
Abstract
Background
ALKBH7 is a mitochondrial protein, involved in programmed necrosis, fatty acid metabolism, cell cycle regulation, and prostate cancer disease. However, the exact roles of ALKBH7 and the underlying molecular mechanisms remain mysterious. Thus, investigations of the interactome and proteomic responses of ALKBH7 in cell lines using proteomics strategies are urgently required.
Methods
In the present study, we investigated the interactome of ALKBH7 in mitochondria through immunoprecipitation-mass spectrometry/mass spectrometry (IP-MS/MS). Additionally, we established the ALKBH7 knockdown and overexpression cell lines and further identified the differentially expressed proteins (DEPs) in these cell lines by TMT-based MS/MS. Two DEPs (UQCRH and HMGN1) were validated by western blotting analysis.
Results
Through bioinformatic analysis the proteomics data, we found that ALKBH7 was involved in protein homeostasis and cellular immunity, as well as cell proliferation, lipid metabolism, and programmed necrosis by regulating the expression of PTMA, PTMS, UQCRH, HMGN1, and HMGN2. Knockdown of ALKBH7 resulted in upregulation of UQCRH and HMGN1 expression, and the opposite pattern of expression was detected in ALKBH7 overexpression cell lines; these results were consistent with our proteomics data.
Conclusion
Our findings indicate that the expression of UQCRH and HMGN1 is regulated by ALKBH7, which provides potential directions for future studies of ALKBH7. Furthermore, our results also provide comprehensive insights into the molecular mechanisms and pathways associated with ALKBH7.
Similar content being viewed by others

Background
Genomic stability, which is threatened by a variety of endogenous and exogenous factors, is vital for the physiological activities of organisms [1]. In bacteria, the defense against alkylating reagents is executed by protein AlkB, which removes alkylating adducts by catalyzing a hydroxylation process utilizing 2-oxoglutarate and ferrous ion [2]. Nine AlkB homologs are encoded by the human genome: ALKBH1–8 and fat mass- and obesity-associated protein (FTO). The quantity suggests the existence of either functional redundancy or diversity among these eukaryotic AlkB family members [3]. Previous studies revealed that ALKBH1, ALKBH2 and ALKBH3 remove the methyl moiety from 3-methylthymine (3-meT) and/or 3-methylcytosine (3-meC) on DNA or RNA strands [4, 5]; ALKBH5 and FTO catalyze demethylation of the 6-methyladenosine (6-meA) moiety of mRNAs [6, 7]; ALKBH8 mediates the methylation and hydroxylation of tRNAs via its methyltransferase and AlkB domains, respectively [8, 9]. To date, there is no evidence that ALKBH4, ALKBH6 and alpha-ketoglutarate-dependent dioxygenase alkB homolog 7 (ALKBH7) function as nucleic acid hydroxylases [10].
The AlkB family belongs to the dioxygenase superfamily, that utilizes similar catalytic mechanisms but acts on completely different substrates [11]. It has been reported that AlkB family members act on protein substrates, such as ALKBH1 on H2A and ALKBH4 on actin K84me1 [12, 13]. But for ALKBH6 and ALKBH7, hint to their substrate type has yet to be revealed. Previous studies revealed that ALKBH7 is a mitochondrial protein and play a key role in programmed necrosis [14]. Furthermore, ALKBH7-knockout mice showed defects in lipid metabolism [15]. However, the exact roles of ALKBH7 and the underlying molecular mechanisms remain mysterious. Thus, it is urgent to globally investigate the interactome and proteomic responses of ALKBH7 in cell lines using proteomics strategies.
In the present study, we investigated the interactome of ALKBH7 in mitochondria through immunoprecipitation-mass spectrometry/mass spectrometry (IP-MS/MS). Additionally, we established the ALKBH7 knockdown and overexpression cell lines and further identified the differentially expressed proteins (DEPs) in these cell lines by Tandem mass tags (TMT)-based MS/MS.
Materials and methods
Cell lines and antibodies
To establish the stable ALKBH7 knockdown HeLa cells, a series of pBABE retrovirus vectors were constructed. The corresponding target sequences were listed in Additional file 1: Table S1. The retroviruses were then packaged in HEK293T cells with plasmids containing gag and rev. HeLa cells were transduced with harvested retroviruses supernatant and screened with puromycin. Knockdown efficiency was confirmed by qPCR and western blotting. Primer sequences used in qPCR are listed in Additional file 1: Table S2. The polyclonal anti-ALKBH7 was prepared by immunizing rabbits with N-terminal GST-tagged human full-length ALKBH7 in Abgent (Suzhou, China). The serum was harvested and antigen affinity-purified. Anti-FLAG (F3165) was purchased from Sigma-Aldrich. Anti-β-actin (GTX124213) was purchased from GeneTex. Anti-UQCRH (ab154803) was purchased from Abcam. Anti- HMGN1 (CSB-PA010568GA01HU) was purchased from CUSABIO.
Immunoprecipitation (IP)
For IP lysate preparation, HeLa S3 cells were harvested and treated with hypotonic buffer. Then cytoplasm and the nuclei were separated with a tissue grinder and by centrifugation. To obtain nuclear extract, nuclei were resuspended in half the pellet volume of low salt buffer and then mixed with half a pellet volume of high salt buffer, drop by drop and with gentle swirling. After dialysis, the supernatant was collected with by centrifugation at 20,000 g ready for IP. To obtain cytosolic fraction, cytoplasm fraction was centrifuged at 17,000 g for 15 min. The supernatant was harvested and then dialyzed and centrifuged at 17,000 g for 15 min. To obtain crude mitochondrial fraction, pellet from cytoplasm fraction was lysed and centrifuged at 20,000 g for 30 min and the supernatant was harvested for further use. For IP assays, the subcellular fractions were incubated with antibody-conjugated agarose for 4 h at 4 °C. The immune-complexes were finally eluted with 0.1 M glycine, pH = 2, and then resolved in a denatured gel.
In-gel digestion
Proteins in the crude mitochondrial fraction were captured in the IP assay and then separated by SDS-PAGE in a 4–12% gradient Noves Bis-Tris gel (Thermo Fisher Scientific, NP0321BOX). The gels were lightly stained with Coomassie brilliant blue R250 (Thermo Fisher Scientific,20,278) for 15 min. Five regions of gel with distinctive proteins bands were removed and diced into 1 mm3 cube, followed by in-gel digested as previously described [16]. Briefly, each gel slice was desiccated with acetonitrile, treated with 10 mM dithiothreitol (DDT) (GE Healthcare Life Sciences, 17,131,801) for 1 h at 55 °C and then with 25 mM iodoacetamide (IAA) (Amersham Biosciences, RPN6302V) for 30 min in the dark at room temperature. The gel slices were then digested overnight at 37 °C with trypsin (Promega, V5280) at a protein/protease ratio of 12.5:1 for liquid chromatography- MS/MS (LC-MS/MS).
TMT-based quantitative proteomics
Four cell lines were used for TMT-based quantitative proteomics: transient ALKBH7 overexpressed (ALKBH7OE) HEK293T cells, stable ALKBH7 knockdown (shALKBH7) HeLa cells, and their corresponding control cells. Cell lines were separately harvested and treated with fresh lysis buffer (8 M urea in PBS, pH 8–8.5;1 mM PMSF; 1 mM protease inhibitor cocktail). The lysates were then reduced by incubation with 5 mM DTT at 60 °C for 1 h and alkylated by incubation with 25 mM IAA in darkness at room temperature for 30 min. Trypsin and Lys-C (Promega, V5072) were then added at a 25:1(w/w) at 37 °C for 16 h according to the manufacturers’ instructions (Thermo Fisher Scientific, 90,068). Digested peptides were acidified with 1% formic acid and desalted with a reverse-phase column (Oasis HLB, WAT094225). The extracts were dried with a vacuum concentrator and finally dissolved in 200 mM triethylammonium bicarbonate buffer for the TMT labeling. TMT isobaric label reagents (0.8 mg TMT dissolved in 40 μl 99.9% acetonitrile) were separately used to label two biological sets of the cell lines as follows: TMT-126 for shALKBH7 control Hela cell lines (Scramble), TMT-127 for shALKBH7 Hela cell lines, TMT-128 for ALKBH7OE control HEK293T cell lines (Vector), TMT-129 for ALKBH7OE HEK293T cell lines. All labeled peptides were then separately mixed, desalted, dried and dissolved in 100 μl 0.1% formic acid for subsequent high-performance liquid chromatography (HPLC) and LC-MS/MS analysis.
HPLC
The TMT-labelled peptides were fractionated using an UltiMate 3000 HPLC system (Thermo Scientific) with an XBridgeTM BEH300 C18 column (Waters, 4.6 × 250 mm, 2.5 μm). Gradient elution buffers A and B were methanol and acetonitrile, respectively. Peptides were separated by gradient elution as follows: 0–8% phase B (98% acetonitrile + 2% ddH20, pH 10 adjusted with NH3·H2O) for 5 min, 8–18% phase B for 35 min, 18–32% phase B for 22 min, 32–95% phase B for 6 min, and 95–0% phase B for 4 min. Fractions were collected every 90 s into 48 tubes, then dried and combined into 20 fractions. Finally, these fractions were re-dissolved in 20 μl 0.1% formic acid for LC-MS/MS analysis.
LC-MS/MS and protein identification
For LC-MS/MS analysis, peptides were separated by a 120 min gradient elution at a flow rate 0.300 μL/min with the EASY-nLC 1000 system which was directly interfaced with the Thermo Orbitrap Fusion mass spectrometer. The analytical column was a homemade fused silica capillary column (75 μm ID, 150 mm length; Upchurch, Oak Harbor, WA) packed with C-18 resin (300 A, 5 μm; Varian, Lexington, MA). Mobile phase A consisted of 0.1% formic acid, and mobile phase B consisted of 100% acetonitrile and 0.1% formic acid. The Orbitrap Fusion mass spectrometer was operated in the data-dependent acquisition mode using Xcalibur 3.0 software. A single full-scan mass spectrum was done in the Orbitrap (350–1550 m/z, 120,000 resolution). The spray voltage is 2500 V and the automatic gain control (AGC) target is 200,000. This was followed by 3 s data-dependent MS/MS scans in an ion routing multipole at 30% normalized collision energy (HCD). The charge state screening of ions was set at 2–7. The exclusion duration was set at 15 s. Mass window for precursor ion selection was set at 1.6 m/z. The MS2 spectra were acquired with a resolution of 30,000 and maximum injection time of 60 ms.
The IP-MS/MS raw data were analysed through the Proteome Discoverer software (Version 1.4, Thermo Scientific) against the human reviewed Swiss-Prot FASTA database (released on 2016.06.19). The criteria for searching were set according to the software recommendations. Furthermore, the Proteome Discoverer software (Version 2.2, Thermo Scientific) was also used to analyse two sets of TMT-based MS/MS raw data against the human reviewed Swiss-Prot FASTA database (released on 2016.06.19). The search criteria set as follows: A maximum number of two missed trypsin/Lys-C cleavages were allowed. Precursor ion mass tolerances were set at 10 ppm for all MS acquired in an Orbitrap mass analyser and the fragment ion mass tolerance was set at 20 mmu for all MS/MS spectra acquired. Carbamidomethylation (C, + 57.021 Da) and TMT-6plex (K and peptide N-terminus) were set as fixed modifications, and oxidation (methionine, M) was specified as the variable modification. The FDR of peptide and protein identification were both set to 0.01 (1%). Relative protein quantification was based on the intensities of six reporter ion per peptide. Quantification was carried out only for proteins with two or more unique peptides matched. Protein ratios were calculated as the median of all peptide hits belonging to a protein. Finally, the IP-MS/MS and TMT-based MS/MS raw data were deposited in the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifiers PXD008318, PXD008319 and PXD008320, respectively.
Bioinformatic analysis
The ratio of proteins (normalized against GAPDH) ≥1.3 or ≤ 0.77 were identified as DEPs. The Gene Ontology (GO) and pathway enrichment analysis of DEPs was performed using the Funrich tool (Version 3.0) [17] and visualized with JMP software (Version 14.0). Boxplot was also visualized by JMP software. The protein-protein interaction (PPI) analysis of DEPs was performed with the String website (http://string-db.org) and visualized through the Cytoscape software (Version 3.1.1). For protein correlation analysis, the identified proteins from TMT-labelled four type of cell lines were analyzed by the ‘corrplot’ and ‘psych’ package with the Pearson correlation coefficient setting in R. Furthermore, Hierarchical cluster analysis of DEPs was also carried out by the ‘iheatmapr’ package with the Complete and Euclidean method setting in R.
Results
Establishing of ALKBH7 knockdown Hela cell lines
To investigate the biological function of ALKBH7, we constructed several stable ALKBH7 knockdown HeLa cell lines by transfection with pBABE retrovirus shRNA vectors and prepared polyclonal anti-ALKBH7 antibody by immunization of rabbits with full-length recombinant ALKBH7. Cell lines, ‘72’ and ‘73’ were shown to exhibit over 80% knockdown efficiency (Additional file 2: Figure S1C). The anti-ALKBH7 antibody was validated with the knockdown cell lines and proven to work well in immunofluorescence (IF) and western blotting assays (Additional file 2: Figure S1A-B). Though anti-ALKBH7 antibody picked up several non-specific bands in western blotting (Additional file 2: Figure S1B), ALKBH7 was the only one enriched in IP assays with the crude mitochondrial fraction (Additional file 2: Figure S2), which demonstrated the specificity of anti-ALKBH7 antibody in IP assays. Furthermore, The IF results showed that the signal of endogenous ALKBH7 is predominantly in cytoplasm (Additional file 2: Figure S1A), which is consistent with previous studies revealing ALKBH7 localizes in mitochondria in the steady-state.
GO and pathway enrichment analysis of proteins enriched in mitochondrion associated with ALKBH7
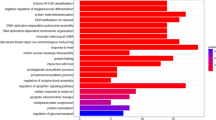
With anti-ALKBH7 antibody, we tried to study the interactome of endogenous ALKBH7 through IP-MS/MS. The anti-ALKBH7 antibody and normal rabbit IgG were covalently conjugated with protein A agarose to prepare the anti-ALKBH7 agarose and the corresponding control, respectively. These agaroses preparations were then incubated with the crude mitochondrial lysate from HeLa S3 cells. As shown in Fig.1a, proteins captured in the IP assay were resolved in reduced denaturing electrophoresis. Five regions of gel with distinctive proteins bands were cut off and underwent in-gel digestion and MS/MS protein identification. A total of 352 proteins were identified, of which two or more unique peptides were identified for 80 proteins with a score ≥ 10. After exclusion of 11 keratins, which are accidental or unavoidable contaminants of proteomics assays, 69 proteins were converted into Entrez Gene IDs and performed for further bioinformatic analysis (Additional file 1: Table S3). Through the GO and pathway enrichment analysis, we found that 27 proteins were identified in the mitochondrion. Furthermore, the other enriched GO and pathway categories included the TRIL signaling pathway, G2/M transition, glypican pathway, protein metabolism, cell growth/maintenance, structural constituent of cytoskeleton, and chaperone activity with adjusted p < 0.01 by Hypergeometric test (Fig.1b, detailed information in the Additional file 1: Table S4). To further investigate the ALKBH7 binding proteins in the mitochondrion, we used the String website to analysis the PPI of the 27 proteins (Fig.1c). The majority of protein interactions converged at two hubs, including several heat shock proteins (HSPA1A, HSPD1, DNAJA3, HSPA5, HSPA8, and HSPA9) and 28S ribosomal proteins (MRPS7, MRPS15, MRPS23, MRPS26, MRPS27, MRPS31, and DAP3), which implied that ALKBH7 may be involved in the protein homeostasis. Additionally, we also considered the possibility of ALKBH7 as a proline hydrolase based on the structural similarity to PHD2 [18]. We designed 71 peptides according to the IP-MS/MS data and performed in vitro demethylation assays, however, no positive results were obtained (Additional file 1: Table S5).

Gene Ontology and pathway enrichment analysis of proteins enriched in mitochondrion associated with ALKBH7. (a) ALKBH7 complexes in the crude mitochondrial extract from HeLa S3 cells were enriched with anti-ALKBH7 agarose. Five regions of gel with distinctive protein bands were excised for in-gel digestion, and further identified by MS. (b) Gene Ontology and pathway enrichment analysis of identified proteins detected through IP-MS/MS. The categories of biological pathway, biological process, cellular component, and molecular function are shown in blue, red, green, and purple, respectively. The percentage of genes per category is represented by the size of the node. (c) Protein-protein interaction network constructed with enriched proteins in mitochondrion identified through IP-MS/MS
Bioinformatic analysis of DEPs in shALKBH7 and ALKBH7OE cell lines through TMT-based MS/MS
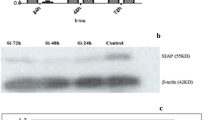
We also conducted a TMT-based proteomic analysis of the effects of knockdown or overexpression of ALKBH7 (two biological replicates). We generated ALKBH7OE HEK293T cells by transient overexpressed ALKBH7-FLAG and the stable ALKBH7 knockdown HeLa cell line ‘73’ (Fig.2a). Using the Proteome Discoverer software, we identified a total of 8234 and 8038 in the two biological TMT-MS/MS replicates, respectively. With the filters of strict protein confidence (q value< 0.05 and unique peptides ≥2), 6948 and 6812 proteins were selected. Notably,14 and 15 were keratins and then excluded from further analysis. Among the remaining proteins, 6369 proteins were found in both two biological TMT-MS/MS replicates and by IP-MS/MS (Fig.2b, Additional file 1: Table S6), and the ratios of these proteins were used for calculating correlations between two biological TMT-MS/MS replicates. Moderate correlation coefficients (r = 0.68) were observed between two shALKBH7/Scramble replicates, whereas high correlation coefficients (r = 0.88) were obtained between the two ALKBH7OE/Vector replicates (Fig.2c). These results suggested the consistency and quality of the two biological TMT-MS/MS replicates, which warranted further analysis. Through using cutoff ratios for DEPs (set as ≥1.3-fold or ≤ 0.77-fold), a total of 326 DEPs were obtained in the two biological replicates (Additional file 1: Table S6). Pairwise comparisons of these DEPs using the hierarchical clustering heatmap revealed that 50 DEPs in cluster 3 were downregulated in two ALKBH7OE/Vector groups and upregulated in two shALKBH7/Scramble groups, while inverse patterns of 100 DEPs were found in cluster 5 (Fig.3a, detailed information in the Additional file 1: Table S6). To gain a better insight into the biological significance of these DEPs, we also performed the GO and pathway enrichment analysis of these 150 DEPs. A total of 20 proteins, mainly participating in respiratory electron transport, were identified in the mitochondrion in the cellular component enrichment analysis (Fig.3b). Furthermore, the other enriched GO and pathway categories included the citric acid cycle and respiratory transport, metabolism of lipids and lipoproteins, triacylglycerol biosynthesis, energy pathways, regulation of cell proliferation, antioxidant activity, and hydrolase activity with adjusted p < 0.05 by Hypergeometric test (Additional file 1: Table S7). These results were consistent with the IP-MS/MS results. Additionally, we found that the categories of interferon signaling and immune system were also enriched, implying ALKBH7 may be involved in regulating immune function. To further clarify biologically meaningful DEPs probably involved in these categories, the gene names of all outlier DEPs were displayed in the boxplot (Fig.4a), and these outlier DEPs in cluster 3 or 5, especially PTMA, may be involved in the physiological or pathological processes of ALKBH7, albeit some proteins changing in only one replicates. Therefore, to further investigated the PPI network among the proteins changed in both two biological TMT-MS/MS replicates, a comprehensive interaction network of the 55 DEPs was performed through the String website and Cytoscape software (Fig.4b, detail information in the Additional file 1: Table S8). We found that the following DEPs have different changing patterns in the shALKBH7 and ALKBH7OE cell lines, including PTMA, PTMS, UQCRH, HMGN1 and HMGN2, especially UQCRH located in the mitochondrion. To further validate our proteomics data, we performed western blotting analysis of UQCRH and HMGN1 in ALKBH7 knockdown and overexpression cell lines. Knockdown of ALKBH7 overexpressed the UQCRH and HMGN1 expression, and vice versa in ALKBH7 overexpression cell lines (Fig.5a-b), The patterns of UQCRH and HMGN1 expression in the western blotting were concordant with our proteomics data, which further validate our bioinformatic results.

Venn diagram and correlation analysis of identified proteins in shALKBH7 and ALKBH7OE cell lines through TMT-based MS/MS. (a) transient overexpressed ALKBH7-FLAG HEK293T cells and the stable ALKBH7 knockdown HeLa cell line ‘72’ and ‘73’. (b) The number of proteins unique to, or shared by, both biological replicates of TMT-based MS/MS and IP-MS/MS is shown in a Venn diagram. (c) Correlation matrix showing all the abundance of the identified proteins in two biological replicates of TMT-based MS/MS. Red boxes indicate correlations between biological duplicates

Bioinformatic analysis of differentially expressed proteins (DEPs) in shALKBH7 and ALKBH7OE cell lines through TMT-based MS/MS. (a) Hierarchical clustering of DEPs in one or two biological replicates of TMT-based MS/MS by Heatmap. Red indicates upregulated DEPs, whereas green indicates downregulated DEPs. The panel on the left shows the five different clusters. (b) GO and pathway enrichment analysis of DEPs in Clusters 3 and 5. The categories of biological pathway, biological process, cellular component, and molecular function are shown in blue, red, green, and purple, respectively. The percentage of genes per category is represented by the size of the node

A comprehensive interaction network among the proteins changed in two biological TMT-MS/MS replicates. (a) Box plot denotes the fold change of DEPs in Cluster 3 and 5. Statistical outlier proteins are colored and labeled. (b) Protein-protein interaction network constructed with DEPs in two biological replicates of TMT-based MS/MS; no connective DEPs were excluded. DEPs are represented by colored nodes labeled with the gene name. The average fold changes of DEPs in two biological replicates of TMT-based MS/MS are reflected by color intensity. Upregulated and downregulated DEPs are shown in red and green, respectively. V-shape nodes denote DEPs located in mitochondrion. Node size correlates with the number of interactions

Validation of TMT-based proteomics data. (a) Two DEPs (UQCRH and HMGN1) identified in proteomics data were validated by western blotting. Knockdown of ALKBH7 resulted in upregulation of UQCRH and HMGN1 expression, and the opposite pattern of expression was detected in ALKBH7 overexpression cell lines, which were consistent with our proteomics data. (b) The relative intensity of UQCRH and HMGN1 in western blotting (normalized by corresponding β-actin expression)
Discussion
We performed IP-MS/MS and TMT-based MS/MS analyses to further investigate the physiological or pathological processes of ALKBH7 in mammals. GO and pathway enrichment analyses revealed that ALKBH7 is associated with protein metabolism, cell growth and/or maintenance, the citric acid cycle and respiratory electron transport, triacylglycerol biosynthesis, antioxidant activity, and structural constituent of the cytoskeleton. These results were consistent with the previous reports that ALKBH7 is associated with programmed necrosis, fatty acid metabolism, cell cycle regulation, and prostate cancer disease [14, 15, 19, 20] and suggest the reliability of our proteomics data. Furthermore,13 identified interacting proteins of ALKBH7 in the mitochondrion by IP-MS/MS were heat shock proteins and 28S ribosomal proteins, suggesting that ALKBH7 may be involved in the protein homeostasis. Notably, biology pathway analysis revealed that interferon signaling and immune system were enriched with DEPs upon different ALKBH7 expression in cell lines by TMT-based MS/MS, implying ALKBH7 may also be involved in cellular immunity. However, these results need to be in-depth investigated in the future.
In TMT-based MS/MS, we also found that the expression of PTMA, PTMS, UQCRH, HMGN1, and HMGN2 proteins were upregulated in shALKBH7 cell lines whereas downregulated in ALKBH7OE cell lines. PTMA and PTMS have similar sequences, comprising 101 and 111 amino acid residues, respectively [21, 22]. Previous study has demonstrated that PTMA is associated with cell division and proliferation, immunomodulatory activity, and acetylation of histones [23,24,25], while PTMS was involved in modulating cellular immunity, binding to the linker histone H1 and remodeling chromatin structure [26,27,28]. UQCRH, which is located in the mitochondria, is required for the electron transfer between cytochrome c and cytochrome c1 during oxidative phosphorylation [29]. It has also been demonstrated that UQCRH is important for the intracellular production of reactive oxygen species (ROS) [30, 31]. These results were reminiscent of the previous study that ALKBH7 is required for programmed necrosis through inhibiting mitochondrial function, such as the formation of ROS [14]. Interesting, HMGN1 and HMGN2 are a family of nuclear proteins that bind specially to nucleosomes and modulate chromatin structure [32]. Several review articles summarized that HMGN proteins also affect cellular differentiation and development, as well as the ability to promote a variety of DNA-related processes, such as transcription, replication, recombination and DNA damage due to both ultraviolet light and ionizing irradiation [32,33,34]. All these results indicate that ALKBH7 may participate in cell proliferation, metabolism of lipids and lipoproteins, and programmed necrosis possibly by directly or indirectly influencing the expression of the five DEPs.
Conclusion
In summary, to the best our knowledge, this is the first study to globally investigate the interactome and proteomic responses of ALKBH7 in cell lines using proteomics strategies. IP-MS/MS and TMT-based MS/MS analyses indicated that ALKBH7 may be involved in both the protein homeostasis and cellular immunity, as well as cell proliferation, lipid metabolism, and programmed necrosis through regulating the expression of PTMA, PTMS, UQCRH, HMGN1, and HMGN2 proteins. Additionally, the expression of UQCRH and HMGN1 was validated in ALKBH7 knockdown and overexpression cell lines by western blotting. All these results provide comprehensive insights into the molecular mechanisms and pathways associated with ALKBH7, as well as highlighting potential directions for future research into ALKBH7.
Availability of data and materials
All data generated or analyzed during this study are included in this published article [and its Additional files].
Abbreviations
- ALKBH7:
-
Alpha-ketoglutarate-dependent dioxygenase alkB homolog 7, mitochondrial
- DDT:
-
Dithiothreitol
- DEPs:
-
Differentially expressed proteins
- GO:
-
Gene ontology
- HMGN1:
-
Non-histone chromosomal protein HMG-14
- HMGN2:
-
Non-histone chromosomal protein HMG-17
- HPLC:
-
High-performance liquid chromatography
- IAA:
-
Iodoacetamide
- IP:
-
Immunoprecipitation
- LC-MS/MS:
-
Liquid chromatography-tandem mass spectrometry
- PPI:
-
Protein-protein interaction
- PTMA:
-
Prothymosin alpha
- PTMS:
-
Parathymosin
- TMT:
-
Tandem mass tags
- UQCRH:
-
Cytochrome b-c1 complex subunit 6, mitochondrial
References
Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40(2):179–204.
Nieminuszczy J, Grzesiuk E. Bacterial DNA repair genes and their eukaryotic homologues: 3. AlkB dioxygenase and Ada methyltransferase in the direct repair of alkylated DNA. Acta Biochim Pol. 2007;54(3):459–68.
Gerken T, Girard CA, Tung YC, Webby CJ, Saudek V, Hewitson KS, Yeo GS, McDonough MA, Cunliffe S, McNeill LA, Galvanovskis J, Rorsman P, Robins P, Prieur X, Coll AP, Ma M, Jovanovic Z, Farooqi IS, Sedgwick B, Barroso I, Lindahl T, Ponting CP, Ashcroft FM, O'Rahilly S, Schofield CJ. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science (New York, N.Y.). 2007;318(5855):1469–72.
Falnes PO. Repair of 3-methylthymine and 1-methylguanine lesions by bacterial and human AlkB proteins. Nucleic Acids Res. 2004;32(21):6260–7.
Westbye MP, Feyzi E, Aas PA, Vagbo CB, Talstad VA, Kavli B, Hagen L, Sundheim O, Akbari M, Liabakk NB, Slupphaug G, Otterlei M, Krokan HE. Human AlkB homolog 1 is a mitochondrial protein that demethylates 3-methylcytosine in DNA and RNA. J Biol Chem. 2008;283(36):25046–56.
Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, Vagbo CB, Shi Y, Wang WL, Song SH, Lu Z, Bosmans RP, Dai Q, Hao YJ, Yang X, Zhao WM, Tong WM, Wang XJ, Bogdan F, Furu K, Fu Y, Jia G, Zhao X, Liu J, Krokan HE, Klungland A, Yang YG, He C. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49(1):18–29.
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG, He C. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–7.
Fu D, Brophy JA, Chan CT, Atmore KA, Begley U, Paules RS, Dedon PC, Begley TJ, Samson LD. Human AlkB homolog ABH8 is a tRNA methyltransferase required for wobble uridine modification and DNA damage survival. Mol Cell Biol. 2010;30(10):2449–59.
Fu Y, Dai Q, Zhang W, Ren J, Pan T, He C. The AlkB domain of mammalian ABH8 catalyzes hydroxylation of 5-methoxycarbonylmethyluridine at the wobble position of tRNA. Angewandte Chemie (International ed. in English). 2010;49(47):8885–8.
Lee DH, Jin SG, Cai S, Chen Y, Pfeifer GP, O'Connor TR. Repair of methylation damage in DNA and RNA by mammalian AlkB homologues. J Biol Chem. 2005;280(47):39448–59.
Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet. 2006;7(9):715–27.
Ougland R, Lando D, Jonson I, Dahl JA, Moen MN, Nordstrand LM, Rognes T, Lee JT, Klungland A, Kouzarides T, Larsen E. ALKBH1 is a histone H2A dioxygenase involved in neural differentiation. Stem Cells. 2012;30(12):2672–82.
Li MM, Nilsen A, Shi Y, Fusser M, Ding YH, Fu Y, Liu B, Niu Y, Wu YS, Huang CM, Olofsson M, Jin KX, Lv Y, Xu XZ, He C, Dong MQ, Rendtlew Danielsen JM, Klungland A, Yang YG. ALKBH4-dependent demethylation of actin regulates actomyosin dynamics. Nat Commun. 2013;4:1832.
Fu D, Jordan JJ, Samson LD. Human ALKBH7 is required for alkylation and oxidation-induced programmed necrosis. Genes Dev. 2013;27(10):1089–100.
Solberg A, Robertson AB, Aronsen JM, Rognmo O, Sjaastad I, Wisloff U, Klungland A. Deletion of mouse Alkbh7 leads to obesity. J Mol Cell Biol. 2013;5(3):194–203.
Shevchenko A, Tomas H, Havli J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc. 2006;1(6):2856–60.
Pathan M, Keerthikumar S, Ang CS, Gangoda L, Quek CY, Williamson NA, Mouradov D, Sieber OM, Simpson RJ, Salim A, Bacic A, Hill AF, Stroud DA, Ryan MT, Agbinya JI, Mariadason JM, Burgess AW, Mathivanan S. FunRich: an open access standalone functional enrichment and interaction network analysis tool. Proteomics. 2015;15(15):2597–601.
Wang G, He Q, Feng C, Liu Y, Deng Z, Qi X, Wu W, Mei P, Chen Z. The atomic resolution structure of human AlkB homolog 7 (ALKBH7), a key protein for programmed necrosis and fat metabolism. J Biol Chem. 2014;289(40):27924–36.
Bjornstad LG, Meza TJ, Otterlei M, Olafsrud SM, Meza-Zepeda LA, Falnes PO. Human ALKBH4 interacts with proteins associated with transcription. PloS one. 2012;7(11):e49045.
Walker AR, Silvestrov P, Muller TA, Podolsky RH, Dyson G, Hausinger RP, Cisneros GA. ALKBH7 Variant Related to Prostate Cancer Exhibits Altered Substrate Binding. PLoS Comput Biol. 2017;13(2):e1005345.
Komiyama T, Pan LX, Haritos AA, Wideman JW, Pan YC, Chang M, Rogers I, Horecker BL. The primary structure of rat parathymosin. Proc Natl Acad Sci U S A. 1986;83(5):1242–5.
Frangou-Lazaridis M, Clinton M, Goodall GJ, Horecker BL. Prothymosin alpha and parathymosin: amino acid sequences deduced from the cloned rat spleen cDNAs. Arch Biochem Biophys. 1988;263(2):305–10.
Pineiro A, Cordero OJ, Nogueira M. Fifteen years of prothymosin alpha: contradictory past and new horizons. Peptides. 2000;21(9):1433–46.
Cotter MA 2nd, Robertson ES. Modulation of histone acetyltransferase activity through interaction of epstein-barr nuclear antigen 3C with prothymosin alpha. Mol Cell Biol. 2000;20(15):5722–35.
Mosoian A, Teixeira A, Burns CS, Sander LE, Gusella GL, He C, Blander JM, Klotman P, Klotman ME. Prothymosin-alpha inhibits HIV-1 via toll-like receptor 4-mediated type I interferon induction. Proc Natl Acad Sci U S A. 2010;107(22):10178–83.
Szabo P, Clinton M, Macera M, Horecker BL. Localization of the gene coding for parathymosin to chromosome 17 in humans. Cytogenet Cell Genet. 1989;50(2–3):91–2.
Martic G, Karetsou Z, Kefala K, Politou AS, Clapier CR, Straub T, Papamarcaki T. Parathymosin affects the binding of linker histone H1 to nucleosomes and remodels chromatin structure. J Biol Chem. 2005;280(16):16143–50.
Kondili K, Tsolas O, Papamarcaki T. Selective interaction between parathymosin and histone H1. Eur J Biochem. 1996;242(1):67–74.
Letts JA, Sazanov LA. Clarifying the supercomplex: the higher-order organization of the mitochondrial electron transport chain. Nat Struct Mol Biol. 2017;24(10):800–8.
Wellen KE, Thompson CB. Cellular metabolic stress: considering how cells respond to nutrient excess. Mol Cell. 2010;40(2):323–32.
Darrouzet E, Valkova-Valchanova M, Moser CC, Dutton PL, Daldal F. Uncovering the [2Fe2S] domain movement in cytochrome bc1 and its implications for energy conversion. Proc Natl Acad Sci U S A. 2000;97(9):4567–72.
Bustin M. Chromatin unfolding and activation by HMGN(*) chromosomal proteins. Trends Biochem Sci. 2001;26(7):431–7.
Gerlitz G. HMGNs, DNA repair and cancer. Biochim Biophys Acta. 2010;1799(1–2):80–5.
Furusawa T, Cherukuri S. Developmental function of HMGN proteins. Biochim Biophys Acta. 2010;1799(1–2):69–73.
Acknowledgements
We thank all staff in Center of Biomedical Analysis, Tsinghua University for their help in Mass Spectrum.
Consent of publication
Not applicable.
Funding
This work was supported by the CAMS Innovation Fund for Medical Sciences (CIFMS) (2016-I2M-1-004) and National Key R&D Program of China (2018YFA0108600).
Author information
Authors and Affiliations
Contributions
Conception and design: Wanchen Dou and Wei Ge. Acquisition of data: Shu Meng and Shaohua Zhan. Interpretation of data: Shu Meng and Shaohua Zhan. Draft manuscript: Shu Meng and Shaohua Zhan. Wanchen Dou and Wei Ge take full responsibility for the integrity of the data analysis. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Target sequences of ALKBH7. Table S2. Primer sequences of ALKBH7 and GAPDH used in qPCR. Table S3. Identified proteins in IP assay for bioinformatic analysis. Table S4. Enriched Gene ontology and pathway categories in IP assay. Table S5. Sequences of 71 peptides performed in vitro demethylation assays. Table S6. Identified proteins and corresponding bioinformatic analysis in TMT-based MS/MS. Table S7. Enriched Gene ontology and pathway categories in TMT-based MS/MS. Table S8. Detailed information of proteins for protein-protein interaction.
Additional file 2: Figure S1.
ALKBH7 knockdown cell lines and custom anti-ALKBH7. Figure S2. anti-ALKBH7 enriched only ALKBH7 in the crude mitochondrial lysate of HeLa S3 cells
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Meng, S., Zhan, S., Dou, W. et al. The interactome and proteomic responses of ALKBH7 in cell lines by in-depth proteomics analysis. Proteome Sci 17, 8 (2019). https://doi.org/10.1186/s12953-019-0156-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12953-019-0156-x