
Abstract
Background
Several fusion oncogenes showing a higher incidence in pediatric acute myeloid leukemia (AML) are associated with heterogeneous megakaryoblastic and other myeloid features. Here we addressed how developmental mechanisms influence human leukemogenesis by ETO2::GLIS2, associated with dismal prognosis.
Methods
We created novel ETO2::GLIS2 models of leukemogenesis through lentiviral transduction and CRISPR-Cas9 gene editing of human fetal and post-natal hematopoietic stem/progenitor cells (HSPCs), performed in-depth characterization of ETO2::GLIS2 transformed cells through multiple omics and compared them to patient samples. This led to a preclinical assay using patient-derived-xenograft models to test a combination of two clinically-relevant molecules.
Results
We showed that ETO2::GLIS2 expression in primary human fetal CD34+ hematopoietic cells led to more efficient in vivo leukemia development than expression in post-natal cells. Moreover, cord blood-derived leukemogenesis has a major dependency on the presence of human cytokines, including IL3 and SCF. Single cell transcriptomes revealed that this cytokine environment controlled two ETO2::GLIS2-transformed states that were also observed in primary patient cells. Importantly, this cytokine sensitivity may be therapeutically-exploited as combined MEK and BCL2 inhibition showed higher efficiency than individual molecules to reduce leukemia progression in vivo.
Conclusions
Our study uncovers an interplay between the cytokine milieu and transcriptional programs that extends a developmental window of permissiveness to transformation by the ETO2::GLIS2 AML fusion oncogene, controls the intratumoral cellular heterogeneity, and offers a ground-breaking therapeutical opportunity by a targeted combination strategy.
Similar content being viewed by others


Introduction
Pediatric acute myeloid leukemia (AML) accounts for 20% of childhood leukemia patients [1]. As clearly demonstrated for pediatric acute lymphoblastic leukemia [2], some de novo pediatric AML mutations are also detected at birth [3,4,5,6]. Despite morphological and phenotypical similarities with adult AML, specific chromosomal alterations are strongly age-related, indicating differences in the underlying molecular bases [7, 8]. These associations could result from the unique properties of fetal hematopoiesis [9,10,11], providing a permissive ground for leukemia development in young patients.
Acute megakaryoblastic leukemia (AMKL) accounts for at least 10% of pediatric AML but only 1% of adult AML patients [12]. As compared to Down Syndrome patients who are predisposed to AMKL development [13], de novo pediatric AMKL patients harboring recurrent chromosomal abnormalities leading to oncogenic fusions involving transcription regulators are associated with poor prognosis [14, 15]. The most frequent alteration in de novo pediatric AMKL is the ETO2::GLIS2 (a.k.a. CBFA2T3::GLIS2) fusion (abbreviated here EG) resulting from a chromosome 16 inversion [inv(16)(p13.3q24.3)] in 20–25% of patients [16, 17]. These patients are characterized by a particularly dismal prognosis, a younger age at diagnosis compared to other AML subtypes, and a low mutational burden with only rare additional mutations [14,15,16]. Molecular studies on AMKL patient-derived blasts showed that EG provides a self-renewal vs differentiation imbalance through higher ERG and lower GATA1 activities [18, 19], an increased proliferation through activation of enhancers (e.g. KIT and PDGFRA) [20] and a dependence on BCL2 family members [21,22,23].
Several observations support that ontogeny plays a significant role in the development of pediatric AML. Indeed, some fusions like EG and NUP98::KDM5A are detected in AMKL or other AML subtypes with an age-dependent incidence [15, 24, 25]. Also, EG expression in fetal hematopoietic stem cells (HSC) and multipotent progenitors (MPP) rapidly induced a megakaryoblastic leukemia in animals, while expression in adult bone marrow (ABM) cells was associated with delayed leukemia development and a shift toward a more myeloid phenotype in an inducible transgenic murine model [15]. Therefore, expression in fetal hematopoietic cells is highly relevant to understand the mechanisms underlying leukemia initiation. While EG expression in human cord blood (CB) confirmed its capacity to induce transformation [22, 26], the specificities of its expression in human pre-natal cells, as recently modeled for Down syndrome AMKL-associated mutations [27], remain unknown.
Here, we engineered EG expression in primary human CD34+ hematopoietic cells from different ontogenic origins (fetal, newborn and adult) by lentiviral delivery and by precise CRISPR/Cas9 genome editing of the chromosome 16 inversion. We show that human fetal HSPCs are the most permissive to EG-driven leukemia development, with reproducible disease development in immune-deficient mouse models. While post-natal CB could also generate leukemia in vivo upon EG expression, adult cells were not permissive. Notably, CB-derived leukemogenesis depended on ectopic human cytokines expression in vivo, while fetal-derived leukemia efficiently emerged independently. Most importantly, targeting of downstream effectors, through combined inhibition of MEK and BCL2, provides a novel therapeutic perspective. Collectively, this study uncovers ontogenic differences for human pediatric AML fusion-driven leukemia development and an impact of the cytokine microenvironment on leukemia propagation/maintenance, which opens the avenue for novel therapies.
Material and methods
Cell manipulations
CD34+ cell isolation
Fetal livers (FL) were minced and gently smashed then filtered through a 70 µm cell strainer. The cartilaginous ends of the human fetal femurs were removed and the bone marrow was flushed with DPBS (Gibco) using a 18G gauge needle and a 10 mL syringe. Bones were cut in half and scratched to recover more hematopoietic mononucleated cells. Fetal Bone Marrow (FBM) cells were filtered using 70 μm cell strainers and washed in DPBS.
FL, FBM, umbilical cord blood (CB), adult bone marrow (ABM) and mobilized peripheral blood (mPB) cells were subjected to Ficoll gradient and enriched for CD34+ cells using the human CD34 MicroBeads Kit according to manufacturer instructions (Miltenyi Biotec). Purity (> 60%) of CD34+ cell suspensions was tested using immunostaining (anti-CD34 antibody, clone 581, BioLegend). Cells were used fresh or after freezing/thawing process in fetal bovine serum (FBS, Sigma) supplemented with 10% DMSO (Sigma). Only samples with ≥ 70% viable cells were used.
Lentiviral vector production and transduction
Concentrated lentiviral supernatants were generated and produced at the Genetic engineering and protein biochemistry platform of CEA/IRCM [28]. The lentiviral pTrip-ΔU3-MND-Luciferase-hPGK-eGFP and pTrip-ΔU3-MND-Luc/eGFP were used as control vectors and the pTrip-MND-HsETO2/GLIS2-hPGK-eGFP and pTrip-MND-EG/eGFP constructs were used as EG vectors [29].
Purified CD34+ cells (or hematopoietic subfractions, 1 × 106 cells/mL) were seeded in BIT 9500 Serum Substitute (StemCell Technologies) supplemented with 1% P/S, 100 ng/mL hSCF, 100 ng/mL hFLT3L, 60 ng/mL hIL3 and 200 ng/mL hTPO (all from Miltenyi Biotec) [28]. Transduction lasted for 2 to 3 days at 37°C 5% CO2 in presence of control and EG vectors (MOI = 10–20). Transduced cells were washed twice using DPBS before further usage. Cells were injected after 4–11 days cultures when performing xenotransplantation assays.
Generation and validation of inv(16) by CRISPR/Cas9
To reproduce the inv(16)(p13.3;q24.2) leading to the ETO2::GLIS2 fusion found in pediatric AML patients, freshly isolated CD34+ were CRISPR/Cas9-engineered using gRNA targeting ETO2 intron 11 and GLIS2 intron 2 (Table S1) to recreate the most common fusion [17]. Cells were cultured in transduction conditions for 24 h before electroporation, counted and 3 × 105 CD34+ cells were electroporated with the RNP/Cas9 complex using the 4D Nucleofector Amaxa technology (Lonza) using the gRNAETO2 and gRNAGLIS2 and the Cas9 protein (quantity ratio 2:1) as previously described in Babin et al., [30]. The gRNA design and off-target identification were determined using CRISPOR software (http://crispor.tefor.net). Primers and gRNA sequences are provided in Table S1. Serial dilutions of DNA from electroporated cells enabled the assessment of inv(16) frequency as previously described in Renouf et al., [31]. For the long-term follow-up of inv(16), DNA were isolated using a DNA Micro kit (Zymo) and quantified using a NanoDrop (ThermoScientific). PCR was performed using a GoTaq polymerase (Promega). The primer sequences are provided in Table S1.
FISH analyses of Inv(16) fusion
Cells were grown in complete medium for 24 h before addition of Colcemid (50-75µL for 10 mL of culture medium) for 45 min at 37°C before to be submitted to a hypotonic shock (KCL 0.075 M) at 37°C for 30 min. The fixation mixture (methanol / acetic acid, ratio 3:1) was gently added and cells were immediately spun down for 5 min at 1500 rpm, resuspended in 10 mL of fixation mixture and kept twice for 15 min at room temperature before spinning down and either directly spread on slides or kept several days at 4°C before deposition on slides. FISH analysis was performed on chromosome preparations, using the GLIS2/CBFA2T3 Dual Fusion OR-GR FISH probe (Empire Genomics, two probes, one for GLIS2 gene labeled with an orange fluorochrome, and one for CBFA2T3 gene labeled with FITC). The protocol of hybridization was performed according to the manufacturer’s instructions. Denaturation was done at 73°C during two minutes. The nuclei pictures were automatically detected on the fully motorized ZEISS Axio Imager.Z2 light microscope, equipped with a motorized stage high-resolution CCD camera. A total of at least 50 interphase nuclei were scored, using the Metafer software (MetaSystems).
Long-term liquid culture (LT-LC)
Transduced or CRISPR-engineered cells were seeded in IMDM + Glutamax supplemented with 15% FBS and 1% P/S completed with 100 ng/mL hSCF, 100 ng/mL hFLT3L, 60 ng/mL hIL3, 200 ng/mL hTPO and 2 U/mL of hEPO (PeproTech) at 1–2 × 105 cells/mL to promote myeloid/erythroid/megakaryocytic differentiation. Cells were counted, replated and immunophenotyped weekly. M07e cells were seeded in liquid culture (2–5 × 105 cells/mL) in RPMI 1640 (Gibco) supplemented with 10% FBS and 1% P/S/G (Glutamine, Gibco) completed with 100 ng/mL hSCF and/or 60 ng/mL hIL3 (Miltenyi Biotec).
Colony forming units assay
Transduced cells were plated at 5 × 102 cells/35mm2 dish in methylcellulose StemMACS HSC-CFU medium (Miltenyi Biotec) completed with 20 ng/mL hTPO. Colonies were scored after 12–14 days and 5 × 104 total cells were replated every 14 days to assess self-renewal capacities in vitro. At each replating, cells were immunophenotyped by flow cytometry.
Xenotransplantation
8 to 12-week-old NSG and NSGS females were sublethally irradiated at 2 and 1.5 Gy respectively, before intra-venous retro-orbital cell transplant under isoflurane anesthesia. NBSGW mice were not irradiated, accordingly to McIntosh et al., [32]. Mice received 0.1–5 × 105 GFP+-transduced cells and 0.35–5.106 gene-edited cells depending on the experiments and samples. Mice were monitored daily and euthanized when exhibiting disease symptoms (hind limbs paralysis, associated with anemia and loss of weight) or > 1 year after transplant if not sick to explore human hematopoietic cell development. Tibiae/femora (4 long bones, BM) and vertebral columns (VC) were harvested and hematopoietic cells were recovered from flushing BM with DPBS using a 26G gauge needle or gently crushing the VC. The isolated cells were kept at 4°C on ice during the process. Serial transplantation was carried out after flow cytometry analysis, by intra-venous injection of 1 × 106 hCD45+(GFP+) cells (1 × 105 hCD45+GFP+ cells for NSG vs NSGS experiments) recovered from primary recipient mice into secondary sublethally irradiated animals.
Flow cytometry
Cells were stained with human specific antibodies purchased from BD Biosciences, BioLegend or eBioscience (Table S2, dilution 1:100 except when indicated), in DPBS for 15-30 min at 4°C. Labelled cells were analyzed on a FACSCanto™II or LSR™II using the FACS Diva software (BD Biosciences). Compensation controls were performed using single-stained compensation beads (UltraComp eBeads™ Plus Compensation Beads, Invitrogen). After acquisition, live cell analysis was done using FlowJo™ v10, excluding debris and doublets using forward and light scatter and dead cells by exclusion of the Zombie Aqua™ Viability Dye (1:500, BioLegend).
Cell proliferation assay
Cell proliferation was assessed using the CellTrace™ Violet Cell Proliferation kit (Invitrogen), following manufacturer instructions. Briefly, control or EG-transduced cells were cultured at 1 × 106 cells/mL in DPBS supplemented with CellTrace™ Violet dye (5 µM), and incubated for 20 min at 37°C in the dark. Cells were washed with five times the original staining volume of medium with 10% FBS and incubated in fresh pre-warmed complete LT-LC medium (see before). Cell proliferation was measured 2 h after staining (Day 0) and after 4 days of culture (Day 4) and the CTV staining dilution was assessed. Day 0 and day 4 time points corresponded to day 4 and day 9 of LT-LC post-transduction.
Cell treatment and viability assay
Control and EG-transduced cells were plated in 12-well plates (5 × 105 cells per well) and M07e cells in 48-well plates (5 × 104 cells per well) in defined LT-LC culture media supplemented or not with BCL2 inhibitor Venetoclax (0.1 µM, MedChemExpress) and/or MEK inhibitor Trametinib (0.1 µM, MedChemExpress) during 4 days. Cells were counted every day of the culture period. Cell viability was measured after 48 h of treatment using an Annexin V Staining Protocol (eBioscience) with some modifications. Briefly, cells were washed with DPBS, spun down and washed with Annexin V-binding buffer 1X (eBioscience). Up to 3 × 105 cells were suspended in 100 µL binding buffer and stained with Annexin V – APC (1:50, BioLegend) for 15 min at room temperature. Cells were washed and incubated in 200µL of the binding buffer. 100µL of Hoechst 33258 fluorescent nuclear DNA stain (Invitrogen) was added just before flow cytometry analysis at a final concentration of 4 µg/ml. Cells were analyzed on a FACSCanto™II or LSR™II with the FACS Diva software within 4 h.
Cytospin preparation and coloration
Cytospins were prepared using the Shandon Cytospin 4 cytocentrifuge (ThermoFisher). ~ 50 000 cells were spun down at 700 rpm during 5 min. Air-dried slides were then manually colored using the May-Grünwald Giemsa coloration method. Briefly, slides were subjected to 2 min of a May-Grünwald solution bath, 2 min of a May-Grünwald / Buffer Solution (ratio 1:1) bath, 14 min of a Giemsa / Buffer Solution (ratio 3:11) bath and 1 min of the Buffer Solution bath (all solutions from Sysmex), before drying and covering with a cover-slip. Colored cytospins were photographed using the Olympus BX51 microscope (Olympus Life Science).
Murine preclinical models
Xenograft of primary human PDX-Luc+ samples was done as described previously [17]. PDX-Luc+ cells were transduced with the FUW-Luc-mCherry-puro lentiviral construct (kindly provided by Dr A Kung, DFCI). In short, 5 × 104 PDX-Luc+ cells were intravenously injected into mice. After leukemia infiltration measurement by bioluminescence imaging (IVIS Lumina III In Vivo Imaging System, PerkinElmer), mice were randomly assigned to groups and treated for 4 weeks with vehicle, Trametinib (1 mg/kg) or Venetoclax (100 mg/kg) alone or in combination by oral gavage 5 times per week. Mice were regularly monitored by IVIS imaging. Mice were followed for survival and sacrificed when disease symptoms were observed.
Molecular analyses
RNA extraction & quantitative RT-PCR analysis
mRNA was isolated using a RNeasy Mini/Micro kit (Qiagen) and quantified using a NanoDrop. Reverse transcription was carried out with SuperScript II (Invitrogen). Q-PCR was performed with SYBR Select Master Mix (Applied Biosystems) using a 7500HT Fast Real-Time PCR System (Applied Biosystems) according to the manufacturer's recommendations. The primer sequences for analyzed genes are provided in Table S1. Raw data were obtained in terms of Ct values and normalized to the Ct values of the housekeeping gene RPLPO.
ATAC sequencing
ATACseq was performed as previously described [33] with some modifications. Briefly, 5 × 104–1 × 105 GFP+ live-sorted cells were spun at 500 g for 5 min, washed with cold DPBS, lysed in cold lysis buffer (10 mM Tris–HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, and 0.1% NP40, 3 min on ice) and immediately spun at 500 g for 10 min. The pelleted nuclei were resuspended in 50 µL of transposase reaction mix (Tagment DNA TDE1 Enzyme and Buffer Kit, Illumina) for 30 min at 39°C. Transposed DNA was purified using a MinElute PCR purification kit (Qiagen) in 15µL of nuclease-free H2O and amplified with NEBNext® Ultra™ II Q5® Master Mix and Nextera XT DNA Library Preparation Kit (Illumina), using the following PCR conditions: 72°C for 5 min, 98°C for 30 s, then 12 cycles of 98°C for 10 s, 63°C for 30 s, and 72°C for 1 min. Libraries were purified with AMPure XP beads (Beckman Coulter) and then subjected to high-throughput paired-end sequencing (50 bp) using the Illumina HiSeq 4000 sequencer (Illumina). Bigwigs files and PCA representations as well as differential analysis were generated using the nfcore pipeline for ATACseq (nf-core/atacseq v1.2.1) with default parameters (with genome hg38). Only differential peaks associated to an FDR lower than 1% and an absolute value of LFC2 (log2 Fold Change) greater than 1 were considered for subsequent analysis of the differential results. Annotations and motifs analysis (with default parameters) were performed with Homer (v4.11). Heatmaps of interest genomic regions were performed with Deeptools (v3.4.0).
RNA sequencing
RNA from FL or CB CD34+ cells subjected to lentiviral expression of EG, selection in vitro or in vivo and GFP+-sorted was extracted using the RNeasy Plus Mini Kit (Qiagen). Poly(A)-selected, first-stranded Illumina libraries were prepared with a modified TruSeq protocol using deoxyuridine triphosphate (dUTP) method [34]. Three biological replicates per cell type were prepared. AMPure XP size–selected libraries were amplified by PCR (maximum 16 cycles), purified with AMPure XP beads, and paired-end–sequenced (50 bp) on the Illumina HiSeq 4000 sequencer. A first assessment of the sequencing reads quality was performed using fastqc (v0.11.9) and an overview of the results obtained for all samples was obtained with multiqc (v1.10.1). Sequencing reads were trimmed and low-quality reads were removed with trim-galore (v0.6.6 with parameters –phred33 –fastqc –quality 20 –paired). In order to keep only human reads and avoid murine reads (essentially for samples coming from NSG mice) the program Xenome (v1.0.0) was used with parameters –pairs –host-name mouse –graft-name human to obtain new fastqs containing only reads considered as human reads. The index used to run Xenome was obtained with the following command: xenome index -T $nbthread -P human_graft_mouse_host -CONTROL $pathgenomemurin -G $pathgenomehuman -K 25 –verbose. The value associated to the parameter K is the k-mer size (25 is the default value).The file used for the human genome was Homo_sapiens.GRCh38.dna.primary_assembly.fa (GRCh38—release 106) while the file used for the murine genome was Mus_musculus.GRCm39.dna.primary_assembly.fa (release 106). Then fastqs corresponding to reads considered as human reads were used for quantification. This step was performed with Salmon (v0.14.2). The index used by Salmon to do the quantification of each fastqs was generated by running: salmon index -t gentrome.fa -i human_97_ensembl_salmon_0142_decoys_index –decoys decoys.txt -k 31 (where the parameter k corresponds to the k-mer size). The decoy transcriptome was generated using the script generateDecoyTranscriptome.sh available at https://github.com/COMBINE-lab/SalmonTools/blob/master/scripts/ (files used to generate the decoy transcriptome are genome: https://ftp.ensembl.org/pub/release-97/fasta/homo_sapiens/dna/; transcriptome: https://ftp.ensembl.org/pub/release-97/fasta/homo_sapiens/cdna/; gtf: https://ftp.ensembl.org/pub/release-97/gtf/homo_sapiens/).
The quantification of reads for each fastq pairs was performed using the command salmon quant (with parameters –seqBias –gcBias -l A –validateMappings). Subsequent steps of analysis were performed using the R software (v4.2.1). First, an R TxDB object was generated using the function makeTxDbFromGFF of the R package GenomicFeatures (v1.48.4). The adress used as value of the argument file of this function was: ftp://ftp.ensembl.org/pub/release-97/gtf/homo_sapiens/. The function tximport of the R package tximport (v1.24.0) was used to import transcript-level estimates and summarizes abundances, counts and transcript length to the gene level (with arguments type = “salmon”, txOut = FALSE, tx2gene = tx2gene, ignoreTxVersion = TRUE with tx2gene corresponding to the TxDB object previously generated). An R object of class DESeqDataSet was generated using the function DESEqDataSetFrom Tximport of the R package DESeq2 (v1.36.0). For further analyses (e.g. the clustering in Fig. 2) only genes associated with 10 or more reads were kept (rowSums(counts(dds)) ≥ 10), resulting in a matrix of 20,745 transcripts. Size factors were computed using the function estimateSizeFactors of DESeq2. The vst (variance stabilizing transformation) normalization method was then applied to the count matrix using the vst function (with argument blind equal to FALSE). The PCA of the normalized count matrix was performed using the function PCA of the package FactoMineR (v2.6).This matrix was also used for violinplot and heatmap representations of the expression of genes of interest as well as for GSEA analysis. Differential expression analysis was performed with the function DESeq (with default parameters). The function lfcShrink (the value of the parameter type was apeglm) was employed to get estimation of the LFC2 and of the associated posterior standard deviation (called lfcSE) for each gene between conditions. GSEA analysis were performed with the GSEA software (v4.1.0). The differentially expressed genes (filtered on fold change ≥ 2 and p-value ≤ 0.05) between FL CTRL and FL EG or between CB CTRL and CB EG or between CB EG and FL EG are provided in Supplementary Tables 3–5.
Single Cell RNA sequencing
The pipeline of Gustave Roussy (https://github.com/gustaveroussy/single-cell; v1.3) was used to handle single cell analysis.
First, each sample was processed with the following method. Raw BCL-files were demultiplexed and converted to Fastq format using bcl2fastq (v2.20.0.422 from Illumina). Reads quality control was performed using fastqc (v0.11.9) and assignment to the expected genome species evaluated with fastq-screen (v0.14.0). Reads were pseudo-mapped to the Ensembl reference transcriptome v99 corresponding to the homo sapiens GRCH38 build with kallisto (v0.46.2) using its 'bus' subcommand and parameters corresponding to the 10X Chromium 3' scRNA-Seq v3 chemistry. The index was made with the kb-python (v0.24.4) wrapper of kallisto. Barcode correction using whitelist provided by the manufacturer (10X Genomics) and gene-based reads quantification was performed with BUStools (v0.40.0). Cell barcode by symbol count table were loaded in R (v4.1.0) using the BUSpaRse package (v1.7.1). To call real cells from empty droplets, we used the emptyDrops() function from the dropletUtils package (v1.12.2), which assesses whether the RNA content associated with a cell barcode is significantly distinct from the ambient background RNA present within each sample. Barcodes with p-values < 0.001 (Benjamini-Hochberg-corrected) were considered as legitimate cells for further analysis. The count matrix was filtered to exclude genes detected in less than 5 cells, cells with less than 1000 UMIs or less than 200 detected genes, as well as cells with mitochondrial transcripts proportion higher than 20%. The proportion of ribosomal gene counts was also estimated but not used to filter cells. Cell cycle scoring of each cell was performed using two methods: the CellcycleScoring() function from the Seurat package (v4.0.4), and the cyclone() function from Scran (v1.20.1). Barcodes corresponding to doublet cells were identified and discarded using the union of two methods: scDblFinder (v1.6.0) using default parameters, and scds (v1.8.0) with its hybrid method using default parameters. We manually verified that the cells identified as doublets did not systematically correspond to cells in G2M phase. Seurat (v4.0.4) was applied for further data processing. The SCTransform normalization method [35] was used to normalize, scale, select 3000 Highly Variable Genes. The number of PCA dimensions to keep for further analysis was evaluated by assessing a range of reduced PCA spaces using 3 to 90 dimensions, with a step of 2. For each generated PCA space, Louvain clustering of cells was performed using a range of values for the resolution parameter from 0.1 to 1.6 with a step of 0.1. The optimal space was manually evaluated as the one combination of kept dimensions and clustering resolution resolving the best structure (clusters homogeneity and compacity) in a Uniform Manifold Approximation and Projection space (UMAP). Additionally, we used the clustree method (v0.4.3) to assess if the selected optimal space corresponded to a relatively stable position in the clustering results tested for these dimensions / resolution combinations.
Dimensionality and resolution (for clustering) chosen for each sample:
-
FL EG NSG : 85 ; 1.4
-
FBM EG NSG : 73 ; 1.3
-
CB EG NSGS : 85 ; 0.1
-
FL EG NSGS : 89 ; 1.6
-
FL EG D7 : 87 ; 1.0
-
CB EG D7 : 77 ; 1.3
-
Patient EG : 73 ; 1.4
The integration of all the samples was performed as follows. Datasets were integrated using the Harmony method. Datasets were merged by the merge() function from Seurat (v4.0.4), and the SCTransform normalization method [35] was used to normalize, scale, select 3000 Highly Variable Genes. The reduced PCA spaces are used as input for the HarmonyMatrix() function implemented in Harmony package (v0.1.0) where the batch effect (orig.ident: CB_EG_J7_GE, CB_EG_NSG_S_GE, CONECT_110_GE, FBM_EG_GE, FF_EG_GE, FL_EG_J7_GE, NSG_S_FLEG_GE) was regressed. The batch-corrected shared space output by Harmony is used for clustering. The number of Harmony dimensions to keep for further analysis was evaluated by assessing a range of reduced Harmony spaces using 3 to 90 dimensions, with a step of 2. For each generated Harmony space, Louvain clustering of cells was performed using a range of values for the resolution parameter from 0.1 to 1.5 with a step of 0.1. The optimal space was manually evaluated as the one combination that kept dimensions and clustering resolution resolving the best structure (clusters homogeneity and compacity) in a Uniform Manifold Approximation and Projection space (UMAP). Additionally, we used the clustree method (v0.4.3) to assess if the selected optimal space corresponded to a relatively stable position in the clustering results tested for these dimensions / resolution combinations.
Assessment of the enrichment of set of genes of interest (for example ETO2::GLIS2 signature) was performed with the method described in Arkoun et al., [36]. Differential analysis to compare gene expression between clusters were performed with the function FindMarkers (default options) of Seurat.
Statistical analysis
Statistical analyses were performed with GraphPad Prism 9.0. Data were analyzed by Mann–Whitney or Kruskal–Wallis tests followed by a multiple comparison test (as indicated).
For xenotransplantation analysis, Kaplan–Meier survival curves were performed and the statistical significance was determined with the Log-rank test. Overall survival was defined as the time from cell injection to euthanasia or death at predefined endpoints. Animals were censored if the cause of death/sacrifice was not leukemia related.
Data are presented as mean ± SEM for in vitro studies, and as medians for in vivo experiments. Results were considered statistically significant when p < 0.05. p values are indicated by "ns" when not significant, * when p ≤ 0.05, ** when p ≤ 0.01, *** when p ≤ 0.001 and **** when p ≤ 0.0001.
Results
ETO2::GLIS2 transforms fetal liver and cord blood, but not adult, CD34+ HSPCs in vitro
To investigate the consequences of ETO2::GLIS2 (EG) on hematopoietic cells from different ontogenic stages, primary human CD34+ cells were isolated from fetal liver (FL; 8–24 weeks of gestation) and bone marrow (FBM; 17–24 weeks of gestation), early post-natal CB and ABM/mobilized peripheral blood (ABM/mPB). Cells were transduced with lentiviral vectors encoding EG and the GFP fluorescent reporter or GFP alone (CTL) and cultured with human FLT3L, SCF, TPO, IL3 and EPO for 6 weeks (Fig. 1A-B, Fig. S1A-C). EG-transduced FL, FBM and CB cells, but not ABM/mPB cells, progressively outgrew control-transduced cells (Fig. 1B, Fig. S1C). The EG-driven enhanced growth was accompanied by the progressive apparition and selection of abnormal CD33+KIT+ cells expressing different levels of CD56 and CD41 during cultures (Fig. 1C). Similar results were obtained with FL CD34+ cells in which the most prevalent inv(16)(p13.3q24.3) found in patients [16, 17, 24] was reproduced by CRISPR/Cas9 gene-editing with an estimated frequency of 1/48 to 1/107 edited cell (Fig. 1D, Fig. S1D). Indeed, inv(16) FL cells bearing the fusion transcript (Fig. S1E) outgrew the control cells (Fig. 1E) and the selected cells also expressed CD33, KIT, CD56 and CD41 surface markers (Fig. 1F). The proliferative advantage of EG-transduced cells took place early in culture as CellTrace Violet staining loss from day 4 to day 9 post-transduction was higher in FL EG compared to FL control cells (Fig. S1F). Of note, the morphology of cells indicated that a myelo-monocytic differentiation was taking place in FL control condition, whereas EG-expressing FL cells were homogenously smaller monomorphic blast cells (Fig. 1G).

ETO2::GLIS2 induces AMKL features in fetal liver and bone marrow, cord blood but not adult HSPCs. A Experimental design to address the impact of the ETO2::GLIS2 (EG) fusion gene on fetal, early post-natal and adult HSPCs. CD34+ cells were isolated from fetal liver (FL), fetal bone marrow (FBM), cord blood (CB), allogenic bone marrow (ABM) and mobilized peripheral blood (mPB) mononuclear cells. EG expression was induced by lentiviral vector delivery (GFP+ cells) or engineered from the endogenous loci by CRISPR/Cas9. Control (CTL) and EG-expressing cells were tested in long-term liquid culture (LT-LC) and colony forming unit assay (CFU), and transcriptomic analysis was performed at different stages of LT-LC. B Cumulative number of total cells (upper panels) and percentage of GFP+ cells (lower panels) of CTL and EG-transduced FL (n = 9 samples, ≥ 3 biological replicates/time point, except %GFP in CTL, 42 days, n = 1), FBM (n = 4 samples, ≥ 2 biological replicates/time point, except %GFP in CTL, 35 days, n = 1), CB (n = 9, ≥ 5 biological replicates/time point), ABM (n = 2, 2 biological replicates/time point, except 7, 28, 42 days, n = 1) and mPB (n = 2, 2 biological replicates/time point) CD34+ cells during LT-LC. Mean ± SEM of biological replicates is shown. C Representative flow cytometry analysis of CD33, KIT, CD56 and CD41 expression in EG and CTL GFP+ FL, FBM and CB cells at day 21 of LT-LC. CD56 and CD41 expression are gated in CD33+KIT+ cells (D) EG fusion frequency assessed by PCR amplification of EG derivative at 2 days (Donor (FL) 1) and 3 days (Donor (FL) 2) after electroporation. PCR were performed in duplicates on DNA dilutions starting from 50 ng DNA (≈ 8 000 cells) to 0.3 ng DNA (≈ 50 cells) [30]. E Cumulative number of CD33+KIT+ cells along culture of CRISPR-edited inv(16) FL CD34+ cells. Mean of technical triplicates is shown for each FL donor (CTL n = 1, EG n = 2). F Representative flow cytometry analysis of CD33, KIT, CD56 and CD41 expression on CRISPR-edited inv(16) FL EG CD34+ cells at day 40–44 of LT-LC. CD56 and CD41 expression are gated in CD33+KIT+ cells (G) May-Grünwald Giemsa stained cytospins from FL cultures (CTL and EG-transduced and EG-engineered cells) at day 20 of LT-LC. Magnification: × 40. H Colony potential assessment upon culture of 500 (P1, CD34+) or 50,000 (P2-4) CTL and EG-FL (n = 8), FBM (n = 3), CB (n = 4) and ABM (n = 2) cells in semi-solid methylcellulose culture medium. Serial replating was performed every 12 to 14 days. P1: first passage, P2: secondary, P3: tertiary, P4: quaternary. Mean ± SEM of biological replicates is shown. Statistical significance is indicated as p values (B and H: Mann–Whitney non parametric test). *: p < 0.05, **: p < 0.01
To further investigate the early functional impact of EG in human HSPCs, we plated EG-transduced and CRISPR-ed CD34+ cells from FL, CB and ABM origin in clonogenic semi-solid medium and assessed their colony-forming unit (CFU) capacity. While EG did not impact the number of CFU at the first passage (P1), it maintained CFU formation in FL, FBM and CB progenitors over 4 passages compared to CTL cells. In contrast, EG did not perpetuate CFU ability of ABM cells (Fig. 1H).
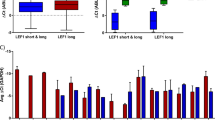
Previous studies associated EG with a distinct gene expression signature, including a high ERG, KIT and NCAM1 (encoding CD56) but low GATA1 and SPI1 expression [15, 17, 18, 26]. We compared expression of selected genes by quantitative RT-PCR analyses in sorted control GFP+ cells at day 0 and EG-selected abnormal cells between day 41 and 75 of liquid culture, when cells were ≥ 90% GFP+, in both fetal and CB contexts. In FL and FBM cells, both lentivirally-transduced and CRISPR-edited EG cells showed higher ERG and lower GATA1 expression compared to parental control cells (Fig. 2A). However, CB EG cells showed no significant difference in ERG and GATA1 expression compared to parental control cells (Fig. 2A). We then performed RNAseq analyses to investigate more globally the gene expression signature (Fig. 2B). Confirming quantitative RT-PCR data, ERG, GATA1 and SPI1 expression were more drastically altered in FL cells (Fig. 2C). Gene Set Enrichment Analysis (GSEA), using three independent gene signatures more highly expressed in EG patients [8, 16, 37], showed an enrichment in both CB EG vs CB CTL and FL EG vs FL CTL (Fig. 2D), supporting that EG alters gene expression in both CB and FL cells. Importantly, the signatures were also significantly enriched in FL EG vs CB EG (Fig. 2D-E), supporting that FL EG cells more closely resemble patient leukemic cells than CB EG cells. Overall, the results show that the sole expression of EG in HSPCs from fetal and CB origin selects for abnormal cells with enhanced growth in vitro that exhibit transcriptional features found in patients' leukemic cells.

ETO2::GLIS2 differentially modifies the gene expression profile of fetal liver and cord blood HSPCs in long-term liquid culture. A Quantitative RT-PCR expression of EG, ERG and GATA1. Upper panel: control (CTL) and EG-transduced CD34+ FL cells cultured for 22 days. Middle panel: CRISPR-edited CD34+ FL cells cultured for 21 and 42 days. Lower panel: CTL and EG-transduced CD34+ CB cells cultured for 47 days. Mean ± SEM of technical triplicates is shown (n = 1). B Clustering of RNAseq data obtained from EG-transduced FL and CB CD34+ cells after 37–78 days of culture. Control (CTL) cells are 0–3 days cultured CD34+ FL or CB cells. C Violin plot representation of gene expression extracted from the RNAseq normalized expression table. Same conditions as in (B). D Gene Set Enrichment Analysis (GSEA) using EG patient highly expressed gene signatures from 3 independent expression analysis [8, 16, 37]. The compared conditions are indicated on each side of the panel, respectively. Same conditions as in (B). E Within genes common to at least two of the three patients signature lists indicated in (D), the heatmap represents those that are more highly expressed in EG FL vs EG CB cells
ETO2::GLIS2-expressing fetal, but not CB, HSPCs efficiently induce leukemia in NSG mice
We next evaluated the in vivo transforming potential of EG expression by transplanting control and EG-transduced FL, FBM, and CB CD34+ cells, as well as CD34+ CRISPR/Cas9-edited for EG, in immune deficient NSG mice (Fig. 3A). Transduced (14–60% GFP+) or EG-CRISPR-ed (1/48–1/107 edited cells in FL and 1/120 in FBM; 1/96 edited cells in CB) cells were injected in mice and animals were carefully monitored for disease onset. Strikingly, mice injected with FL and FBM EG-expressing cells developed a lethal disease with a respective penetrance of 17/26 (65%) and 5/8 (63%) when injected with EG-transduced cells, and 7/11 (64%) and 6/7 (86%) respectively when injected with CRISPR-edited EG cells (Fig. 3B). Overall, median survival of FL EG-injected mice was 141 days for EG-transduced cells (n = 26 mice, in 9 biological replicates) and 302 days for CRISPR-edited EG cells (n = 11, in 3 biological replicates). In contrast, only 4/35 (11%) mice injected with EG-transduced (4/31 mice) and CRISPR-edited (0/4 mice) CB HSPCs developed symptoms of disease (Fig. 3B, lower panel), indicating major differences in EG-driven disease onset in vivo between fetal and CB cells. The most common symptoms observed in FL and FBM EG-injected mice were weight loss and hind limb paralysis (Fig. S2A). Careful analyses showed that the mice consistently had long bone and vertebral column BM infiltration of human CD45+ cells (Fig. 3C, Fig. S2B) along with variable spleen and liver invasion (not shown). Some mice showed low percentages of abnormal cells, consistent with the low number of blood blasts reported in AMKL and EG-expressing AMKL patients [14, 38,39,40]. Histological analyses revealed meningeal and nervous infiltration in the spinal cord that were associated with brain spongiosis and multifocal hemorrhages (Figure S2C), in accordance with reported CNS involvement in PDX models and in some patients [17, 39]. Phenotypically, human FL EG grafts were homogeneously constituted by CD33+KIT+ abnormal cells carrying variable levels of CD56 and CD41 expression, in line with the in vitro cultures (Fig. 3D-E, Fig. S2D). In the case of CB grafts, human EG GFP+ cells were detected in 12/31 mice, even though 22/31 mice carried human CD45+ cells (> 1%) in their long bones (Fig. 3C) and vertebral column (Fig. S2B) BM. Of these 12 CB EG cell engrafted mice, only 6 mice (19% of total mice) exhibited cells with substantial levels of the abnormal markers (Fig. 3E, Fig. S2D). Besides, many CB EG-transplanted mice carried human GFP+/GFP− cells with normal hematopoietic B, T and myeloid lineage cell markers, as found in control mice (Fig. S3A-B). We validated the CRISPR editing by FISH analysis, finding > 90% and > 98% of positive cells for EG fusion in the spleen and vertebral column from a sick mouse injected with FL EG-engineered CD34+ cells (Fig. 3F).

ETO2::GLIS2 expression induces leukemic transformation in FL HSPCs in vivo. A Experimental design of in vivo experiments. EG-expressing FL, FBM and CB HSPCs (via lentiviral vectors delivery or CRISPR/Cas9-editing) were injected into NSG mouse strains following 2–11 days of culture. Mice were monitored daily and sacrificed when exhibiting disease symptoms. Recovered cells from primary (1ry) recipients were serially transplanted into secondary (2ry) and tertiary (3ry) recipients. Phenotypic and transcriptomic analysis were performed on the recovered cells and murine organs were assessed by histological analysis. B Kaplan–Meier survival plot of 1ry recipients injected with FL, FBM and CB CTL vs EG cells. Median survival of mice with FL EG-transduced and CRISPR-edited cells are 141 days and 302 days respectively, 275.5 and 371 days for FBM EG-transduced and CRISPR-edited cells respectively. C Percent of hCD45+ (left panel) and GFP+ cells (gated in hCD45+ cells) (right panel) measured in the BM of 1ry mice injected with CTL and EG FL and CB cells. Data from mice with CRISPR/Cas9-edited (GFP−) cells are presented in light red (left panel). D Representative flow cytometry analysis of cells recovered from the vertebral column (VC) of 1ry recipients injected with FL and CB EG cells (lentiviral delivery). E Percent of CD33+KIT+, KIT+CD56+ and KIT+CD41+ cells in the BM of 1ry mice, gated in hCD45+(GFP+) cells. CRISPR/Cas9-edited (GFP−) cells are shown in light red. F Fluorescent in-situ hybridization (FISH) analysis of human cells recovered from a 1ry sick NSG recipient injected with FL EG-CRISPR/Cas9-edited cells. G Kaplan–Meier survival plot of mice serially transplanted with 1 × 106 FL EG cells and FBM EG cells. Median survival of 2ry recipients was 85 and 84 days for FL and FBM groups respectively. Median survival of 3ry recipients was 50 and 96 days for FL and FBM groups respectively. H Percent of CD33+KIT+, KIT+CD56+ and KIT+CD41+ cells in BM of 2ry and 3ry mice injected with FL EG cells, gated in hCD45+(GFP+ or GFP− (Inv(16) engineered)) cells. Data of mice with CRISPR/Cas9-edited (GFP−) cells are presented in light red. Statistical significance is indicated as p values (B and G: Log-rank test, C and E: Kruskal–Wallis test, H: Mann–Whitney test). *: p < 0.05, **: p < 0.01, ***: p < 0.001. ns, non significant
We next performed serial transplantation of primary EG-abnormal cell grafts. FL and FBM EG cells from primary mice promoted leukemia in secondary (FL: 23/33 (70%), FBM: 4/7 (57%)) and tertiary (FL: 17/18 (94%), FBM: 9/10 (90%)) recipients with a similar phenotype (Fig. 3G-H, Fig. S2E). Disease developed with a significantly shorter latency in tertiary (50 days) compared to secondary (85 days) FL EG recipients injected with the same number of cells (106 equivalent GFP+ cells) (Fig. 3G), confirming transformation of fetal EG cells to bona fide acute leukemia. Importantly, EG GFP+ CB grafts, both from healthy or sick mice, that were transplanted into secondary recipients, induced a disease in 2/14 (14%) injected mice, further highlighting the low EG-transforming efficiency in CB HSPCs. Collectively, this in vivo model recapitulates the EG-driven leukemogenesis with symptoms observed in patients, and points to higher sensitivity of fetal HSPCs to EG transformation in the human context in line with a previously reported murine model [15].
Efficient transformation of CB HSPCs by ETO2::GLIS2 is dependent on human cytokine stimulation in vivo
As EG expression in CB reproducibly induces abnormal cell growth in vitro but rare/poor in vivo leukemia development, we hypothesized that human cytokines, present in the culture media and non-cross reactive or absent in NSG mouse recipients, may play a role in EG-driven leukemogenesis. We thus transplanted EG CD34+ cells from CB and FL (105 EG GFP+ cells/mouse) into regular NSG or NSGS recipients [41] that constitutively produce human IL3, SCF (both provided in the culture medium) and GM-CSF (absent from the culture medium) (Fig. 4A). FL EG cells generated fatal leukemia in both mouse strains but with a significantly shorter latency in NSGS mice, with 13/13 (100%) NSGS vs 7/8 (88%) NSG mice becoming sick with median survivals of 25 and 75.5 days, respectively (Fig. 4B), showing that EG-driven leukemogenesis in FL HSPCs is accelerated but not dependent on human cytokines. Strikingly, EG CD34+ CB cells also generated a lethal disease in 12/12 (100%) injected NSGS mice with a median survival of 44 days whereas only 1/10 (10%) NSG mice injected with the same cell suspension developed a pathology (Fig. 4C), indicating a remarkable human cytokine dependency of EG-driven transformation in CB HSPCs. Phenotypically, abnormal FL EG and CB EG cells were alike in a given recipient but different in NSG vs NSGS recipients. In particular percentages of KIT+CD56+ cells were lower in EG FL and EG CB cells from NSGS compared to NSG mice and low to no CD41 expression was reproducibly observed in cells recovered from NSGS mice, indicating that the cytokine environment during leukemogenesis impacts the cell phenotype of leukemic cells (Fig. 4D). FL EG and CB EG grafts were serially transplanted into side-by-side secondary NSG and NSGS mice (Fig. 4E). FL EG propagating cells induced faster disease development upon injection into secondary NSGS vs NSG mice with median survivals of 45.5 vs 83 days, respectively (Fig. 4F). NSGS-derived CB EG propagating cells generated a lethal disease in all (8/8) secondary NSGS and 71% (5/7) secondary NSG mice, with median survivals of 44 and 174 days, respectively. Although the penetrance was not absolute in NSG recipients, CB EG propagating cells became less dependent to cytokines once transformed in an NSGS background (Fig. 4G). Similar to secondary recipients, CB EG leukemia-propagating cells induced faster disease development in NSGS vs NSG tertiary recipients. Serially transplanted FL and CB cells retained an abnormal phenotype (Fig. 4H).

EG-expressing cord blood cells are dependent on IL3 and/or SCF signaling for leukemic transformation in vivo, while FL EG cells show sensitivity. A Experimental design: EG-transduced FL or CB CD34+ cells (105 GFP+ equivalent cells) were injected side-by-side in NSG and NSGS mice. Mice were monitored daily and sacrificed when exhibiting disease symptoms as in Fig. S2. B-C Kaplan–Meier survival plot of primary recipient mice injected with FL EG (B) and CB EG (C). Median survival of mice injected with FL EG cells was 75.5 days in NSG and 25 days in NSGS recipients. For the CB EG group, the median survival was 44 days in NSGS recipients. Only 1/10 mice got sick in the NSG group. D Percent of CD33+KIT+, KIT+CD56+ and KIT+CD41+ cells gated in hCD45+GFP+ cells in the BM of primary recipient mice injected with FL EG (left panel) and CB EG (right panel) cells. E Design of serial transplantation, 105 GFP+ equivalent cells from primary NSGS recipients were transplanted side-by-side into secondary NSG and NSGS mice. Propagation of CB EG leukemia was further tested by transplantation of cells from 2ry NSG mice into tertiary NSG or NSGS mice. F-G Kaplan–Meier survival plot of serially transplanted mice injected with FL-derived (F) and CB-derived (G) grafts. Median survivals were 83 and 45.5 days for NSG and NSGS recipients of 2ry FL EG cells. Median survivals were 174 and 44 days for NSG and NSGS recipients of 2ry CB EG recipients, and 73 and 38 days for NSG and NSGS recipients of 3ry CB EG recipients. H Same analyses as in (D), but in the BM of vertebral columns (VC) of mice from (F-G). Statistical significance is indicated as p values (B-C, F-G: Log-rank test, D, H: Mann–Whitney test). *: p < 0.05, **: p < 0.01, ***: p < 0.001
These results demonstrate that CB CD34+ cells are strongly dependent on human cytokines for full-blown EG leukemic transformation. For FL cells, human cytokines were dispensable for transformation by EG but significantly accelerated the leukemia development.
EG-driven leukemia initiation and growth is sensitive to cytokines
To narrow down the contribution of cytokines, we compared the effect of human cytokines on in vitro CB EG cell growth, including in presence or absence of IL3 and SCF, the two common cytokines between in vitro and NSGS conditions (Fig. 5A). We observed that CB EG GFP+ cells were selected in culture only when IL3 and SCF were present in the medium, regardless of TPO/FLT3L/EPO supplementation (Fig. 5B, Fig. S4A). Phenotypically, the CB EG cells selected with IL3 and SCF were CD33+KIT+ (Fig. 5C-D), indicating that IL3 and SCF are important for abnormal EG cell expansion. Some residual phenotypically abnormal cells were detected in absence of IL3 and SCF (Fig. 5C-D), thus both cytokines were added back to cultures at 21 days, leading to the expansion of the remaining abnormal cells after IL3/SCF supplementation (Fig. 5E), regardless of the other cytokine presence (Fig. S4B). Inversely, CB EG cells cultured with IL3 and SCF during 21 days stopped growing after IL3/SCF removal even in presence of TPO/FLT3L/EPO (Fig. S4C). Finally, we tested the relative contribution of IL3 and SCF to either EG transformation initiation from FL and CB cells or to EG leukemic cell propagation, through changing to cytokine supplementation either right after EG transduction or after 5 days of culture, respectively (Fig. 5F). GFP+ cell numbers significantly dropped for CB EG cells after transduction, when both IL3 and SCF were absent, while individual cytokine retrieval had milder intermediate effects (Fig. 5G), indicating a cumulative impact of both IL3 and SCF deprivation. A same trend was observed for FL cells, but without reaching significance (Fig. 5G), consistent with a lower dependency of FL EG cells to exogenous cytokines. Besides, retrieving both IL3 and SCF from already selected EG cell cultures induced the most significant decrease in cell numbers for both FL and CB origins (Fig. 5H). Together these data indicate a major role of IL3 and SCF cytokines in the susceptibility of CB and, to a lesser extent, of FL HSPCs to EG-induced leukemogenesis. Of note, in two experiments, we cultured FL EG cells without IL3 and/or SCF during 35 days after transduction. Even though a drop of cell numbers and GFP+ cells was observed shortly after culture initiation as in Fig. 5G, EG/GFP+ cells recovered and grew in cultures devoid of added cytokines, especially in one sample, developing abnormal CD33+/KIT+ cells (Fig. S4D-F). These results further support the lowest cytokine dependency of FL EG cells for abnormal cell production, in line with in vivo NSG and NSGS data.

EG-expressing FL and CB cells have a growth dependency on IL3 and SCF human cytokines. A Experimental design: CB CD34+ cells were transduced with CTL/EG-GFP lentiviral vectors and cultured for 21 days in the indicated cytokine conditions. B Percent of GFP+ cells during the culture for CTL (upper panel) and EG (lower panel) cells. Mean ± SEM of 3 biological replicates is shown. C Example of flow cytometry dot plots showing the percent of CD33+KIT+ cells obtained after 21 days of culture in the different cytokine conditions. D Cumulative cell numbers of CD33+KIT+ cells in GFP+ CB EG cells at day 21 of culture. Mean ± SEM of 3 biological replicates is shown. E Experimental design: freshly transduced CB EG CD34+ cells were cultured for 21 days with TPO/FLT3L/EPO and without IL3/SCF. At that time point, IL3 and SCF were added or not to the culture medium and cells were grown for 14 more days (upper panel). Lower panel shows the mean ± SEM cumulative number of GFP+ cells of 3 biological replicates. F Experimental design: FL and CB EG cells were deprived of IL3 and/or SCF during 5 days of culture, after transduction or after their in vitro selection. The number of cells was counted at day 5 of culture ± cytokines. G Absolute numbers of EG GFP+ cells generated from transduced cells after transduction (condition with TPO/FLT3L/EPO). Mean ± SEM of 3 biological replicates. H Absolute numbers of EG GFP+ cells from in vitro selected EG cells. Mean ± SEM of 3 biological replicates. Statistical significance is indicated as p values (B,D,G,H: Kruskal–Wallis test, E: Mann–Whitney test). *: p < 0.05, **: p < 0.01
Cytokines control ETO2::GLIS2 leukemia cellular and molecular features
To characterize the consequences of cytokine stimulation, we then compared EG cells from different origins by single cell transcriptomes (scRNAseq). We analysed FL and CB EG CD34+ progeny after 7 days in vitro with cytokines, human cells from diseased NSG (FL- and FBM-derived) and NSGS (CB- and FL-derived) recipients and also cells from 4 ETO2::GLIS2 patients (Fig. 6A). Cells with appropriate quality controls from each condition (Fig. S5A) were integrated into 24 clusters (Fig. 6B, Fig. S5B), including some associated with cell cycle status (Fig. S5C). By computing a score of expression of an EG patient positively-associated gene signature [8], we confirmed significant enrichment in cells of most clusters (Fig. 6C, Fig. S5D). Human cytokine stimulated cells from in vitro and NSGS leukemia conditions occupied different territories compared to NSG leukemia, both overlapping with patient cells (Fig. 6D, Fig. S5E). NSG leukemia were enriched more specifically in clusters 0/3/5/14/15 (Fig. 6E), showing higher expression of several EG patient leukemia markers (e.g. NCAM1) [17] (Fig. 6F) and of megakaryocyte progenitor genes (Fig. 6G). Human cytokine stimulated cells were enriched in clusters 2/6/7/8/9 (Fig. 6H), presenting some myeloid gene expression (Fig. 6I, Fig. S5F). Enrichment in cytokine/receptor related genes and myeloid features in the NSGS context was confirmed using GSEA on bulk transcriptomes from CB NSGS vs FL NSG leukemia showing differential expression, including for myeloid markers CD14, CSF1R, FLT3, ITGAM (Fig. S5G-I). Enrichment in stemness gene expression was observed in most cells from EG conditions irrespective of cytokine stimulation (Fig. 6J).

The cytokine stimulation controls ETO2::GLIS2 leukemia cellular and molecular features. A Experimental design: Fetal or CB CD34+ EG transduced or CRISPR-edited cells were either cultured for 7 days in the LT-LC medium (± IL3/SCF) or injected into sublethally irradiated NSG or NSGS mice. After 7 days of culture or at disease development in vivo, EG cells were collected and live hCD45+(GFP+) cells were sorted prior to single cell and bulk transcriptome analysis. Patient cells (n = 4) were also analysed by scRNAseq. B UMAP representation of the integration of cells from all conditions and cluster identification. Clusters were defined using the Louvain algorithm. C Score of enrichment of the gene signature [8] upregulated in ETO2::GLIS2 patients projected on the UMAP of the integration (red: cells presenting an enrichment of the signature; blue: cells with a depletion of the signature; grey: lack of significance). D Cells from each condition are highlighted in red on the UMAP of the integration. E Percentage of cells in clusters 0 + 3 + 5 + 14 + 15 for each condition. F Projection of the level of expression of NCAM1 on cells of the integration. G Left panel: score of enrichment of a megakaryocyte progenitor gene signature (MSigDB, Hay et al., [63]) on the integration. Middle panel: percentage of cells presenting enrichment of the signature (red). Right panel: projection of the level of expression of ITGA2B on cells of the integration. H Percentage of cells in clusters 2 + 6 + 7 + 8 + 9 for each condition. I Projection of the level of expression of AIF1 on cells of the integration. J eft panel: score of enrichment of a HSC gene signature (MSigDB, Hay et al., [63]) on the integration. Middle panel: percentage of cells presenting enrichment of the signature (red). Right panel: projection of the level of expression of CD44 on cells of the integration. K Projection of the level of expression of KIT and IL3RA on cells of the integration. L Upper panel: experimental design of the ATACseq approach. Lower panel: histogram representation of the intensity at loci that present gained accessibility in + IL3 + SCF vs –IL3-SCF cells. M Peaks differentially gained in (L) were annotated to the closest gene. The corresponding gene list was used to compared bulk RNAseq data from leukemic cells in CB NSGS vs FL NSG using GSEA. N Score of enrichment of the gene list used in (M) projected on the integration
KIT and IL3RA showed higher expression in clusters specific for human cytokine-stimulated conditions (Fig. 6K). To understand the early consequences of cytokine stimulation, we hypothesized that IL3 and SCF may impact chromatin accessibility. Therefore, we performed ATACseq analyses on primary human CD34+ fetal and cord blood cells exposed to IL3 and SCF for 5 days in vitro. 688 peaks significantly gained accessibility in + IL3 + SCF vs –IL3–SCF conditions (Fig. 6L) and showed an enrichment in GATA and AP1 motifs (Fig. S5J). Differential peaks within the promoter or gene body (exons and introns) defined a list of 383 early cytokine-responsive target genes (e.g. SCN9A, Fig. S5K). To investigate whether expression of these early target genes was maintained at a later stage of transformation, their expression was investigated in bulk and scRNAseq from leukemic cells obtained in human cytokine-expressing NSGS vs. NSG recipients. In bulk RNAseq data, GSEA showed an enrichment of this list in the NSGS CB EG vs NSG FL EG conditions (Fig. 6M). In scRNAseq data, this list also showed a higher enrichment score in clusters and conditions associated with NSGS leukemia (Fig. 6N). Notably, among the differential genes more expressed in NSGS vs NSG leukemia is CCND2 (Fig. S5L), which is also highly expressed in EG patient transcriptomes (Fig. S5M), represents a selective vulnerability in EG+ M07e cells as defined by DepMap (Fig. S5N) and was previously implicated in RUNX1::RUNX1T1 leukemia [42].
Together, these data indicate that leukemic cells obtained from NSG and NSGS follow different trajectories, toward either megakaryoblastic or more myeloid features respectively, and reproduce cellular features found in EG patients. These data also show that early cytokine stimulation leads to changes in chromatin accessibility and in expression of a subset of genes that remain expressed at the full-blown leukemia stage, supporting a functional contribution to EG leukemia.
Human EG leukemic cell vulnerability is efficiently targeted using pharmalogical inhibitors of survival and MAPK signaling pathways
We next reasoned that downstream signaling pathways induced by this extrinsic cytokine stimulation could represent a vulnerability for EG leukemic cells. IL3 and GM-CSF receptors share a common ß-chain subunit which translates to the downstream activation of the MAPK signaling [43, 44]. SCF signaling also converges on MAPK activation [44, 45]. Therefore, we tested whether targeting of the MEK/ERK pathway with the MEK inhibitor Trametinib [46] may impair EG leukemia. Evidences suggest that signaling pathway inhibitors are of interest but may not sufficiently inhibit leukemic cell progression when used as a single agent [20] Since EG has recently been described to interfere with cell survival pathways [21], we explored the combination of Trametinib with Venetoclax, a widely clinically-used BCL2 inhibitor. FL and CB EG cells selected in cultures were treated with DMSO, Venetoclax, Trametinib or the combination of both inhibitors (Fig. 7A). Venetoclax or Trametinib alone showed a rather cytostatic effect on both FL and CB EG cells (Fig. 7B). The combination however significantly reduced cell numbers of both FL and CB EG cells (Fig. 7B) associated with enhanced apoptosis (Fig. 7C), indicating a major effect on EG cell maintenance. These observations were corroborated in EG+ M07e cells which growth was sensitive to IL3 and SCF (Fig. S6A). Also, regardless of the cytokine complementation, EG+ M07e cells were extremely sensitive to the combination of both inhibitors, whereas single inhibitor treatment had variable effects, depending on cytokine stimulation (Fig. S6B). In light of the observed effect on the EG cell proliferative capacity and the induction of apoptosis by Venetoclax and Trametinib, we next evaluated the drug effects in preclinical NSG or NSGS mouse models injected with luciferase-expressing (Luc+) EG+ patient-derived xenografted cells. Mice were treated during four weeks, five times a week, with vehicle, Venetoclax (100 mg/kg) and Trametinib (1 mg/kg) alone or in combination and leukemia development was monitored by bioluminescence imaging (BLI) (Fig. 7D-E). We observed that treatment with Trametinib alone was not sufficient to inhibit leukemia development, as measured by luciferase intensity (Fig. 7E-F) nor to enhance mice survival in both NSG and NSGS preclinical models (Fig. 7G-H). On the contrary, the leukemic burden was significantly decreased during and after the combination of Venetoclax and Trametinib, as compared to vehicle or Trametinib alone (Fig. 7E-F). The effect of Venetoclax alone was only tested in NSG mice and had early (5 weeks) effects on the leukemic burden that did not last as long as the combination ones (Fig. 7E, Fig. S6C-D), resulting in a significantly increased mouse survival in the drug combination group (Fig. 7G). Interestingly, the effects of the combinatory treatment were higher in NSG vs NSGS mice as NSGS mice did not survive over 80 days as compared to 44% (8/18) NSG mice being cleared of leukemic cells after Venetoclax/Trametinib combination treatment during ≥ 8.5 months (Fig. 7G-7H, Fig. S6C). The resistance to Trametinib and Venetoclax combination can be related to a NSGS mouse context in which there is important cytokine stimulation. In vitro, we observed that adding GM-CSF to IL3 and/or SCF revealed leukemic cell growth redundancies (Fig. S6E), suggesting GM-CSF was not fully neutral for the leukemic growth, especially in absence of IL3 and/or SCF. Moreover the presence of the 3 cytokines tuned down the leukemic cell response to Trametinib and Venetoclax in comparison with IL3 + SCF combination (Fig. S6B and S6F). Together, these results conclusively show that a combination of Venetoclax and Trametinib has a significantly stronger effect on in vivo EG leukemia progression than either molecule alone. However, cytokine stimulation can undermine the observed drug effects, leading to leukemic relapse.

EG leukemogenic dependency on cytokines can be targeted by inhibiting signal transduction and survival pathways. A In vitro experimental design: FL and CB EG in vitro selected cells were treated with DMSO, Venetoclax (0.1 µM), Trametinib (0.1 µM) or the combination of both. Cells were counted every 24 h after treatment and cell death was measured at 48 h. B Cumulative number of FL EG (left panel) and CB EG (right panel) cells during 96 h control in presence of the inhibitors. Mean ± SEM of 3 biological replicates is shown. C Relative percentage of apoptotic (AnnV+) cells at 48 h of treatment. Mean ± SEM of 3 biological replicates. D In vivo experimental design: 5 × 104 PDX luciferase-expressing cells injected NSG (3 pooled experiments) or NSGS (1 experiment) mice were treated for four weeks, five times a week, with Venetoclax (100 mg/kg), Trametinib (1 mg/kg) or the combination of both molecules. Arrowheads indicate timing of the imaging of the luciferase-expressing PDX leukemic cells. Leukemia development was monitored by bioluminescence tracking and mice were euthanized upon disease symptoms detection. E–F Follow up of luciferase intensity in photon per second (p/s) during and after treatment of NSG (E) and NSGS (F) mice. Mean ± SEM of the indicated number of mice per group is shown. G Kaplan–Meier survival plot of NSG mice after treatment. Median survival was 81 days for the sham treated (CTL) group, 129 days for Venetoclax alone, 95 days for the Trametinib alone and 164 days for the combination group. H Kaplan–Meier survival plot of NSGS mice after treatment. Median survival was 52.5 days for the CTL group, 56 days for the Trametinib alone group and 75 days for the combination group. Statistical significance is indicated as p values (B-C-E–F: Kruskal–Wallis test, G-H: Log-rank test). *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001
Discussion
Here, we present unique models of ETO2::GLIS2-induced leukemogenesis starting from primary human cells of three different developmental stages: fetal, newborn and adult. Firstly, we found that EG induces drastic transcriptional changes and leukemia development more efficiently in fetal than newborn and adult primary human HSPCs. Secondly, we uncovered that cytokines, including SCF and IL3, strongly influence EG-leukemia development in human HSPCs and are able to modulate the leukemogenic potential determined by ontogeny. Thirdly, we exploited this functional interplay through combined targeting of the downstream MEK pathway and the recently identified vulnerability of EG cells on BCL2 cell survival [21] and propose a novel targeting strategy for this poor outcome leukemia.
In this study, we developed EG-leukemia models either by lentiviral delivery or by precise CRISPR/Cas9-mediated engineering of the ETO2::GLIS2 chromosomal inversion found in patient cells. EG-expressing fetal HSPCs generated a transplantable leukemia in NSG mice, even with CRISPR-edited cells where the estimated number of EG-expressing HSPCs was lower (~ 1/48–1/107 edited cells) compared to the lentiviral delivery (≥ 14% of transduced cells). Conversely, EG was less efficient in transforming post-natal HSPCs. In our setting, CB HSPCs with EG rarely generated a fatal leukemia in NSG mice even though it was reported to be possible [22, 26, 47]. In addition, EG-transduction of BM HSPCs did not generate any outgrowth or selection of abnormal cells, indicating EG transforming ability declines in older post-natal HSPCs. These differences in EG transformation susceptibility between fetal and post-natal cells are in line with data from a murine model in which EG expressing fetal immature HSPCs develop aggressive megakaryoblastic leukemia while the leukemia shifted toward a more myeloid phenotype and a less aggressive presentation upon expression in cells downstream the murine cellular hierarchy and ontogeny [15]. Therefore, these data furthermore underline that ontogeny/tissue of origin play an essential role in pediatric leukemia pathogenesis. Whether the more aggressive disease development from fetal tissues is due to different cellular compositions or hematopoietic hierarchy within CD34+ cells requires additional studies. Differences between fetal, newborn and adult HSPCs transformation potential is relevant to other pediatric leukemia. MLL::AF4, commonly found in infant B-cell acute leukemia, has been shown recently to transform human CD34+ cells in vivo [48, 49]. Interestingly, CRISPR-edited MLL::AF4 CB HSPCs showed higher permissiveness to transformation than fetal liver and adult BM cells [48,49,50]. This observation suggests the existence of fusion-specific transformation properties that preferentially affect different HSPCs ontogenic stages.
In line with this, we observed that more pronounced transcriptional alterations are imposed by EG in fetal than in newborn HSPCs. Notably, our in vitro models showed that expression of EG in CB did not alter expression of some master regulators of hematopoietic differentiation, including ERG, GATA1 and SPI1, while they were significantly deregulated in FL cells. This data support that EG induces a higher hematopoietic differentiation blockage in the fetal context. Also, the upregulated gene expression signature characteristic of patient cells is more strongly enriched upon EG expression in FL cells, supporting that induction of EG targeted genes is dependent on the development stage of the targeted cells. Together, these data support that intrinsic differences between FL and CB progenitors are associated with differential impact of the EG fusion on transcription deregulations and transforming ability.
Our data also reveal a major effect of the cytokine microenvironment on the efficiency and latency of EG leukemia development depending on the developmental stage of the cell of origin. Indeed, while fetal cells can efficiently induce leukemia in regular immunodeficient mice, human cytokine supplementation, including IL3 and SCF, strikingly increased EG-driven leukemogenesis from CB HSPCs. Phenotypically, a small proportion of human abnormal cells can be maintained in cultures without IL3 and SCF, and these cells may proliferate again upon addition of IL3 and SCF. In vivo, CB EG cells rarely developed into a full-blown leukemia in human cytokine-free NSG mice but occasional abnormal cells were detected in the surviving mice BM, long after transplant. Together, these results show that EG alone can initiate an abnormal cell development in CB HSPCs but expansion is blunted without appropriate extrinsic factors. Interestingly, EG CB leukemia established in NSGS mice can perpetuate upon injection into secondary and tertiary NSG mice, indicating that, after the transformation process has taken place, leukemic cell propagation is either cytokine-independent or dependent on other mouse cross-reactive factors.
These data provide important insights for both leukemia initiation and patient clinical presentations. They further support the existence of a developmental window of permissiveness to transformation provided by intrinsic differences in the cell of origin [15, 51]. Most importantly, they uncover that human pediatric leukemia can be generated from cells of different developmental origin according to the microenvironmental conditions. Indeed, they demonstrate that the biological determinants required for EG human leukemogenesis can be met in post-natal progenitors provided that some cytokines, including IL3 and SCF, are present in their niche. Interestingly, the requirement for microenvironmental support could explain that a recent EG transformation model from CB cells was dependent on an endothelial stroma [47].
SCF is secreted from various cells including fibroblasts, endothelial cells, stellate cells and bone marrow adipocytes [52,53,54], while IL3 is produced by hematopoietic (e.g. activated T-cells, basophils, dendritic and mast cells) but also by non-hematopoietic cells like microglial cells and astrocytes [55, 56]. In addition, IL3 has been associated with leukemogenic processes in AML [57,58,59,60] along with other inflammatory cytokines [61, 62]. And this is not to exclude other cytokines, such as GM-CSF, which presence may substitute IL3 or SCF in situations where they would be lacking, and infers stimulating growth effects on leukemic cells.Thus, both physiological cytokine secretion territories but also altered production in inflammatory situations may drive leukemia initiation and sustain expansion in heterogeneous locations (including CNS involvement) as observed in patients [17, 39].
While single cell transcriptomics on EG leukemic cells from human cytokine-stimulated and non-stimulated conditions showed a global enrichment of the EG patient signature, they also revealed at least two different states. Leukemia developing in regular NSG showed an enrichment in megakaryocytic markers, while human cytokines stimulation was associated with a myeloid shift. Importantly, early exposition to cytokines has an impact on the chromatin accessibility that translated into the expression of a group of genes, which remained more highly expressed in the bona fide EG leukemia. This interplay between the microenvironmental conditions and transcriptional programs of the leukemic cells could represent a basis for the large array of phenotypic presentations observed in patients [24]. It could also represent a mechanism of adaptation and resistance to treatment. Together, our results support that the phenotype of the leukemia is controlled at least in part by the cytokine milieu.
Finally, we obtained a proof of concept that the interplay between extrinsic and intrinsic vulnerabilities may be therapeutically exploited for the treatment of this aggressive leukemia. Indeed, the combined targeting of a BCL2 dependency with Venetoclax [21] and the cytokine-induced MAPK signaling pathway with the MEK inhibitor Trametinib, demonstrates a better control of the leukemic progression than individual molecules in regular NSG mouse models. Leukemia developing in humanized cytokines recipients was however more resistant to the treatment and our results formally confirmed that the cytokine microenvironment may contribute to treatment resistance. Besides, the presence or absence of human cytokines, here in the two strains of mice, led to different leukemia phenotypes that may also influence drug sensitivity. Therefore, addressing the relative importance of cytokines, including their levels, and the microenvironment niches in general, the biodistribution of therapeutic compounds and the best combinations and doses for therapeutic application will be key for the treatment of ETO2::GLIS2 and other pediatric hematological malignancies.
To sum up, our work shows how the oncogenic ETO2::GLIS2 fusion, diagnosed in very young children leukemia, efficiently promotes leukemia development when expressed in human fetal but not in post-natal HSPCs. It uncovers that human cytokine stimulation strongly increases the permissiveness to ETO2::GLIS2 leukemogenesis in post-natal HSPCs and enhances leukemia development from fetal HSPCs. Single cell analyses revealed that cytokine stimulation leads to distinct phenotypes and expression patterns in transformed cells. Importantly, we show that the combined targeting of the MAPK pathway downstream of cytokine signaling using Trametinib, together with the recently identified BCL2-dependent survival mechanism involved in ETO2::GLIS2 leukemia, displays important leukemic growth suppression in preclinical patient-derived xenograft models.
Conclusions
Altogether, we demonstrate that leukemogenesis modeling starting from normal human cells offers the unique opportunity to directly compare the transcriptome and chromatin state profiles of models with those from patients' leukemia, which was not previously possible with murine cell-based models. The cytokine microenvironment strongly increases the developmental window of permissiveness of post-natal cells to pediatric oncogenes and changes the phenotype of the resulting leukemia. This important observation supports that local or systemic changes in cytokine homeostasis during post-natal life may impact leukemia initiation and aggressiveness. Most importantly, this approach provides new insights and perspectives, in particular for patient treatments. Thus, such precise reproduction and characterization of the trajectory from normal human cells to leukemia in models and its comparison with patients' leukemia should be of interest for the development of novel therapeutic perspectives for other aggressive pediatric cancers.
Availability of data and materials
Sequencing data were submitted to EBI (Array Express) and are available under the accession numbers E-MTAB-14124 (AtacSeq), E-MTAB-14131 (Bulk RNAseq) and E-MTAB-14140 (scRNAseq-experiments).
Abbreviations
- ABM:
-
Adult Bone Marrow
- AML:
-
Acute Myeloid Leukemia
- AMKL:
-
Acute Megakaryoblastic Leukemia
- BLI:
-
Bioluminescence Imaging
- BM:
-
Bone Marrow
- CB:
-
Umbilical Cord Blood
- CFU:
-
Colony Forming Unit
- CTL:
-
Control
- CTV:
-
CellTrace Violet
- CNS:
-
Central Nervous System
- EG:
-
ETO2::GLIS2
- FBM:
-
Fetal Bone Marrow
- FBS:
-
Fetal Bovine Serum
- FL:
-
Fetal Liver
- GFP:
-
Green Fluorescent Protein
- GM-CSF:
-
Granulo-Macrophage Colony Stimulating Factor
- GSEA:
-
Ene Set Enrichment Analysis
- HSC:
-
Hematopoietic Stem Cell
- HSPCs:
-
Hematopoietic Stem and Progenitor Cells
- (h)EPO:
-
(Human) Erythropoietin
- (h)IL3:
-
(Human) Interleukine 3
- (h)FLT3L:
-
(Human) FLT3 Ligand
- (h)SCF:
-
(Human) Stem Cell Factor
- (h)TPO:
-
(Human) Thrombopoietin
- inv(16):
-
Inversion 16
- LFC2:
-
Log2 Fold Change
- Luc:
-
Luciferase
- LT-LC:
-
Long-Term Liquid Culture
- MOI:
-
Multiplicity of Infection
- mPB:
-
Mobilized Peripheral Blood
- MPP:
-
Multipotent Progenitor
- ns:
-
Non significant
- P(1):
-
Plating (1)
- PDX:
-
Patient-Derived Xenograft
- P/S(/G):
-
Peniciline/Sterptomycine(/Glutamine)
- RPKM :
-
Reads per kilo base per million mapped reads
- RT-qPCR:
-
Real-Time quantitative Polymerase Chain Reaction
- SEM:
-
Standard Error of the Mean
- UMAP:
-
Uniform Manifold Approximation and Projection space
- VC:
-
Vertebral Column
References
de Rooij JDE, Zwaan CM, van den Heuvel-Eibrink M. Pediatric AML: From Biology to Clinical Management. J Clin Med. 2015;4:127–49.
Greaves M. A causal mechanism for childhood acute lymphoblastic leukaemia. Nat Rev Cancer. 2018;18:471–84.
Wiemels JL, Xiao Z, Buffler PA, Maia AT, Ma X, Dicks BM, et al. In utero origin of t(8;21) AML1-ETO translocations in childhood acute myeloid leukemia. Blood. 2002;99:3801–5.
McHale CM, Wiemels JL, Zhang L, Ma X, Buffler PA, Feusner J, et al. Prenatal origin of childhood acute myeloid leukemias harboring chromosomal rearrangements t(15;17) and inv(16). Blood. 2003;101:4640–1.
Schlosser M-P, Bucking S, Lemyre B, Grynspan D, Padmore R, Johnston D. Prenatally Diagnosed Infant AML. J Pediatr Hematol Oncol. 2018;40:238.
Marcotte EL, Spector LG, Mendes-de-Almeida DP, Nelson HH. The Prenatal Origin of Childhood Leukemia: Potential Applications for Epidemiology and Newborn Screening. Front Pediatr. 2021;9:639479. Available from: https://www.frontiersin.org/articles/10.3389/fped.2021.639479. Cited 2024 Apr 10.
Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, et al. The Origin and Evolution of Mutations in Acute Myeloid Leukemia. Cell. 2012;150:264–78.
Bolouri H, Farrar JE, Triche T Jr, Ries RE, Lim EL, Alonzo TA, et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med. 2018;24:103–12.
Notta F, Zandi S, Takayama N, Dobson S, Gan OI, Wilson G, et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science. 2016;351:aab2116.
Roy A, Wang G, Iskander D, O’Byrne S, Elliott N, O’Sullivan J, et al. Transitions in lineage specification and gene regulatory networks in hematopoietic stem/progenitor cells over human development. Cell Rep. 2021;36:109698.
Sommarin MNE, Olofzon R, Palo S, Dhapola P, Soneji S, Karlsson G, et al. Single-cell multiomics of human fetal hematopoiesis defines a developmental specific population and a fetal signature. Blood Advances. 2023;7(18):5325–40.
Pagano L, Pulsoni A, Vignetti M, Mele L, Fianchi L, Petti MC, et al. Acute megakaryoblastic leukemia: experience of GIMEMA trials. Leukemia. 2002;16:1622–6.
McNulty M, Crispino JD. Acute Megakaryocytic Leukemia. Cold Spring Harb Perspect Med. 2020;10:a034884.
de Rooij JDE, Branstetter C, Ma J, Li Y, Walsh MP, Cheng J, et al. Pediatric non–Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat Genet. 2017;49:451–6.
Lopez CK, Noguera E, Stavropoulou V, Robert E, Aid Z, Ballerini P, et al. Ontogenic Changes in Hematopoietic Hierarchy Determine Pediatric Specificity and Disease Phenotype in Fusion Oncogene-Driven Myeloid Leukemia. Cancer Discov. 2019;9:1736–53.
Gruber TA, Larson Gedman A, Zhang J, Koss CS, Marada S, Ta HQ, et al. An Inv(16)(p13.3q24.3)-Encoded CBFA2T3-GLIS2 Fusion Protein Defines an Aggressive Subtype of Pediatric Acute Megakaryoblastic Leukemia. Cancer Cell. 2012;22:683–97.
Thiollier C, Lopez CK, Gerby B, Ignacimouttou C, Poglio S, Duffourd Y, et al. Characterization of novel genomic alterations and therapeutic approaches using acute megakaryoblastic leukemia xenograft models. J Exp Med. 2012;209:2017–31.
Thirant C, Ignacimouttou C, Lopez CK, Diop M, Le Mouël L, Thiollier C, et al. ETO2-GLIS2 Hijacks Transcriptional Complexes to Drive Cellular Identity and Self-Renewal in Pediatric Acute Megakaryoblastic Leukemia. Cancer Cell. 2017;31:452–65.
Thirant C, Lopez C, Malinge S, Mercher T. Molecular pathways driven by ETO2-GLIS2 in aggressive pediatric leukemia. Molecular & Cellular Oncology. 2017;4:e1345351.
Benbarche S, Lopez CK, Salataj E, Aid Z, Thirant C, Laiguillon M-C, et al. Screening of ETO2-GLIS2–induced Super Enhancers identifies targetable cooperative dependencies in acute megakaryoblastic leukemia. Science Adv. 2022;8:eabg9455.
Aid Z, Robert E, Lopez CK, Bourgoin M, Boudia F, Le Mene M, et al. High caspase 3 and vulnerability to dual BCL2 family inhibition define ETO2::GLIS2 pediatric leukemia. Leukemia. 2023;37:571–9.
Gress V, Roussy M, Boulianne L, Bilodeau M, Cardin S, EL-Hachem N, et al. CBFA2T3::GLIS2 Pediatric Acute Megakaryoblastic Leukemia is Sensitive to BCL-XL Inhibition by Navitoclax and DT2216. Blood Adv. 2023;8(1):112–29.
Neault M, Lebert-Ghali C-É, Fournier M, Capdevielle C, Garfinkle EAR, Obermayer A, et al. CBFA2T3-GLIS2-dependent pediatric acute megakaryoblastic leukemia is driven by GLIS2 and sensitive to navitoclax. Cell Rep. 2023;42:113084.
Masetti R, Pigazzi M, Togni M, Astolfi A, Indio V, Manara E, et al. CBFA2T3-GLIS2 fusion transcript is a novel common feature in pediatric, cytogenetically normal AML, not restricted to FAB M7 subtype. Blood. 2013;121:3469–72.
Noort S, Wander P, Alonzo TA, Smith J, Ries RE, Gerbing RB, et al. The clinical and biological characteristics of NUP98-KDM5A in pediatric acute myeloid leukemia. Haematologica. 2021;106:630–4.
Smith JL, Ries RE, Hylkema T, Alonzo TA, Gerbing RB, Santaguida MT, et al. Comprehensive Transcriptome Profiling of Cryptic CBFA2T3–GLIS2 Fusion-Positive AML Defines Novel Therapeutic Options: A COG and TARGET Pediatric AML Study. Clin Cancer Res. 2020;26:726–37.
Wagenblast E, Araújo J, Gan OI, Cutting SK, Murison A, Krivdova G, et al. Mapping the cellular origin and early evolution of leukemia in Down syndrome. Science. 2021;373:eabf6202.
Sirven A, Ravet E, Charneau P, Zennou V, Coulombel L, Guétard D, et al. Enhanced Transgene Expression in Cord Blood CD34+-Derived Hematopoietic Cells, Including Developing T Cells and NOD/SCID Mouse Repopulating Cells, Following Transduction with Modified TRIP Lentiviral Vectors. Mol Ther. 2001;3:438–48.
Gerby B, Armstrong F, de la Grange PB, Medyouf H, Calvo J, Verhoeyen E, et al. Optimized gene transfer into human primary leukemic T cell with NOD-SCID/leukemia-initiating cell activity. Leukemia. 2010;24:646–9.
Babin L, Darchen A, Robert E, Aid Z, Borry R, Soudais C, et al. De novo generation of the NPM-ALK fusion recapitulates the pleiotropic phenotypes of ALK+ ALCL pathogenesis and reveals the ROR2 receptor as target for tumor cells. Mol Cancer. 2022;21:65.
Renouf B, Piganeau M, Ghezraoui H, Jasin M, Brunet E. Creating cancer translocations in human cells using Cas9 DSBs and nCas9 paired nicks. Methods Enzymol. 2014;546:251–71.
McIntosh BE, Brown ME, Duffin BM, Maufort JP, Vereide DT, Slukvin II, et al. Nonirradiated NOD,B6.SCID Il2rγ−/−KitW41/W41 (NBSGW) mice support multilineage engraftment of human hematopoietic cells. Stem Cell Rep. 2015;4:171–80.
Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213–8.
Parkhomchuk D, Borodina T, Amstislavskiy V, Banaru M, Hallen L, Krobitsch S, et al. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009;37:e123.
Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296.
Arkoun B, Robert E, Boudia F, Mazzi S, Dufour V, Siret A, et al. Stepwise GATA1 and SMC3 mutations alter megakaryocyte differentiation in a Down syndrome leukemia model. J Clin Invest. 2022;132(14):e156290.
Bourquin J-P, Subramanian A, Langebrake C, Reinhardt D, Bernard O, Ballerini P, et al. Identification of distinct molecular phenotypes in acute megakaryoblastic leukemia by gene expression profiling. Proc Natl Acad Sci USA. 2006;103:3339.
Paredes-Aguilera R, Romero-Guzman L, Lopez-Santiago N, Trejo RA. Biology, clinical, and hematologic features of acute megakaryoblastic leukemia in children. Am J Hematol. 2003;73:71–80.
Ishibashi M, Yokosuka T, Yanagimachi MD, Iwasaki F, Tsujimoto S, Sasaki K, et al. Clinical Courses of Two Pediatric Patients with Acute Megakaryoblastic Leukemia Harboring the CBFA2T3-GLIS2 Fusion Gene. Turk J Haematol. 2016;33:331–4.
Hara Y, Shiba N, Ohki K, Tabuchi K, Yamato G, Park M, et al. Prognostic impact of specific molecular profiles in pediatric acute megakaryoblastic leukemia in non-Down syndrome. Genes Chromosom Cancer. 2017;56:394–404.
Wunderlich M, Chou F-S, Sexton C, Presicce P, Chougnet CA, Aliberti J, et al. Improved multilineage human hematopoietic reconstitution and function in NSGS mice. PLoS ONE. 2018;13: e0209034.
Martinez-Soria N, McKenzie L, Draper J, Ptasinska A, Issa H, Potluri S, et al. The Oncogenic Transcription Factor RUNX1/ETO Corrupts Cell Cycle Regulation to Drive Leukemic Transformation. Cancer Cell. 2018;34:626-642.e8.
Hara T, Miyajima A. Function and Signal Transduction Mediated by the Interleukin 3 Receptor System in Hematopoiesis. Stem Cells. 1996;14:605–18.
Wilkes MC, Shibuya A, Sakamoto KM. Signaling Pathways That Regulate Normal and Aberrant Red Blood Cell Development. Genes (Basel). 2021;12:1646.
Liang J, Wu Y-L, Chen B-J, Zhang W, Tanaka Y, Sugiyama H. The C-Kit Receptor-Mediated Signal Transduction and Tumor-Related Diseases. Int J Biol Sci. 2013;9:435–43.
Yoshida T, Kakegawa J, Yamaguchi T, Hantani Y, Okajima N, Sakai T, et al. Identification and Characterization of a Novel Chemotype MEK Inhibitor Able to Alter the Phosphorylation State of MEK1/2. Oncotarget. 2012;3:1533–45.
Le Q, Hadland B, Smith JL, Leonti A, Huang BJ, Ries R, et al. CBFA2T3-GLIS2 model of pediatric acute megakaryoblastic leukemia identifies FOLR1 as a CAR T cell target. J Clin Invest. 2022;132(22):e157101. Available from: https://www.jci.org/articles/view/157101. Cited 2024 Feb 21.
Rice S, Jackson T, Crump NT, Fordham N, Elliott N, O’Byrne S, et al. A human fetal liver-derived infant MLL-AF4 acute lymphoblastic leukemia model reveals a distinct fetal gene expression program. Nat Commun. 2021;12:6905.
Bueno C, Torres-Ruíz R, Velasco-Hernandez T, Molina O, Petazzi P, Martinez-Moreno A, et al. A human genome editing-based MLL::AF4 B-cell ALL model recapitulates key cellular and molecular leukemogenic features. Blood. 2023;142(20):1752–6.
Secker K-A, Bruns L, Keppeler H, Jeong J, Hentrich T, Schulze-Hentrich JM, et al. Only Hematopoietic Stem and Progenitor Cells from Cord Blood Are Susceptible to Malignant Transformation by MLL-AF4 Translocations. Cancers. 2020;12:1487.
Li Z, Godinho FJ, Klusmann J-H, Garriga-Canut M, Yu C, Orkin SH. Developmental stage–selective effect of somatically mutated leukemogenic transcription factor GATA1. Nat Genet. 2005;37:613–9.
Zhou BO, Yu H, Yue R, Zhao Z, Rios JJ, Naveiras O, et al. Bone marrow adipocytes promote the regeneration of stem cells and haematopoiesis by secreting SCF. Nat Cell Biol. 2017;19:891–903.
Xu C, Gao X, Wei Q, Nakahara F, Zimmerman SE, Mar J, et al. Stem cell factor is selectively secreted by arterial endothelial cells in bone marrow. Nat Commun. 2018;9:2449.
Lee Y, Leslie J, Yang Y, Ding L. Hepatic stellate and endothelial cells maintain hematopoietic stem cells in the developing liver. J Exp Med. 2021;218:e20200882.
Varricchi G, Poto R, Marone G, Schroeder JT. IL-3 in the development and function of basophils. Semin Immunol. 2021;54:101510.
Kiss MG, Mindur JE, Yates AG, Lee D, Fullard JF, Anzai A, et al. Interleukin-3 coordinates glial-peripheral immune crosstalk to incite multiple sclerosis. Immunity. 2023;56:1502-1514.e8.
Wunderlich M, Chou F-S, Link KA, Mizukawa B, Perry RL, Carroll M, et al. AML xenograft efficiency is significantly improved in NOD/SCID-IL2RG mice constitutively expressing human SCF, GM-CSF and IL-3. Leukemia. 2010;24:1785–8.
Sadras T, Perugini M, Kok CH, Iarossi DG, Heatley SL, Brumatti G, et al. Interleukin-3-mediated regulation of β-catenin in myeloid transformation and acute myeloid leukemia. J Leukoc Biol. 2014;96:83–91.
Bulaeva E, Pellacani D, Nakamichi N, Hammond CA, Beer PA, Lorzadeh A, et al. MYC-induced human acute myeloid leukemia requires a continuing IL-3/GM-CSF costimulus. Blood. 2020;136:2764–73.
Fan AC, Nakauchi Y, Bai L, Azizi A, Nuno KA, Zhao F, et al. RUNX1 loss renders hematopoietic and leukemic cells dependent on IL-3 and sensitive to JAK inhibition. J Clin Invest. 2023;133:e167053.
Carey A, Edwards DK, Eide CA, Newell L, Traer E, Medeiros BC, et al. Identification of Interleukin-1 by Functional Screening as a Key Mediator of Cellular Expansion and Disease Progression in Acute Myeloid Leukemia. Cell Rep. 2017;18:3204–18.
Meisel M, Hinterleitner R, Pacis A, Chen L, Earley ZM, Mayassi T, et al. Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature. 2018;557:580–4.
Hay SB, Ferchen K, Chetal K, Grimes HL, Salomonis N. The Human Cell Atlas bone marrow single-cell interactive web portal. Exp Hematol. 2018;68:51–61.
Acknowledgements
We thank the patients and their parents for giving their consent for their participation in this study. We acknowledge C Robin, ML Arcangeli, C Lopez, J Schwaller, B Gerby and OA Bernard for their constructive input and the whole team “Niche and Cancer in Haematopoiesis” for excellent discussions during lab meetings. We are undebted to T Domet and L Faivre from the Centre de Thérapie Cellulaire in Hôpital Saint Louis, Paris, France, and Dr Y Rouquet and the midwives from Clinique des Noriets in Vitry-sur-Seine, France, for their devotion in providing cord blood and bone marrow samples. Fetal/embryonic human samples were obtained thanks to the great help of the unit of fetal pathology from Hôpital A Béclère in Clamart, France. We thank P Rameau and the Gustave Roussy cytometry platform for sorting cells of interest during the project and V Menard, IRCM irradiation facility, for managing irradiation doses. Q Pascal from the IBFJ anatomo-pathology platform performed the histologic analysis of sick mice. We will always remember Dr C Bilhou-Naberra who was so eager to help with FISH analyses. This study was supported by Inserm, CEA, Université Paris Cité and Université Paris Saclay.
Funding
Grants were obtained from Enfants Cancer Santé (ECS), Société Française de Lutte Contre les Cancers et les Leucémies de l’Enfant et de l’Adolescent (SFCE, RAK22052LLA), Institut National Du Cancer (PLBIO-2018–169, CONECT-AML: Subvention INCA-ARC-LIGUE_11905, PEDIAC: INCA_15670, PEDIAMOD22-008), Ligue Contre le Cancer (LNCC, TM: Equipe labellisée; FP: Equipe labellisée), VAP was supported by INCA and ECS/SFCE, KG received a PhD fellowship from the French Ministry of Research and higher Education and from LNCC, ZA was supported by Fondation pour la Recherche Médicale (FRM-ING20150532273), OPALE Carnot institute and Gustave Roussy (PMS Crescendo). FB was supported by FRM (FDT202106012809) and Gustave Roussy Genomic Core Facility (Taxe apprentissage 2022 FABO).
FP, AP and TM teams are part of the OPALE Carnot institute (Grant obtained in 2022) and the Paris Kids Cancer program. FP team is part of Institut Hospitalo-Universitaire THEMA-2.
Author information
Authors and Affiliations
Contributions
Conceptualization: FP and TM; Methodology: VAP, KG, FB, ER, EB, FP, TM; Software: ER; Formal analysis: ER, FB, VAP and KG; Data curation: VAP, KG, FB, ER, ZA; Investigation: VAP, KG, FB, ER, ZA, CM, VB, SD, LB, LR; Writing-original draft : VAP, KG, TM and FP; Resources: VRF, DM, DB, GP, NA, PB, PH, RH, JM, AP, HL, EB; Supervision: FP, TM and EB; Project Administration and funding acquisition: FP and TM.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Human sample collection: Umbilical cord blood (CB) samples were collected from healthy infants at Clinique des Noriets, Vitry-sur-Seine, France and Cell Therapy Department in Hôpital Saint Louis, Paris, France with informed consent of the mothers based on the declaration of Helsinki. Samplings and experiments were acknowledged by the Institutional Review Board of INSERM (Opinion number 12–079, IRB00003888).
Human fetal liver (FL) and bone marrow (FBM) samples were collected after pregnancy termination (8-14w and 17-24w of amenorrhea) with informed consent of the parents in accordance to guidelines approved by the French Agence de Biomédecine (PFS18-009).
Allograft bone marrow (ABM) and mobilized peripheral blood (mPB) samples were collected from healthy individuals at Necker Enfants Malades hospital and Centre de Recherche Biologique of Hôpital Saint Louis, Assistance Publique-Hôpitaux de Paris (AP-HP), Paris, France, respectively, with informed consent of the patients and samples were used for research only according to the Material Transfer Agreement VAL 08/08–081/01.
Mouse experiments: All procedures were done in accordance with the recommendations of the European Community and French Ministry of Agriculture regulations (animal facility registration number: A920322) for the care and use of laboratory animals. Experimental procedures were specifically approved by the local Ethical Committee (CEEA 26: A18_105 and APAFIS #20538–2019050710555633). NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) and NOD.Cg-KitW−41JPrkdcscidIl2rgtm1Wjl/KitW-41 J (NBSGW) mice were originally obtained from the Jackson Laboratory (Bar Harbor, Maine, USA), housed and bred in specific pathogen-free animal facilities (IRCM, CEA, Fontenay-aux-Roses, France). NOD.Cg-PrkdcscidIl2rgtm1WjlTg(CMV-IL3,CSF2,KITLG)1Eav/MloySzJ (NSGS) were housed and bred in the animal colony of Gustave Roussy Institute, France (CEEA 26: #2017122111548235_v2 and APAFIS #43692–2023053116192480 v3).
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Alonso-Pérez, V., Galant, K., Boudia, F. et al. Developmental interplay between transcriptional alterations and a targetable cytokine signaling dependency in pediatric ETO2::GLIS2 leukemia. Mol Cancer 23, 204 (2024). https://doi.org/10.1186/s12943-024-02110-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-024-02110-y