analysis. c Forest plot after the elimination of GSE40738

Pooled sensitivity, specificity, positive likelihood ratio, negative likelihood ratio, diagnostic score, and odds ratio obtained using MetaDisc 1.4 based on GEO datasets and in-house RT-qPCR results

Summarized receiver operating characteristic (SROC) curve of GEO datasets and in-house RT-qPCR results
Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway annotation, and protein-protein interaction (PPI) network
In total, 542 genes were considered putative targets of miR-198-5p in LUSC (Fig. 13). Based on Gene Ontology (GO) analysis, the putative target genes of miR-198-5p were predominantly related to epidermis development with respect to biological processes (P = 4.84E−04) (Fig. 14, Table 4), cell junction in the case of cellular components (P = 7.60E−05) (Fig. 15, Table 4), and protein dimerization activity regarding molecular functions (P = 8.03E−05) (Fig. 16, Table 4). In terms of the KEGG pathway, the putative target genes of miR-198-5p were associated with non-small cell lung cancer, pathways in cancer and pancreatic cancer (Fig. 17, Table 5). The protein-protein interaction (PPI) network is shown in Fig. 18. The seven chosen genes in the KEGG pathway “non-small cell lung cancer” were all significantly upregulated in the LUSC samples compared to the non-tumor samples based on TCGA data (Fig. 19, Fig. 20).

Venn diagram based on the 12 predicting tools and the differentially expressed genes (DEGs) in TCGA

Biological processes (BP) were analyzed using the BiNGO plugin in Cytoscape

Cellular components (CC) were analyzed using the BiNGO plugin in Cytoscape

Molecular functions (MF) were analyzed using the BiNGO plugin in Cytoscape

Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was performed using the EnrichmentMap plugin in Cytoscape

Protein-protein interaction (PPI) networks with 537 nodes and 848 edges were constructed using STRING. The PPI enrichment P value was 1.7E−14. Disconnected nodes were hidden in the network

Scatterplots of the seven chosen genes from the Cancer Genome Atlas (TCGA). a CDK4. b CDK6. c E2F2. d E2F3. e EGF. f PRKCG. g TGFA

Receiver operating characteristic (ROC) curves of the seven chosen genes from the Cancer Genome Atlas (TCGA). a CDK4. b CDK6. c E2F2. d E2F3. e EGF. f PRKCG. g TGFA
Discussion
MiR-198-5p was clearly under-expressed in LUSC tissues in comparison with non-cancer lung tissues. The cases with LUSC in Asia expressed lower levels of miR-198-5p than did the healthy controls; however, the expression pattern in other regions was unclear. Our RT-qPCR indicated that the expression of miR-198-5p might be related to the tumor TNM stage, which suggests that miR-198-5p likely plays a role in tumor growth, lymph node metastasis, or distant metastasis. However, the downregulation of miR-198-5p was not obvious in our in-house RT-qPCR analysis. The diagnostic validation and meta-analysis based on GEO and RT-qPCR data indicated that miR-198-5p might be a biomarker of LUSC, but the sensitivity and specificity were not sufficiently high. Similarly, study region may be a source of heterogeneity, which suggested that the diagnostic screening might be suitable for Asian populations but not for others. Apart from LUSC, several studies have explored the expression pattern and mechanism of miR-198-5p in other diseases. MiR-198-5p has been reported to be upregulated in multiple myeloma [14], chronic pancreatitis or pancreatic ductal adenocarcinoma [15], Parkinson’s disease [16], esophageal cancer [17], preeclampsia [18], pancreatic adenocarcinoma, ampullary adenocarcinoma [19], lupus nephritis [20], retinoblastoma [21], anencephaly [22], and squamous cell carcinoma of tongue [23]. On the other hand, low expression of miR-198-5p has been found in prostate cancer [24], breast cancer [25], glioblastoma [26, 27], hepatocellular carcinoma [28], especially hepatitis C virus-associated hepatocellular carcinoma [29, 30], osteosarcoma [31], gastric cancer [32], colorectal cancer [33], pancreatic cancer [34], and respiratory syncytial virus (RSV) infection [35]. The overexpression of miR-198-5p has also been documented in CD8+ T cells in renal cell carcinoma [36]. In prostate cancer, a recent study indicated that miR-198-5p is targeted by the long noncoding RNA SChLAP1, leading to the activation of the MAPK1 pathway, thereby promoting cancer cell proliferation and metastasis [24]. Another study suggested that miR-198-5p may be involved in prostate cancer [37]. In hepatocellular carcinoma, miR-198-5p has been shown to target the HGF/c-MET pathway [38]. Several studies have revealed that the expression of miR-198-5p is greatly related to lymph node metastasis or distant metastasis in different malignant diseases, such as breast cancer [25], osteosarcoma [31], gastric cancer [32], and colorectal cancer [33]. Some studies have also shown that miR-198-5p is closely related to cell proliferation, apoptosis, and migration [12, 14, 39, 40]. The relationship between miR-198-5p and cancer prognosis is controversial [15, 17, 27, 32,33,34]. Thus, we investigated whether miR-198-5p plays an important role in biological processes in various diseases, both malignant and benign.
In the case of lung adenocarcinoma, two reports have verified that miR-198-5p is under-expressed [12, 41]. However, studies on the characteristic of miR-198-5p in LUSC are lacking. One study assessed the diagnostic significance of miR-198-5p in lung adenocarcinoma, with sensitivity = 71.1%, specificity = 95.2%, and AUC = 0.887 (95% CI 0.801, 0.945) [41]. Our study highlighted the diagnostic value of miR-198-5p in LUSC. Yang et al. showed that miR-198-5p was capable to suppress proliferation and promote apoptosis in lung cancer cells by targeting FGFR1 [12], and Wu et al. showed that miR-198-5p promotes apoptosis, represses cell proliferation, and leads to cell cycle arrest in lung adenocarcinoma cells by directly targeting SHMT1 [39]. Our study showed that expression pattern and diagnostic value of miR-198-5p varied according to the race of the patient population, which should be further validated in larger samples.
Although many studies use prediction tools to determine miRNA target genes, the inadequate number of available prediction tools can lead to unreliable data. We used 12 online prediction tools based on miRWalk 2.0, and this method had not been previously utilized in LUSC. The predicted genes were cross-referenced with the differentially expressed genes in TCGA, which further enhanced the specificity and accuracy of our investigation. Because miR-198-5p is downregulated in LUSC, we chose the upregulated genes from TCGA. Via bioinformatics analyses, the putative target genes of miR-198-5p were most significantly enriched in the KEGG pathways of non-small cell lung cancer, pathways in cancer, pancreatic cancer, glioma, and ECM-receptor interactions. For the GO biological processes, the putative target genes of miR-198-5p were involved in epidermis development, negative regulation of neuron apoptotic processes, positive regulation of cell proliferation, the canonical Wnt signaling pathway, and nervous system development, which indicated that the putative target genes might regulated epidermis proliferation, cell apoptosis, or carcinoma of nervous tissues. In addition, for the GO cellular components, the putative target genes of miR-198-5p were enriched in cell junction, transcription factor complex, synaptic vesicle, cell surface proteins, and synapses, which are related to migration, metastasis, and intercellular exchange of molecules. In terms of the GO molecular functions, the putative target genes of miR-198-5p were associated with protein dimerization activity, calcium ion binding, hydrolase activity on carbon-nitrogen (but not peptide) bonds, frizzled binding, and RNA polymerase II transcription co-activator activity. To verify the accuracy of our analysis, we selected several genes and determined its expression based on TCGA. The genes included the pathway non-small cell lung cancer were E2F2, E2F3, TGFA, PRKCG, CDK6, EGF, and CDK4, which were all expressed at significantly higher levels in LUSC tissues in comparison to that in the non-cancer group. Thus, these genes are probably the targets of miR-198-5p. The putative target genes of miR-198 should be validated further in the future.
Conclusions
MiR-198-5p might be downregulated in LUSC, especially in Asia region. The putative target genes of miR-198-5p were closely related to tumorigenesis and progress in LUSC.
Abbreviations
- LUSC:
-
Lung squamous cell carcinoma
- GEO:
-
Gene Expression Omnibus
- FFPE:
-
Formalin-fixed, paraffin-embedded
- mRNA:
-
Messenger RNA
- miRNA:
-
Micro RNA
- TCGA:
-
The Cancer Genome Atlas
- AUC:
-
Area under the curve
- SROC:
-
Summarized receiver operating characteristic
- NSCLC:
-
Non-small cell lung cancer
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- BP:
-
Biological processes
- CC:
-
Cellular components
- MF:
-
Molecular functions
- PPI:
-
Protein-protein interaction
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108.
An HJ, Lee YJ, Hong SA, Kim JO, Lee KY, Kim YK, Park JK, Kang JH. The prognostic role of tissue and serum MMP-1 and TIMP-1 expression in patients with non-small cell lung cancer. Pathol Res Pract. 2016;212:357–64.
Siegfried JM, Lin Y, Diergaarde B, Lin HM, Dacic S, Pennathur A, Weissfeld JL, Romkes M, Nukui T, Stabile LP. Expression of PAM50 genes in lung cancer: evidence that interactions between hormone receptors and HER2/HER3 contribute to poor outcome. Neoplasia. 2015;17:817–25.
Tajima S, Takanashi Y, Koda K. Squamous cell carcinoma of the lung with highly proliferating fibromatosis-like stroma: a rare phenomenon. Int J Clin Exp Pathol. 2015;8:5870–6.
Chiang Y, Yang JC, Hsu FM, Chen YH, Shih JY, Lin ZZ, Lan KH, Cheng AL, Kuo SH. The response, outcome and toxicity of aggressive palliative thoracic radiotherapy for metastatic non-small cell lung cancer patients with controlled extrathoracic diseases. PLoS One. 2015;10:e0145936.
Lee E, Jin D, Lee BB, Kim Y, Han J, Shim YM, Kim DH. Negative effect of cyclin D1 overexpression on recurrence-free survival in stage II-IIIA lung adenocarcinoma and its expression modulation by vorinostat in vitro. BMC Cancer. 2015;15:982.
Lin SY, Peng F. Association of SIRT1 and HMGA1 expression in non-small cell lung cancer. Oncol Lett. 2016;11:782–8.
Ming F, Sun Q. Epigenetically silenced PTPRO functions as a prognostic marker and tumor suppressor in human lung squamous cell carcinoma. Mol Med Rep. 2017;16:746–54.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97.
Cho WC. MicroRNAs: potential biomarkers for cancer diagnosis, prognosis and targets for therapy. Int J Biochem Cell Biol. 2010;42:1273–81.
Dou Z, Lin S, Dai C, Lu Y, Tian T, Wang M, Liu X, Zheng Y, Xu P, Li S, et al. Pooling-analysis for diagnostic and prognostic value of MiRNA-100 in various cancers. Oncotarget. 2017;8:62703–15.
Yang J, Zhao H, Xin Y, Fan L. MicroRNA-198 inhibits proliferation and induces apoptosis of lung cancer cells via targeting FGFR1. J Cell Biochem. 2014;115:987–95.
Peltier HJ, Latham GJ. Normalization of microRNA expression levels in quantitative RT-PCR assays: identification of suitable reference RNA targets in normal and cancerous human solid tissues. RNA. 2008;14:844–52.
Bi C, Chung TH, Huang G, Zhou J, Yan J, Ahmann GJ, Fonseca R, Chng WJ. Genome-wide pharmacologic unmasking identifies tumor suppressive microRNAs in multiple myeloma. Oncotarget. 2015;6:26508–18.
Vychytilova-Faltejskova P, Kiss I, Klusova S, Hlavsa J, Prochazka V, Kala Z, Mazanec J, Hausnerova J, Kren L, Hermanova M, et al. MiR-21, miR-34a, miR-198 and miR-217 as diagnostic and prognostic biomarkers for chronic pancreatitis and pancreatic ductal adenocarcinoma. Diagn Pathol. 2015;10:38.
Cardo LF, Coto E, Ribacoba R, Menendez M, Moris G, Suarez E, Alvarez V. MiRNA profile in the substantia nigra of Parkinson’s disease and healthy subjects. J Mol Neurosci. 2014;54:830–6.
Qi B, Yao WJ, Zhao BS, Qin XG, Wang Y, Wang WJ, Wang TY, Liu SG, Li HC. Involvement of microRNA-198 overexpression in the poor prognosis of esophageal cancer. Asian Pac J Cancer Prev. 2013;14:5073–6.
Choi SY, Yun J, Lee OJ, Han HS, Yeo MK, Lee MA, Suh KS. MicroRNA expression profiles in placenta with severe preeclampsia using a PNA-based microarray. Placenta. 2013;34:799–804.
Schultz NA, Werner J, Willenbrock H, Roslind A, Giese N, Horn T, Wojdemann M, Johansen JS. MicroRNA expression profiles associated with pancreatic adenocarcinoma and ampullary adenocarcinoma. Mod Pathol. 2012;25:1609–22.
Lu J, Kwan BC, Lai FM, Tam LS, Li EK, Chow KM, Wang G, Li PK, Szeto CC. Glomerular and tubulointerstitial miR-638, miR-198 and miR-146a expression in lupus nephritis. Nephrology (Carlton). 2012;17:346–51.
Zhao JJ, Yang J, Lin J, Yao N, Zhu Y, Zheng J, Xu J, Cheng JQ, Lin JY, Ma X. Identification of miRNAs associated with tumorigenesis of retinoblastoma by miRNA microarray analysis. Childs Nerv Syst. 2009;25:13–20.
Zhang Z, Chang H, Li Y, Zhang T, Zou J, Zheng X, Wu J. MicroRNAs: potential regulators involved in human anencephaly. Int J Biochem Cell Biol. 2010;42:367–74.
Wong TS, Liu XB, Wong BY, Ng RW, Yuen AP, Wei WI. Mature miR-184 as potential oncogenic microRNA of squamous cell carcinoma of tongue. Clin Cancer Res. 2008;14:2588–92.
Li Y, Luo H, Xiao N, Duan J, Wang Z, Wang S. Long noncoding RNA SChLAP1 accelerates the proliferation and metastasis of prostate cancer via targeting miR-198 and promoting the MAPK1 pathway. Oncol Res. 2017;
Hu Y, Tang Z, Jiang B, Chen J, Fu Z. miR-198 functions as a tumor suppressor in breast cancer by targeting CUB domain-containing protein 1. Oncol Lett. 2017;13:1753–60.
Nie E, Jin X, Wu W, Yu T, Zhou X, Shi Z, Zhang J, Liu N, You Y. MiR-198 enhances temozolomide sensitivity in glioblastoma by targeting MGMT. J Neuro-Oncol. 2017;133:59–68.
Man HB, Bi WP, Man HH. Decreased microRNA-198 expression and its prognostic significance in human glioma. Genet Mol Res. 2016;15
Huang WT, Wang HL, Yang H, Ren FH, Luo YH, Huang CQ, Liang YY, Liang HW, Chen G, Dang YW. Lower expressed miR-198 and its potential targets in hepatocellular carcinoma: a clinicopathological and in silico study. Onco Targets Ther. 2016;9:5163–80.
Elfimova N, Sievers E, Eischeid H, Kwiecinski M, Noetel A, Hunt H, Becker D, Frommolt P, Quasdorff M, Steffen HM, et al. Control of mitogenic and motogenic pathways by miR-198, diminishing hepatoma cell growth and migration. Biochim Biophys Acta. 1833;2013:1190–8.
Varnholt H, Drebber U, Schulze F, Wedemeyer I, Schirmacher P, Dienes HP, Odenthal M. MicroRNA gene expression profile of hepatitis C virus-associated hepatocellular carcinoma. Hepatology. 2008;47:1223–32.
Zhang S, Zhao Y, Wang L. MicroRNA-198 inhibited tumorous behaviors of human osteosarcoma through directly targeting ROCK1. Biochem Biophys Res Commun. 2016;472:557–65.
Cui Z, Zheng X, Kong D. Decreased miR-198 expression and its prognostic significance in human gastric cancer. World J Surg Oncol. 2016;14:33.
Wang M, Wang J, Kong X, Chen H, Wang Y, Qin M, Lin Y, Chen H, Xu J, Hong J, et al. MiR-198 represses tumor growth and metastasis in colorectal cancer by targeting fucosyl transferase 8. Sci Rep. 2014;4:6145.
Marin-Muller C, Li D, Bharadwaj U, Li M, Chen C, Hodges SE, Fisher WE, Mo Q, Hung MC, Yao Q. A tumorigenic factor interactome connected through tumor suppressor microRNA-198 in human pancreatic cancer. Clin Cancer Res. 2013;19:5901–13.
Bakre A, Mitchell P, Coleman JK, Jones LP, Saavedra G, Teng M, Tompkins SM, Tripp RA. Respiratory syncytial virus modifies microRNAs regulating host genes that affect virus replication. J Gen Virol. 2012;93:2346–56.
Gigante M, Pontrelli P, Herr W, Gigante M, D'Avenia M, Zaza G, Cavalcanti E, Accetturo M, Lucarelli G, Carrieri G, et al. miR-29b and miR-198 overexpression in CD8+ T cells of renal cell carcinoma patients down-modulates JAK3 and MCL-1 leading to immune dysfunction. J Transl Med. 2016;14:84.
Ye L, Li S, Ye D, Yang D, Yue F, Guo Y, Chen X, Chen F, Zhang J, Song X. Livin expression may be regulated by miR-198 in human prostate cancer cell lines. Eur J Cancer. 2013;49:734–40.
Tan S, Li R, Ding K, Lobie PE, Zhu T. miR-198 inhibits migration and invasion of hepatocellular carcinoma cells by targeting the HGF/c-MET pathway. FEBS Lett. 2011;585:2229–34.
Wu S, Zhang G, Li P, Chen S, Zhang F, Li J, Jiang C, Chen X, Wang Y, Du Y, et al. miR-198 targets SHMT1 to inhibit cell proliferation and enhance cell apoptosis in lung adenocarcinoma. Tumour Biol. 2016;37:5193–202.
Wang J, Dan G, Shangguan T, Hao H, Tang R, Peng K, Zhao J, Sun H, Zou Z. miR-198 represses the proliferation of HaCaT cells by targeting cyclin D2. Int J Mol Sci. 2015;16:17018–28.
Han HS, Yun J, Lim SN, Han JH, Lee KH, Kim ST, Kang MH, Son SM, Lee YM, Choi SY, et al. Downregulation of cell-free miR-198 as a diagnostic biomarker for lung adenocarcinoma-associated malignant pleural effusion. Int J Cancer. 2013;133:645–52.
Acknowledgements
None.
Funding
Open fund of Guangxi Colleges and Universities Key Laboratory of Biological Molecular Medicine Research (GXBMR201601), the Youth Science Foundation of Guangxi Medical University (GXMUYSF2014032), and Future Academic Star of Guangxi Medical University (WLXSZX17001) funded this study.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Author information
Authors and Affiliations
Contributions
YF, GC, and YG participated in the design of this study. YL and JH drafted the manuscript. GC, SH, YF, and YG revised the manuscript. YL conducted the RT-qPCR. JH performed the statistical analysis. YL, JH, RT, WC, PC, WC, KS, LG, XG, AL, and XP contributed to the retrieval of database. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Medical Ethics Committee of First Affiliated Hospital of Guangxi Medical University (Approval Number: 2015 (KY-E-041)), and written informed consent was also obtained from each participant.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Liang, Yy., Huang, Jc., Tang, Rx. et al. Clinical value of miR-198-5p in lung squamous cell carcinoma assessed using microarray and RT-qPCR. World J Surg Onc 16, 22 (2018). https://doi.org/10.1186/s12957-018-1320-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12957-018-1320-y