
Abstract
Rheumatoid arthritis is a chronic inflammatory debilitating disease triggered by a complex interaction involving genetic and environmental factors. Active smoking and occupational exposures such as silica increase its risk, suggesting that initial inflammation and generation of rheumatoid arthritis-related autoantibodies in the lungs may precede the clinical disease. This hypothesis paved the way to epidemiological studies investigating air pollution as a potential determinant of rheumatoid arthritis. Studies designed for epidemiology of rheumatoid arthritis found a link between traffic, a surrogate of air pollution, and this disease. Furthermore, a small case–control study recently found an association between wood smoke exposure and anticyclic citrullinated protein/peptide antibody in sera of patients presenting wood-smoke-related chronic obstructive pulmonary disease. However, reports addressing impact of specific pollutants on rheumatoid arthritis incidence and severity across populations are somewhat conflicting. In addition to the link reported between other systemic autoimmune rheumatic diseases and particulate matters/gaseous pollutants, experimental observation of exacerbated rheumatoid arthritis incidence and severity in mice models of collagen-induced arthritis after diesel exhaust particles exposure as well as hypovitaminosis D-related autoimmunity can help understand the role of air pollution in rheumatoid arthritis. All these considerations highlight the necessity to extend high quality epidemiological researches investigating different sources of atmospheric pollution across populations and particularly in low-and-middle countries, in order to further explore the biological plausibility of air pollution’s effect in the pathogenesis of rheumatoid arthritis. This should be attempted to better inform policies aiming to reduce the burden of rheumatoid arthritis.
Similar content being viewed by others

Background
Rheumatoid arthritis (RA) is a systemic autoimmune disease that primarily targets joints leading to progressive joint erosions, and which affects 0.24 % [95 % confidence interval (CI) 0.23 to 0.25 %] of the world population. RA patients continue to face steep rises of the burden of disease, with disability-adjusted life years estimates increased from 3 335 000 (95 % CI 2 573 000 to 4 192 000) in 1990 to 4 815 000 (95 % CI 3 705 000 to 6 056 000) in 2010 [1, 2]. Based on serologic features, two forms are identified: seronegative and seropositive RA [3]. Hereditability of the disease accounts for 50–60 % of its variance, the HLA-DRB1 shared epitope–containing allele being the strongest genetic risk factor [4–6]. Some environmental risk factors are playing out on this genetic background to determine the occurrence of RA. Besides active and heavy tobacco smoking which is the most important environmental risk factor for RA with its attributable risk sustained for up to 20 years after discontinuation, other potential risk factors include estrogens, low socioeconomic status, silica dust, abestoses, mineral oil, diet and infections [5, 6]. However, the exact etiopathogenesis of RA is still not clearly elucidated.
Air pollution, a mixture of suspended particulate matters (PM) of different diameters (<10 μm [PM10], <2.5 μm [PM2.5] and <0.1 μm [PM0.1]) and gases (nitrates [NO2], sulphur dioxide [SO2], ozone [O3], and carbon monoxide) has recently been paid more attention in the field of RA [7]. Main sources include: traffic, industry, stationary fuel burners, forest fires, and solid fuel combustion [7]. Tropospheric pollutants drive climate change and adverse health effects including chronic obstructive pulmonary disease (COPD), cancers, cataracts, stillbirths, and cardiovascular diseases [7].
Assuming that active smoking and occupational exposures increase the risk of RA, it was suggested that initial inflammation and production of RA-related autoantibodies in the lungs may lead to RA [4]. This idea together with the inflammation-related burden of disease attributable to atmospheric pollution guided epidemiological studies of air pollution in relation to RA, in view of eventual preventive strategies.
A link between sources of air pollution and rheumatoid arthritis
Currently, the association between sources of atmospheric pollution and RA has been investigated in three large epidemiological studies: the Nurses’ Health Study (NHS) in the United States [8, 9], the British Colombian (BC) study in Canada[10], and the Swedish Epidemiological Investigation of Rheumatoid Arthritis (EIRA) [11, 12]. In the NHS, association of distance to road-a marker of traffic pollution exposure- and incidence of RA was studied in 90297 women. After adjustment for multiple confounders (age, calendar year, race, cigarette smoking, parity, lactation, menopausal status and hormone use, oral contraceptive use, body mass index, physical activity, and census-tract-level median income and house value), women living within 50 m of a road had an incremented risk of RA compared with those living 200 m or farther away (hazard ratio [HR] = 1.31; 95 % CI 0.98–1.74) [8]. This finding was more consistent among nonsmokers (HR = 1.62; 95 % CI, 1.04–2.52). Similarly, the BC study assessed the effects of proximity to traffic, ambient air pollution, and community noise on the risk of developing RA using health records of 640041 subjects at risk of developing RA in the Border Air Quality Study cohort. Residential proximity to traffic five years prior to RA diagnosis, but consistently not noise, was associated with an increased risk of RA; with highest risks noted for highways and with greater proximity. Since health service records did not contain informations on low socioeconomic status, nonwhite race, and smoking which are all risk factors for RA highly prevalent close to roadways, the authors did not adjust for these factors in statistical analyses. Nevertheless, there was little confounding due to neighborhood-level income, and smoking was unlikely to be an important confounder according to sensitivity analyses [10]. Additionally, Sigari et al. [13] recently found significantly elevated anticyclic citrullinated protein/peptide antibody (ACPA) levels in 56 wood smoke-induced COPD patients compared to 56 tobacco-induced COPD patients, and to 56 healthy controls. It is noteworthy that ACPA (the most specific biological markers with predictive and prognostic value in RA patients) are elevated in patients’ sera five to 10 years prior to diagnosis with RA [4, 11, 12, 14], suggesting that factors initiating autoimmunity in RA may act before appearance of symptoms and signs characteristic of clinical disease. In this sense, wood smoke might be a risk factor for RA. However, tropospheric pollutants including PM10, PM2.5, SO2, and NO2 were associated with RA neither in the NHS [9], nor in the BC study [10]. In the same way, the EIRA Swedish population-based study of 1497 incident RA patients compared with 2536 age and sex matched controls investigated the impact of an interquartile range increase (2 μg/m3 for PM10, 8 μg/m3 for SO2, and 9 μg/m3 for NO2) in each pollutant from traffic and home heating sources in the 5th, 10th, and 20th years prior to symptom onset (considering increment of ACPA titers five to 10 years before occurrence of clinical signs and symptoms of RA) and average exposure on the risk of all RA and the risk of RA serologic features. Total RA risks were increased for exposure to the gaseous pollutants (NO2 and SO2) in the 10th year before onset, but were no more statistically significant after adjustment for smoking and education (odds ratio [OR] =1.18, 95 % CI 0.97–1.43] and OR = 1.09, 95 % CI: 0.99–1.19) for SO2 and NO2 respectively). Stronger elevated risks at the same time point were noted for ACPA-negative RA cases even after adjustment for smoking and education (OR = 1.48, 95 % CI 1.13–1.95 and OR = 1.22, 95 % CI 1.07–1.40 for SO2 and NO2 respectively) [12]. Discrepant results about NO2 and SO2 between studies indicate the necessity to better clarify their role in the pathogenesis of RA [8, 10, 12], as well as that of PM which can be formed from these gases by enucleation in the air [7], and considering the high correlation between NO2 and PM10 [12]. Furthermore, results have not been verified across populations including different age groups, especially the ageing population which is more at risk of developing RA [1], and mixed ancestries. Moreover, a major shortcoming of the study by Hart el al. [9] which examined the association between specific pollutants and incident RA within the NHS was the lack of informations necessary for assessment of the impact of each nurses’ total air pollution exposure experience on the incidence of RA (amount of time spent at each home address, exposure to pollutants at locations other than the residence, exposure models that can perfectly predict personal exposures) [9]. In addition, the follow up period of the BC cohort was probably shorter than the induction period of tropospheric pollutants [10]. Taken together, sources of air pollution including traffic and probably solid fuels are associated with RA, but smoking may attenuate this association. Yet, inconclusiveness persists regarding the role of tropospheric pollutants including NO2, SO2 and PM. Indirect arguments can help support the role of these pollutants in the pathogenesis of RA.
Indirect evidence for a potential link between air pollution and rheumatoid arthritis
Despite sparse literature on air pollution and RA per se, tropospheric pollutants have repeatedly been associated with various systemic autoimmune rheumatic diseases (SARDs) [15–18]. In particular, PM2.5 levels 24 h–48 h before visits were significantly associated with anti-double stranded DNA serum specific autoantibodies in 237 Canadian patients followed up for systemic lupus erythematosus (SLE) during a time frame of seven years [15]. Likewise, a Brazilian study reported a link between SO2 exposures 14 days earlier and increased hospital admissions for pediatric autoimmune rheumatic diseases [16]. Moreover, Bernatsky et al. recently studied associations between local-scale ambient NO2 and PM2.5 in Calgary, Alberta-Canada, using Land Use Regression models (a preferred approach for exposure estimation including individual exposure estimation) and SARDs (SLE, Sjogren’s Syndrom, scleroderma, polymyositis, dermatomyositis, or undifferentiated connective tissue disease) [17]. Resultantly, ambient PM2.5 levels but not NO2 are strongly associated with SARDs in Alberta (OR adjusted for age, sex and income = 1.10, 95 % CI 1.01–1.22).
Based on the aforementioned studies and the small amount of data addressing air pollution in relation to RA together with extrapolations from relevant data in other areas, RA could be associated with PM, but also NO2, SO2 and O3 exposures as well. Indeed, PM pollutants are the most toxic tropospheric pollutants for all-cause morbidity and mortality associated with air pollutants [19–22]. In addition, they have been associated with other SARDs [15, 17]. Still, they can interact with gaseous pollutants that have been associated with RA [11], and even result from their transformation [22]. Most importantly, exposure to diesel exhaust particles (nano PM) during arthritis development exacerbated incidence and severity of RA in mice models of collagen-induced arthritis [23, 24]. However, NO2 and SO2 linked with RA in the EIRA study [11], and O3 linked with risk of RA in the BC study [10] all appear less toxic than PM and their respective concentrations highly depend on their complex interactions. Furthermore, NO2 may essentially serve as a proxy for PM, and O3 increases permeability of the respiratory epithelium to PM, hence exacerbating their actions [22].
Reports on the precise mechanistic link between these specific air pollutants and RA are somewhat conflicting. Anyhow, extrapolating from studies on air pollution and respiratory diseases [21, 22] as well as studies on air pollution and other SARDs [15–17] together with knowledge on mechanism of action of tropospheric pollutants [15–24], local lung and systemic oxidative stress and inflammation may be the central underlying mechanism [4, 18, 19, 25, 26]. In particular, free reactive oxygen species released by fine/ultrafine PM or gaseous pollutants inhaled in the respiratory tract are capable of activating nuclear factor ƙappa B (NF-ƙB), a key regulator for pro-inflammatory cytokine production in RA patients [18, 19, 25], leading to excess T helper lymphocyte type1 (Th1) production of tumor necrosis factor alpha, interleukin-1 and interleukin-6 [25]. These cytokines stimulate resting monocytes to mature dendritic cells which then present autoantigens, co-stimulating self-reactive T lymphocytes that migrate to target tissues (preferentially synovial joints), and cause destruction of cells expressing autoantigens [18]. Hence, there is persistent amplified chronic inflammation, a major prerequisite for protein bounded arginine citrullination and subsequent ACPA production that will later on lead to clinical signs of RA in genetically susceptible individuals [4, 26]. Moreover, reactive oxygen species per se are capable of worsening RA through induction of periarticular bone erosion [26].
Beyond oxidative stress, air pollution might further contribute to RA development and exacerbation through hypovitaminosis D [27–30]. Remarkably, the link between air pollution and low serum vitamin D has been investigated in astonishingly few cross-sectional studies [31–33]. Agarwal et al. [31] compared serum total 25-hydroxycholecalciferol (25-OH-D)-the best indicator of vitamin D storage- of 34 infants (aged 9–24 months) and toddlers in Mori Gate–a highly polluted area- with serum total 25-OH-D of a similar number of age and sex matched controls living in Gurgaon, a less polluted area. Mori Gate inhabitants had significantly lower mean serum 25-OH-D compared with Guargaon inhabitants (11.7 ± 7 ng/ml vs 27.1 ± 7 ng/ml, p < 0.001) [31]. Another study [32] evaluated the characteristics and prevalence of subjects with serum 25-OH- D less than 75 nmol/l among 85 postmenopausal women engaging in outdoor activities in either Brussels (urban highly polluted area) or the countryside (rural less polluted area) and recruited from the rheumatology outpatient clinic. Despite a higher sun exposure index (113 vs. 87, P < 0.001), city dwellers had a higher prevalence of serum 25-OH- D less than 75 nmol/l (84 % vs. 38 %) when compared with rural residents. Furthermore, Hosseinpanah et al. [33] studied the role of air population on serum total 25-OH-D in 200 healthy middle aged Iranian women divided into two groups with respect to the level of pollution in their residence: 100 women living in Tehran (highly polluted) and 100 women living in Ghazvin. After adjustment for important determinants of vitamin D status including age, body mass index, daily dietary vitamin D, calcium, phosphorus, protein, and the levels of sunlight exposure, living in higher polluted area was associated with lower levels of serum 25-OH-D [33]. Particularly relevant here, ~90 % of the body’s requisites for vitamin D are covered by skin synthesis in presence of sun’s ultraviolet B (UVB) radiations [33]. Also, 99 % of UVB radiations with wavelength 291–320 nm are absorbed by the ozone layer which is increased in situations of incremented air pollution levels [30]. Along with O3, NO2 and SO2 can absorb UVB in polluted atmosphere, thus reducing the effectiveness of sun exposure on skin synthesis of vitamin D [30]. Therefore, air pollution lowers serum vitamin D independently of sun exposure and other determinants of vitamin D status, probably through UVB radiations absorption by gaseous pollutants, and could thus increment RA incidence.
In fact, the active form of vitamin D, 1,25-dihydroxyvitamin D3 [1,25(OH)2D3], acts as an immunomodulator by binding to vitamin D receptor (VDR) besides its metabolic activities [27–30, 33]. Remarkably, 1,25(OH)2D3 is capable of inducing natural killer and T regulatory cells, thereby preventing autoimmunity [27]. Furthermore, 1,25 (OH)2D3 suppresses Th1 responses via interferon-γ inhibition, prevents monocyte differentiation into dendritic cells as well as antibody formation, and downregulates NF-ƙB [27].
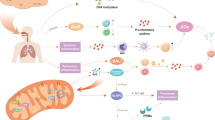
Altogether, local lung and systemic oxidative stress along with hypovitaminosis D may trigger RA in the context of air pollution (Fig. 1).

Schematic representation of mechanisms putatively influencing rheumatoid arthritis through air pollution exposure. Four main mechanistic pathways are represented (blue, brown, red, and green arrows). Blue arrows: free reactive oxygen species released by fine/ultrafine particulate matter (PM) inhaled in the respiratory activate nuclear factor ƙappa B (NF-ƙB) that stimulates the production of tumor necrosis factor alpha (TNF-α), interleukin 1 (IL1) and interleukin 6 (IL6) by T helper lymphocytes type 1 (Th1). These cytokines stimulate resting monocytes to mature dendritic cells which then present autoantigens, co-stimulating self-reactive T lymphocytes that migrate to target tissues (preferentially synovial joints), and cause destruction of cells expressing autoantigens and thereby joint inflammation and erosion. Brown arrows: free reactive oxygen species cause chronic lung and systemic inflammation that enhance citrullination of arginine amino acid residues into citrullinated proteins/peptides. These citrullinated products induce anticyclic citrullinated protein/peptide antibodies (ACPAs) production that will later on lead to immune reaction through binding to cellular Fc receptors and complement activation, and finally cause joint inflammation and erosion. Red arrow: reactive oxygen species per se can worsen joint inflammation and erosion. Green arrows: Reduced ultraviolet B (UVB) radiation levels lead to decreased skin synthesis of 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] which acts as an immunomodulator through vitamin D receptor (VDR) activation. Resultantly, immunomodulatory activities of 1,25(OH)2D3 represented by green doted arrows are not met, and RA can develop. IFN-γ interferon gamma; NK natural killer cells; and Treg T regulatory cells
Air pollution and rheumatoid arthritis with focus on low-and-middle income countries
Considering that majority of the world population resides in low-and-middle-income countries (LMICs), the number of affected people in this part of the world are projected to increase in the upcoming years. For instance, with an additional prevalence of 0.1 %, four million supplemental individuals might be affected [34]. Furthermore, a low socio-economic status has been associated with both increased risk of developing RA and increased mortality of RA patients [35]. Yet, RA is neglected in LMICs undoubtedly because of gaps in estimates of the burden of disease along with lack of communication strategies informing policy makers on the urgency of the problem [34].
As concerns sub-Saharan Africa, potential RA genetic background and environmental risk factors are quite different from those of other parts of the world despite an overall similar phenotype [4, 14, 36–38]. Notheworthy, smoking is not a risk factor for RA in this population [14, 36]. Hence, RA in Black Africans may be determined by specific genetic and environmental factors such as lower socio-economic status, and infections [4, 14, 37]. But the role of infections is controversial [14, 36], with parasitic infections suggested to protect against RA in Sub-Saharan Africa since they rage at endemic levels in that area [14, 39, 40]. Admittedly, parasites cannot resist in a milieu characterized by amplified immuno-inflammatory activation. Subsequently, the protection against RA is probably a consequence of defense mechanisms developed by parasites to survive in the host organism [39, 40]. Along this line, ES-62, a glycoprotein secreted by a filarial nematode called Acanthocheilonema viteae has been shown to restore interleukin-10 producing regulatory B cells (Breg) in collagen-induced arthritis mice models [39]. It is notable that IL-10 is an anti-inflammatory cytokine associated with downregulation of Th1/Th17 responses [39]. Therefore, it appears necessary to pursue research of risk factors for RA in sub-Saharan Africa, and air pollution could be a plausible, if not the most important candidate.
Household air pollution (HAP) defined as “air pollution generated by household fuel combustion, leading to indoor air pollution, and contributing to ambient air pollution” [41] is mostly prevalent in LMICs where nearly three million people still rely on solid fuels for heating, cooking and lighting [21, 41–43] resulting in approximately 4.3 million premature deaths worldwide [41]. Still, peak prevalences of HAP exposure worldwide occur in sub-Saharan Africa (77 % with the number of affected individuals almost doubled from 333 million to 646 million over three decades) and Southeast Asia (61 %) [21]. Of note, this prevalence also varies within countries, with up to 100 % households concerned in some rural areas [44]. Moreover, individuals with a lower socioeconomic status have an incremented risk of exposure to atmospheric pollution compared with individuals who have high incomes (differential exposure) [11, 45, 46]. Likewise, people with a lower socioeconomic status are at increased risk of developing air pollution-related health hazards compared with those who have a higher socioeconomic status (differential susceptibility) [11, 45, 46]. Taken collectively, poverty increases the risk of air pollution and exacerbates its consequences. Hence, Africa and Southeast Asia may carry the heaviest burden of air pollution.
RA has been associated with a lower socioeconomic status [5, 35] which may be a marker for residential exposure to air pollution [11]. Indeed, it is a confounder of the association between tropospheric pollutants and RA [9, 11]. In this light, results from the NHS which failed to demonstrate any association between RA and specific pollutants [9] are unlikely to be generalized in impoverished societies given the high economic status of the included nurses thus residing in less polluted areas [9], unlike populations from LMICs.
In total, the burden of RA and increased atmospheric pollution coincide in LMICs-especially in sub-Saharan Africa and Southeast Asia- with poverty raising the risk of air pollution and exacerbating air pollution. However, there is still a small amount of data on air pollution and RA coming from LMICs.
Conclusion
Traffic, a surrogate of air pollution, has been associated with incident RA. Whether this environmental factor is causally associated with RA across populations is still a matter of debate. Based on indirect arguments, as well as exacerbation of RA incidence and severity in mice models of collagen-induced arthritis after diesel exhaust particles exposure, local lung and systemic oxidative stress together with vitamin D deficiency-related autoimmunity may help to clarify a putative mechanistic link between tropospheric pollutants and RA. Additional well-designed epidemiological studies investigating different sources of air pollution and specific pollutants in relation to RA across populations, especially in LMICs are thus needed to better explore the biological plausibility of air pollutions’ effects in RA. If a causal relationship is confirmed, then health policies should be made to reduce the burden of RA.
Abbreviations
- 25-OH-D:
-
25-hydroxycholecalciferol
- 1,25(OH)2D3 :
-
1,25-dihydroxyvitamin D3
- ACPA:
-
Anticyclic citrullinated protein/peptide antibody
- BC:
-
British Colombian study
- COPD:
-
Chronic obstructive pulmonary disease
- EIRA:
-
Epidemiological Investigation of Rheumatoid Arthritis
- HAP:
-
Household air pollution
- HR:
-
Hazard ratio
- LMICs:
-
Low-and-middle-income countries
- NF-ƙB:
-
Nuclear factor ƙappa B
- NHS:
-
Nurses’ health study
- NO2 :
-
Nitrates
- OR:
-
Odds ratio
- O3 :
-
Ozone
- PM:
-
Particulate matters
- PM0.1 :
-
Particulate matter with an aerodynamic diameter less than 0.1 μ
- PM2.5 :
-
Particulate matter with an aerodynamic diameter less than 2.5 μ
- PM10 :
-
Particulate matter with an aerodynamic diameter less than 10 μ
- RA:
-
Rheumatoid arthritis
- SARDs:
-
Systemic autoimmune rheumatic diseases
- SLE:
-
Systemic lupus erythematosus
- SO2 :
-
Sulphur dioxide
- Th1:
-
T helper lymphocyte type 1
- UVB:
-
Ultraviolet B radiations
- VDR:
-
Vitamin D receptor
References
Cross M, Smith E, Hoy D, Carmona L, Wolfe F, Vos T, et al. The global burden of rheumatoid arthritis: estimates form the Global Burden of Disease 2010 study. Ann Rheum Dis. 2014;73:1316–22.
Dadoun S, Zeboulon-Ktorza N, Combescure C, Elhai M, Rozenberg S, Gossec L, et al. Mortality in rheumatoid arthritis over the last fifty years: systematic review and meta-analysis. Joint Bone Spine. 2013;80:29–33.
Aletaha D, Neogi T, Silman AJ, Funovtis J, Felson DT, Bingham 3rd CO, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis. 2010;69:1580–8.
Ruiz-Esquide V, Sanmartí R. Tobacco and other environmental risk factors in rheumatoid arthritis. Reumatol Clin. 2012;8(6):342–50.
Tobon GJ, Youinou P, Saraux A. The environment, geo epidemiology, and auto-immune disease: Rheumatoid arthritis. J Autoimmun. 2010;35(1):10–4.
Hoovestol RA, Mikuls TA. Envirronmental exposures and rheumatoid arthritis risk. Clin Rheumatol Rep. 2011;13:431–9.
Boama PO, Onumah J, Takase M, Bonsu PO, Salifu T. Air pollution control techniques. Global J Bio-Sci Biotechnol. 2012;1(2):124–31.
Hart JE, Laden F, Puett RC, Kostenbader KH, Karlson WE. Exposure to traffic pollution and increased risk of rheumatoid arthritis. Environ Health Perspect. 2009;117:1065–9.
Hart JE, Kalberg H, Laden F, Kostenbader KH, Yanosky JD, Klareskog R, et al. Ambient air pollution exposures and risk of rheumatoid arthritis in the Nurses’ Health Study. Arthritis Care Res (Hoboken). 2013;65(7):1190–6.
De Roos AJ, Koehoorn M, Tamburic L, Davies HW, Brauer M. Proximity to traffic, ambient air pollution, and community noise in relation to incident rheumatoid arthritis. Environ Health Perspect. 2014;122:1075–80.
Gan RW, Deane KD, Zerbe GO, Demoruelle MK, Weisman MH, Buckner JH, et al. Relationship between air pollution and positivity of RA-related autoantibodies in individuals without established RA: a report on SERA. Ann Rheum Dis. 2013;72(12):2002–5.
Hart JE, Källberg H, Laden F, Bellander T, Costenbader KH, Holmqvist M, et al. Ambient air pollution exposures and risk of rheumatoid arthritis: results from the Swedish EIRA case–control study. Ann Rheum Dis. 2013;72(6):888–94.
Sigari N, Moghimi N, Sharhaki SF, Mohammadi S, Roshani D. Anti‑cyclic citrullinated peptide (CCP) antibody in patients with wood‑smoke‑induced chronic obstructive pulmonary disease (COPD) without rheumatoid arthritis. Rheumatol Int. 2015;35:85–91.
Nogueira L, Cornillet M, Singwe-Ngandeu M, Viatte S, Bas S, Gabay C, et al. In Black Africans with rheumatoid arthritis, ACPA recognize citrullinated fibrinogen and the derived peptides α36-50Cit38,42 and β60-74Cit60,72,74, like in Caucasians. Clin Immunol. 2014;152:58–64.
Bernatsky S, Fournier M, Pineau CA, Clarke AE, Vinet E, Smargiassi A. Associations between ambient fine particulate levels and disease activity in patients with Systemic Lupus Erythematosus (SLE). Environ Health Perspect. 2011;119:45–9.
Vidotto JP, Pereira LA, Braga ALF, Silva CA, Sallum AM, Campos LM, et al. Atmospheric pollution: influence on hospital admissions in paediatric rheumatic diseases. Lupus. 2012;21:526–33.
E Bernatsky S, Smargiassi A, Johnson M, Kaplan GG, Barnabe C, Svenson L, et al. Fine particulate air pollution, nitrogen dioxide, and systemic auto- immune rheumatic disease in Calgary, Alberta. Environment Res. 2015;140:474–8.
Ritz SA. Air pollution as a potential contributor to the ‘epidemic’ of autoimmune disease. Med Hypotheses. 2010;74:110–7.
Farhat SCL, Silva CA, Orione MAM, Campos LMA, Sallum AME, Braga ALF. Air pollution in autoimmune rheumatic diseases: a review. Autoimmunity Rev. 2011;11:14–21.
Balti EV, Echouffo-Tcheugui JB, Yako YY, Kengne AP. Air pollution and risk of type 2 diabetes mellitus: a systematic review and meta-analysis. Diabetes Res Clin Pract. 2014;106:161–72.
Bonjour S, Adair-Rohani H, Wolf J, Bruce NG, Mehta S, Pru-ustun A, et al. Solid fuel use for household cooking: country and regional estimates for 1980–2010. Environ Health Perspect. 2013;121:784–90.
World Health Organization. Health Aspects of Air Pollution with Particulate Matter, Ozone and Nitrogen Dioxide. World Health Organization. Bonn, Germany; 2003. http://www.euro.who.int/__data/assets/pdf_file/0005/112199/E79097.pdf. Accessed 7 June 2015.
Yoshino S, Sagai M. Enhancement of collagen-induced arthritis in mice by diesel exhaust particles. J Pharmacol Exp Ther. 1999;290(2):524–9.
Yoshino S, Hayashi H, Taneda S, Sagai M, Mori Y. Effect of diesel exhaust particle extracts on collagen-induced arthritis in mice. Autoimmunity. 2002;35(1):57–61.
Ying G, Wang Y, Cen XM, Yang M, Liang Y, Xie QB. Lipid peroxidation-mediated inflammation promotes cell apoptosis through activation of NF-κB pathway in rheumatoid arthritis synovial cells. Mediators Inflamm. 2015;2015:460310.
Valesini G, Gerardi MC, Iannuccelli C, Pacucci VA, Pendolino M, Shoenfeld Y. Citrullination and autoimmunity. Autoimmunity Rev. 2015;14:490–7.
Song GG, Bae SC, Lee YH. Association between vitamin D intake and the risk of rheumatoid arthritis: a meta-analysis. Clin Rheumatol. 2012;31:1733–9.
Hiraki LT, Arkema EV, Cui J, Malspeis S, Costenbader KH, Karlson EW. Circulating 25-hydroxyvitamin D level and risk of developing rheumatoid arthritis. Rheumatology. 2014;53:2243–8.
Sahebari M, Mirfeizi Z, Rafatpanah H, Goshyeshi L. 25(OH) vitamin D serum values and rheumatoid arthritis disease activity (DA S28 ESR). Caspian J Intern Med. 2014;5(3):148–55.
Wacker M, Holick MF. Sunlight and Vitamin D: A global perspective for health. Dermatoendocrinol. 2013;5(1):51–108.
Agarwal KS, Mughal MZ, Upadhyay P, Berry JL, Mawer EB, Puliyel JM. The impact of atmospheric pollution on vitamin D status of infants and toddlers in Delhi. India Arch Dis Child. 2002;87(2):111–3.
Manicourt DH, Devogelaer JP. Urban tropospheric ozone increases the prevalence of vitamin D deficiency among Belgian postmenopausal women with outdoor activities during summer. J Clin Endocrinol Metab. 2008;93(10):3893–9.
Hosseinpanah F, Hashemi Pour S, Heibatollahi M, Moghbel N, Asefzade S, Azizi F. The effects of air pollution on vitamin D status in healthy women: A cross sectional study. BMC Public Health. 2010;10:519.
Rudan I, Sidhu S, Papana A, Meng S-J, Xin–Wei Y, Wang W, et al. Prevalence of rheumatoid arthritis in low– and middle–income countries: A systematic review and analysis. J Global Health. 2015;5(1):010409.
Ghawi H, Crowson CS, Rand-Weaver J, Krusemark E, Gabriel SE, Juhn YJ. A novel measure of socioeconomic status using individual housing data to assess the association of SES with rheumatoid arthritis and its mortality: a population-based case–control study. BMJ Open. 2015;5:e006469.
Malemba JJ, Mbuyi-Muamba JM, Mukaya J, Bossuyt X, Emonds MP, Deiteren K. The phenotype and genotype of rheumatoid arthritis in the Democratic Republic of Congo. Arthritis Res Ther. 2013;15:R89.
Jung KR, Choi S-E, Cha CH, Oh HB, Heo SH, Ahn HY, et al. Meta-analysis of the association between HLA-DRB1 allele and rheumatoid arthritis susceptibility in Asian populations. J Korean Med Sci. 2007;22:973–80.
Herraez DL, Martinez-Bueno M, Riba L, Gracia de la Torre I, Sacnum M, Goni M, et al. Rheumatoid Arthritis in Latin Americans Enriched for American Ancestry Is Associated with Loci in Chromosomes 1, 12, 13, and the HLA Class II Region. Arthritis Rheum. 2013;65(6):1457–67.
Rodgers DT, Pineda MA, McGrath MA, Al-Riyami LA, Harnett W, Harnett MM. Protection against collagen-induced arthritis in mice afforded by the parasitic worm product, ES-62, is associated with restoration of the levels of interleukin-10-producing B cells and reduced plasma cell infiltration of the joints. Immunology. 2013;141:457–66.
Rzepecka J, Pineda MA, Al-Riyami LA, Rodgers DT, Huggan JK, Lumb FE, et al. Prophylactic and therapeutic treatment with a synthetic analogue of a parasitic worm product prevents experimental arthritis and inhibits IL-1β production via NRF2-mediated counter-regulation of the Inflammasome. J Autoimmun. 2015;60:59–73.
World Health Organization. WHO indoor air quality guidelines: household fuel combustion. World Health Organization. Geneva, Switzerland; 2014. http://apps.who.int/iris/bitstream/10665/141496/1/9789241548885_eng.pdf. Accessed 07 June 2015.
Bruce N, Pope D, Rehfuess E, Balakrishnan K, Adair-Rohani H, Dora C. WHO indoor air quality guidelines on household fuel combustion: Strategy implications of new evidence on interventions and exposure-risk functions. Atmos Environ. 2015;106:451–7.
Rogalsky DK, Mendola P, Metts TA, Martin 2nd JW. Estimating the number of low-income Americans exposed to household air pollution from burning solid fuels. Environ Health Perspect. 2014;122(8):806–10.
Rehfuess EA, Bruce NG, Smith KR. Solid Fuel Use: Health Effect. In: Nriagu JO, editor. Encyclopedia of Environmental Health. Burlington: Elsevier; 2011. p. 150–61.
Samet JM, White RH. Urban air pollution, health, and equity. J Epidemiol Community Health. 2004;58:3–5.
Deguen S, Zmirou-Navier D. Social inequalities resulting from health risks related to ambient air quality-A European review. Eur J Public Health. 2010;20(1):27–35.
Acknowledgments
The authors are solely responsible for the manuscript preparation, for its intellectual content, and for the decision to submit it for publication. No author received a funding. The manuscript was not revised by any language editor.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ME conceived the plan of the review, drafted the manuscript, and revised it. JJNN was involved in drafting and revised the manuscript. All authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Essouma, M., Noubiap, J.J.N. Is air pollution a risk factor for rheumatoid arthritis?. J Inflamm 12, 48 (2015). https://doi.org/10.1186/s12950-015-0092-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12950-015-0092-1