
Abstract
Ferroptosis is a type of regulated cell death characterized by iron accumulation and uncontrolled lipid peroxidation, leading to plasma membrane rupture and intracellular content release. Originally investigated as a targeted therapy for cancer cells carrying oncogenic RAS mutations, ferroptosis induction now exhibits potential to complement chemotherapy, immunotherapy, and radiotherapy in various cancer types. However, it can lead to side effects, including immune cell death, bone marrow impairment, liver and kidney damage, cachexia (severe weight loss and muscle wasting), and secondary tumorigenesis. In this review, we discuss the advantages and offer an overview of the diverse range of documented side effects. Furthermore, we examine the underlying mechanisms and explore potential strategies for side effect mitigation.
Similar content being viewed by others


Introduction
Cell death is a fundamental biological process inherent in complex organisms, serving as a critical mechanism for the elimination of dysfunctional or aging cells [1]. This process comprises two primary categories: accidental cell death, occurring in response to unexpected insults and injuries, and regulated cell death, a finely tuned machinery susceptible to modulation by drugs or genetic interventions [2]. Regulated cell death plays a pivotal role not only in tissue development and cellular homeostasis, but also contributes to various diseases and pathological conditions, including cancer, neurodegeneration, infections, and ischemia-reperfusion damage. In the context of cancer treatment, the induction of regulated cell death has become a primary goal in various targeted therapies, enabling the precise eradication of tumor cells while minimizing harm to healthy cells [3].
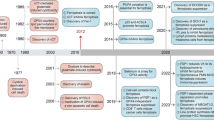
Considering the limitations associated with current drug treatments, often leading to apoptosis resistance, the exploration of non-apoptotic cell death has arisen as a logical strategy in both basic research and clinical trials. Specifically, the term 'ferroptosis,' coined in 2012, delineates a distinctive form of non-apoptotic cell demise marked by uncontrolled lipid peroxidation [4] (Fig. 1). Traditional cell death effectors, such as caspases, GSDMD (gasdermin D), and MLKL (mixed lineage kinase domain like pseudokinase), are not essential for the process of ferroptosis [4]. Ferroptosis can be categorized as a type of regulated necrosis and exhibits some morphological characteristics reminiscent of necrotic cell features, such as plasma membrane rupture [5]. Mechanistically, ferroptosis is mediated by the generation of toxic oxidized lipids, including 4-hydroxynonenal [6], as a result of lipid peroxidation [7]. Additionally, advanced lipid peroxidation end products can lead to oxidative damage to proteins or nucleic acids, causing cellular dysfunction [8]. Conversely, various antioxidant systems, comprising both GPX4 (glutathione peroxidase 4)-dependent and GPX4-independent pathways, play a context-dependent role in defending against ferroptosis [9,10,11,12,13,14,15,16,17,18].

A summary of the process of ferroptosis. Ferroptosis is a form of regulated necrosis primarily driven by lipid peroxidation, resulting in membrane damage and rupture. Increased production of ROS (reactive oxygen species) from endogenous or exogenous sources can initiate lipid peroxidation of PUFA (polyunsaturated fatty acids), which are a major component of cell membranes. The toxic byproducts of lipid peroxidation, including early-stage lipid hydroperoxides and late-stage aldehydes, as well as aldehyde-modified molecules, impair membrane structure and function. In contrast, various defense systems are capable of blocking or delaying this oxidative damage
In recent years, there has been growing interest in using ferroptosis activators as a cancer treatment approach [19]. Tumor cells with metastatic potential or resistance to apoptosis-based therapies have demonstrated susceptibility to ferroptosis. Various interventions, including experimental compounds and clinically used drugs (Table 1), have shown promise in inducing ferroptosis for cancer therapy, even overcoming resistance to traditional treatments [20]. Furthermore, combination therapies involving ferroptosis induction hold potential to enhance the efficacy of conventional treatments (such as radiotherapy and immunotherapy), while preventing tumor recurrence [19, 21]. However, the current ferroptosis induction strategy can lead to significant side effects, highlighting the need for a deeper understanding of its role in cancer therapy [22].
In this review, we discuss the benefits and risks of ferroptosis-based antitumor therapy. Our aim in this introduction is to provide insights into strategies for mitigating these limitations and optimizing ferroptosis-based cancer treatments.
The discovery of ferroptosis
Erastin and RSL3 are two well-known small molecule compounds that are frequently used to trigger ferroptosis and explore the associated mechanisms of this cell death pathway. However, the discovery and utilization of erastin and RSL3 preceded the formal recognition of the term 'ferroptosis'.
In 2003, the laboratory of Brent Stockwell utilized synthetic lethal high-throughput screening, assessing a vast library of 23,550 compounds for their ability to selectively eliminate engineered tumorigenic cells (BJ-TERT/LT/ST/RASV12) while sparing their isogenic normal cell counterparts [51]. Through this screening, they identified erastin as a novel compound capable of selectively killing engineered tumorigenic cells with RAS mutations, but not wild type cells. This cell death induced by erastin was found to be caspase-independent [51]. In 2008, the same research group conducted a similar screening involving 47,725 compounds to target BJ-TERT/LT/ST/RASV12 cells [52]. This effort led to the discovery of RSL3, another compound that selectively kills RAS-mutant cells through a caspase-independent mechanism. In contrast to erastin, RSL3 activates a similar death mechanism, but in a mitochondrial VDAC (voltage-dependent anion channel)-independent manner.
Subsequent studies have shown that erastin-induced cell death in the fibrosarcoma cell line HT1080 and the lung cancer cell line Calu-1 relies on iron accumulation, consequent oxidative damage through the activation of the Fenton reaction, and inhibition of system xc- [4]. System xc- is an amino acid antiporter that facilitates the exchange of extracellular cystine and intracellular glutamate across the cellular plasma membrane. It comprises a heavy chain component, SLC3A2 (solute carrier family 3 member 2), and a transport module, SLC7A11 (solute carrier family 7 member 11) [53]. The term 'ferroptosis' was introduced in 2012 to describe this iron-dependent, non-apoptotic form of cell death. Subsequent research has demonstrated that RSL3 directly binds to and inhibits GPX4, a downstream molecule in the cystine uptake and subsequent GSH (glutathione) synthesis pathway mediated by the system xc-, following the reduction of cystine to cysteine [9]. The current list of GPX4 inhibitors is expanding, with ML210 standing out as a covalent GPX4 inhibitor, utilizing intracellular drug metabolism to target the selenocysteine residue of GPX4 [27]. Nevertheless, ongoing debates persist regarding whether RSL3 and ML210 should selectively target GPX4, rather than other proteins in this context [26]. While GSH is the preferred co-factor for GPX4, it can also utilize other thiol-containing proteins as reductants [54]. In general, classical ferroptosis is triggered by erastin and RSL3, both of which inhibit the system xc--GSH-GPX4 pathway. This inhibition results in the accumulation of ROS (reactive oxygen species) within cells, ultimately leading to unrestricted lipid peroxidation.
It's worth mentioning that the early mechanisms of ferroptosis exhibit similarities to oxytosis, a form of cell death observed in neuronal cells, including the HT-22 cell line [55]. Oxytosis involves glutamate-induced inhibition of system xc-, subsequent depletion of GSH, production of ROS, and activation of lipoxygenases and heat shock proteins [56]. The depletion of cystine or cysteine in the cell culture medium can also trigger ferroptotic cell death [57]. However, our current understanding of ferroptosis extends beyond the oxytotic phenotype, as we will discuss later, due to the discovery of GPX4-independent ferroptotic pathways. Furthermore, the induction of ferroptosis is not limited to cells with RAS mutations but has been observed in normal tissues or RAS wild-type cells [58, 59], highlighting the context-dependent nature of ferroptosis.
Positive regulators of ferroptosis
Given the intricate connection between the ferroptotic process and oxidative stress-induced lipid peroxidation, the synergistic interplay of ROS amplification, lipid provisioning, and activation of lipid peroxidation enzymes collectively contribute to fostering ferroptosis induction or augmenting ferroptosis sensitivity (Fig. 1).
ROS generation
ROS are chemically reactive molecules containing oxygen, typically produced as natural byproducts of cellular metabolism. They are essential for various physiological processes within cells. However, excessive accumulation of ROS can lead to oxidative stress, causing damage to cellular components (e.g., DNA, proteins, and lipids), and potentially contributing to various diseases. There are three main sources that generates ROS for ferroptosis:
-
1) Mitochondria: Mitochondria serve as the primary source of ROS during oxidative phosphorylation, a process vital for cellular energy generation. Electrons escaping from the electron transport chain can react with molecular oxygen, leading to the production of O2·− (superoxide radicals), a type of ROS. Mitochondrial ROS can act as triggers for ferroptosis, while the presence of mitochondrial antioxidant systems, comprised of enzymatic and non-enzymatic antioxidants, can mitigate ferroptosis [50, 57, 60,61,62,63,64,65,66]. Multiple mitochondrial metabolic pathways influence ATP (adenosine triphosphate) and ROS generation, thereby influencing ferroptosis sensitivity. For instance, glutaminolysis assumes particular importance in scenarios with limited glucose availability, such as in rapidly growing tumors. Cancer cells often depend on glutaminolysis to fulfill their high energy and biosynthetic demands. However, glutaminolysis can promote ferroptotic death triggered by deprivation of full amino acids or of cystine alone [57, 67]. The AMPK (AMP-activated protein kinase) functions as a critical cellular energy sensor. When activated in response to declining energy levels, AMPK promotes ATP production by enhancing the activity or expression of catabolic proteins. Simultaneously, it conserves ATP by inhibiting biosynthetic pathways. AMPK activation during energy deficiency can both inhibit ferroptosis, through phosphorylation of ACACA/ACC (acetyl-CoA carboxylase alpha) in MEFs (mouse embryonic fibroblasts) and human renal adenocarcinoma cells [68], and promote ferroptosis, by targeting BECN1 (beclin1)-mediated system xc- inhibition in human colorectal cancer cells [25] or regulating pyrimidinosome assembly in human cervical cancer cells [69]. These findings suggest the presence of a threshold for AMPK activity in regulating ferroptosis levels in cancer cells through various substrates or binding proteins.
-
2) NOXs (NADPH oxidases): These enzymes are specialized proteins that generate ROS as part of their normal function. While they serve crucial roles in immune responses and cellular signaling, they also contribute to ferroptosis. Increased expression of NOX can result in elevated ROS levels, thereby increasing sensitivity to ferroptosis [70,71,72]. The activity of NOX in ferroptosis is subject to regulation by various factors. For example, the tumor suppressor protein TP53/p53 (tumor protein p53) can inhibit ferroptosis in human colorectal cancer cells by binding to DPP4 (dipeptidyl peptidase 4) [70]. Conversely, TP53 deficiency promotes the accumulation of DPP4 on the cell membrane, where it forms a complex with NOX1, resulting in oxidative damage [70]. Arachidonic acid also has the capacity to enhance NOX1 activity through phosphorylation by PRKC/PKC (protein kinase C), thereby promoting ROS production [73]. Furthermore, 4-hydroxynonenal, a byproduct of lipid peroxidation, augments NOX1 activity and induces ferroptosis in HT1080 and Calu1 cells [6]. The ferroptosis-inducing effect of NOX1 activity can be counteracted by ALDH1B1 (aldehyde dehydrogenase 1 family member B1) in HT1080 and Calu-1 cells [6]. ALDH1B1 catalyzes the oxidation of aldehydes, converting them into their corresponding carboxylic acids, a process relevant to the development of colorectal and pancreatic tumors [74, 75].
-
3) The fenton reaction. The fenton reaction is a chemical process that occurs when H2O2 (hydrogen peroxide) interacts with a metal catalyst, usually Fe2+, resulting in the formation of highly reactive and destructive ·OH (hydroxyl radicals) [76]. This reaction is a notable contributor to oxidative stress and can cause damage to cellular components. Hydroxyl radicals target and harm the lipids found in cell membranes, namely lipid peroxidation. Lipid peroxidation can result in membrane destabilization, compromising the integrity of cells and potentially leading to ferroptosis. Apart from its involvement in the fenton reaction, iron can also enhance the activity of enzymes, such as ALOX (arachidonate lipoxygenase) family and POR (cytochrome p450 oxidoreductase), thereby increasing ferroptosis sensitivity. Consequently, alterations in iron metabolism, including processes, such as iron uptake, storage, utilization, and release, can modulate ferroptosis sensitivity. An extensively studied example is the induction of ferritinophagy, a selective form of autophagy that promotes the degradation of the iron storage protein ferritin in MEFs or pancreatic cancer cells, resulting in an increase in labile iron [77, 78]. This process has been demonstrated to enhance ferroptosis sensitivity in various disease models [79,80,81,82,83].
PUFA synthesis
PUFAs (polyunsaturated fatty acids) are a category of dietary fats characterized by having two or more double bonds within their molecular structure. These double bonds introduce kinks into the fatty acid chains, preventing them from closely packing together. Consequently, PUFAs typically maintain a liquid state at room temperature and are commonly referred to as "healthy" or "good" fats. PUFAs are essential for the human body and serve critical roles in various cellular functions. They become integrated into cell membranes, where they exert influence over membrane fluidity and flexibility. This, in turn, has a direct impact on the membrane's ability to function effectively in processes, such as signal transduction, the transport of molecules in and out of the cell, and the regulation of enzymes bound to the membrane [84].
PUFAs are highly susceptible to lipid peroxidation due to the presence of weak C-H bonds at the bis-allylic positions. Recent research has primarily focused on ω-6 PUFAs (e.g., linoleic acid, gamma-linolenic acid, dihomo-gamma-linolenic acid, arachidonic acid, and adrenic acid) and ω-3 PUFAs (e.g., alpha-linolenic acid, eicosapentaenoic acid, and docosahexaenoic acid) in the context of ferroptosis. Among these, arachidonic acid and adrenic acid are the primary substrates of lipid peroxidation during ferroptosis. It's important to note that free PUFAs themselves do not directly drive ferroptosis; they must be esterified into membrane phosphatidylethanolamines or cholesteryl esters to become lethal after peroxidation [84].
The downstream pathways mediated by ACSL4 (acyl-CoA synthetase long chain family member 4) result in the production of different PUFA-related acyl-CoA esters [85,86,87] (Fig. 2), even though ACSL4-independent ferroptosis can still occur [88, 89]. One pathway involves LPCAT3 (lysophosphatidylcholine acyltransferase 3), which incorporates PUFA into phosphatidylethanolamines in various types of cancer cells [85, 90]. The other pathway involves the activation of SOAT1 (sterol O-acyltransferase 1) in pancreatic cancer and leukemia cells, leading to the production of PUFA-cholesteryl esters instead of PUFA-phosphatidylethanolamines [91]. Both pathways contribute to lipid peroxidation, with PUFA-related acyl-CoA esters serving as substrates. The activity of ACSL4 can be enhanced by PRKCB/PKCβII (protein kinase C beta) in MDA-MB-231 (breast cancer cell line) and HT1080 cells, which catalyzes the phosphorylation of ACSL4 at Thr328 [92]. This suggests that ACSL4 phosphorylation is a crucial post-translational modification for regulating ferroptosis sensitivity.

PUFAs and MUFAs in ferroptosis. Both dietary and de novo synthesized PUFAs play a central role in propagating lipid peroxidation during ferroptosis. Notably, free PUFAs themselves do not directly initiate ferroptosis; they must first undergo esterification into membrane PEs (phosphatidylethanolamines) or CEs (cholesteryl esters) following peroxidation to become lethal. ACSL4 regulates PUFA-PE synthesis through LPCAT3 and PUFA-CE synthesis via SOAT1. TMEM164 forms ferroptotic C20:4 ether phospholipids or induces autophagy, whereas TMEM189 is involved in peroxisome-mediated plasmalogen biosynthesis for lipid peroxidation. The POR and ALOX families mediate lipid peroxidation, leading to the production of PUFA-OOH, whereas ACSL3-mediated metabolism of MUFAs can exert an inhibitory effect on ferroptosis. The mitochondrial transporter SLC25A22 inhibits ferroptosis, in part, through the de novo synthesis of MUFAs. Furthermore, in ER+ breast cancer and AR+ prostate cancer cells, MBOAT1 and MBOAT2, upregulated by sex hormone receptors, inhibit ferroptosis through the production of MUFA-PLs, respectively
Additional sources of PUFAs for lipid peroxidation include the peroxisome-mediated biosynthesis of plasmalogens through TMEM189 (transmembrane protein 189) in human ovarian and kidney cancer cell lines [93, 94] and lipophagy-mediated lipid droplet degradation in human liver cancer cell lines [95, 96], although the initiation signals for these alternative pathways are not well understood. Recently, TMEM164 (transmembrane protein 164) was identified as a mediator of ferroptosis, with multifunctional roles as an acyltransferase involved in the synthesis of C20:4 ether phospholipids in renal carcinoma 786-O cells [97] or in driving the formation of phagophores, leading to excessive autophagosome formation in HT1080 and pancreatic cancer cell line PANC1 [98]. These diverse sources of PUFAs during ferroptotic stimuli highlight the heterogeneity and plasticity of ferroptosis machinery.
Notably, not all lipids contribute to increased lipid peroxidation. In contrast, ACSL3 (acyl-CoA synthetase long chain family member 3)-mediated metabolism of MUFAs (monounsaturated fatty acids) can inhibit ferroptosis, possibly due to its structural differences from PUFAs [7, 99,100,101] (Fig. 2). The mitochondrial glutamate transporter SLC25A22 (solute carrier family 25 member 22) inhibits ferroptosis in pancreatic cancer cells by remodeling lipid metabolism pathways and promoting the production of GSH and MUFAs [102]. SLC25A22 achieves this by utilizing NADPH and stimulating the activity of the enzyme SCD/SCD1 (stearoyl-CoA desaturase) [102]. Moreover, MBOAT1 (membrane-bound O-acyltransferase domain-containing 1) and MBOAT2 (membrane-bound O-acyltransferase domain-containing 2), which are transcriptionally upregulated by sex hormone receptors, exert inhibitory effects on ferroptosis in ER+ breast cancer and AR+ prostate cancer cells, respectively [11]. They achieve this by remodeling the cellular phospholipid profile, ultimately leading to the synthesis of phospholipids rich in MUFA [11].
Hence, adjusting the balance between PUFA and MUFA levels is critical for regulating ferroptosis sensitivity. Nonetheless, lipid metabolism is a complex process with numerous interconnected steps. Understanding the bioavailability and absorption of PUFAs and MUFAs from diverse food sources and dietary supplements is vital, as it may play a context-dependent role in preventing or enhancing the ferroptotic process and its health implications.
Lipid peroxidation
Lipid peroxidation stands as the defining characteristic of ferroptosis. The susceptibility of lipids to oxidation hinges on both the surrounding chemical environment and their inherent molecular structure. PUFAs are particularly prone to peroxidation due to the presence of bis-allylic moieties within their structure. The oxidation of PUFA during ferroptosis occurs through two primary mechanisms: enzymatic reactions and non-enzymatic autoxidation, driven by the Fenton reaction.
In enzymatic lipid peroxidation, the oxidation of PUFA is predominantly mediated by enzymes known as ALOXs and POR (Fig. 2). ALOXs, which are nonheme iron-containing enzymes, directly introduce molecular oxygen into PUFAs and PUFA-containing lipids within biological membranes. For instance, ALOX12 plays a crucial role in TP53-dependent ferroptosis [88]. ALOX15 is essential for ferroptosis induced by compounds (e.g., erastin or RSL3), as it forms a complex with PE binding protein 1 (PEBP1), specifically recognizing stearoyl-arachidonoyl-phosphatidylethanolamines and generating lipid peroxides [103]. Additionally, ALOXE3, ALOX5, ALOX12B, and ALOX15B have been implicated in ferroptosis induction, including in various cancer cells, in a context-dependent manner. Certain ALOX inhibitors (e.g., baicalein) protect cells against lipid peroxidation and ferroptosis [104]. However, it is important to note that genetic deletion of Alox15 in Gpx4 conditional knockout mice in kidney did not prevent ferroptosis in vivo [58]. Genetic deletion of Alox12 and Alox15 also failed to restore the viability of Gpx4-deficient T cells [59]. This suggests the existence of an ALOX-independent mechanism leading to ferroptosis in GPX4 knockout mice.
POR, on the other hand, directly provides electrons to P450 enzymes, facilitating the catalysis of PUFA peroxidation in an ALOX-independent manner in skin, ovarian, and kidney cancer cell lines [105, 106]. While POR expression is widespread, the expression of ALOX family members is highly cell- or tissue-specific. These observations collectively underscore the role of various iron-dependent enzymes, including ALOX and POR, in promoting lipid peroxidation and ferroptosis. Further investigations are necessary to explore the potential involvement of other oxygenases, such as cyclooxygenases and peroxygenases, in lipid peroxidation.
Nonenzymatic peroxidation of lipids is catalyzed by redox-active metals, with iron playing a prominent role. In non-enzymatic lipid peroxidation, when initial PLOOH (phospholipid hydroperoxides) are generated and not promptly reduced by the enzyme GPX4, they can react with ferrous ions, leading to the generation of alkoxyl and peroxyl radicals through the Fenton reaction. This process drives the propagation of PLOOH production to neighboring PUFA-phospholipids [23]. Furthermore, these toxic lipid products can react with nucleophilic amino acid residues within various proteins, forming covalent adducts and inducing protein lipoxidation [107]. If the defense system cannot effectively remove these toxic products, it can result in an irreversible point of no return in the progression of ferroptosis. Elucidating the precise mechanisms involved in this intricate process necessitates further investigation.
Negative regulators
The defense against ferroptosis encompasses various antioxidant systems and membrane repair mechanisms, all working in concert to interrupt lipid peroxidation reactions or eliminate compromised membranes.
GPX4-dependent antioxidant pathway
GPX4 stands out as the exclusive member within the GPX family recognized for its role as a phospholipid hydroperoxidase, directly transforming PLOOH into the corresponding PLOH (phospholipid alcohols) [108]. This distinctive function establishes GPX4 as the pivotal negative regulator of ferroptosis [9]. The catalytic mechanism employed by GPX4 adheres to a ping-pong mechanism, characterized by the dynamic interchange of the enzyme's active site between oxidation and reduction states (Fig. 3).

GPX4-dependent pathway in ferroptosis. The GPX4-dependent antiferroptotic pathway is associated with classical ferroptosis, a form of cell death driven by iron-dependent lipid peroxidation of PUFA. In this process, Fe3+ is transported into the intracellular space through binding to TF (transferrin) and its receptor TFRC. Within endosomes, STEAP3 converts intracellular Fe3+ to Fe2+, which can catalyze the production of .OH through the Fenton reaction, leading to lipid peroxidation. Conversely, storing Fe2+ in ferritin or exporting iron into the extracellular space via SLC40A1 can inhibit ferroptosis. Furthermore, the degradation of ferritin through ferritinophagy increases free Fe2+ levels, thereby promoting ferroptosis. GPX4 plays a pivotal role in this pathway by catalyzing the reduction of toxic PLOOH to non-toxic PLOH. The activity of GPX4 is tightly regulated by GSH (glutathione) and Se (selenium). Intracellular GSH levels are primarily controlled by system xc--mediated cystine uptake and subsequent cysteine transformation for GSH synthesis. In addition to the system xc-, methionine metabolism serves as an additional intracellular source of cysteine for the synthesis of GSH. The radical-trapping antioxidants ferrostatin-1 and liproxstatin-1 are the most used ferroptosis inhibitors
Initially, GPX4's active site selenol (GPX4-SeH) undergoes oxidation by PLOOH, resulting in the formation of a selenenic acid intermediate (GPX4-SeOH). Subsequently, this intermediate interacts with GSH, leading to the creation of a selenium-glutathione adduct (GPX4-Se-SG) [10]. The system xc-, which plays a role in importing cysteine for GSH synthesis, is a significant upstream modulator in this process. Finally, through a reaction with a second GSH molecule, GPX4-Se-SG is converted back to GPX4-SeH, simultaneously generating oxidized glutathione (GSSG). A crystal structure analysis of seleno-GPX4 has unveiled the presence of seleninic acid (GPX4-Se-OO-) within the enzyme's active site [109]. This finding suggests an alternative reaction mechanism characterized by three distinct redox states (GPX4-SeH, GPX4-SeOH, and GPX4-Se-OO-) involving the catalytically active selenocysteine. These studies underscore the role of selenocysteine in GPX4 expression and activity [10].
To prevent hydroperoxide-induced ferroptosis, it is imperative to substitute selenocysteine with a cysteine residue (U46C) in GPX4 [110]. Furthermore, the R152H mutation in GPX4 can lead to Sedaghatian-type spinal metaphyseal dysplasia, a rare and fatal disease in newborns [111]. In vitro studies suggest that this R152H mutation doesn't directly impact the enzyme's catalytic activity but rather interferes with its allosteric activation by cardiolipin [112]. In the context of ferroptosis, the expression of GPX4 protein is regulated by either the ubiquitin-proteasome system or the autophagy degradation pathway [36, 113,114,115,116,117]. CKB (creatine kinase B)-mediated phosphorylation of GPX4 at serine residue 104 inhibits autophagy-mediated GPX4 degradation and subsequent ferroptosis in human liver cancer cells [116]. However, the exact checkpoint governing these two degradation pathways of GPX4 remains unclear.
GPX4 exists in three isoforms: mitochondrial, cytosolic, and nuclear GPX4 [108]. All of these isoforms are encoded by the same GPX4 gene, but have different transcription initiation sites. When the cytosolic GPX4 is genetically ablated or rendered inactive, it leads to early embryonic lethality [118]. Interestingly, reintroducing cytoplasmic GPX4, rather than its mitochondrial or nuclear counterparts, can rescue the lethal phenotype in Gpx4-null mice [119]. Generally, cytosolic GPX4 plays a prominent role in preventing ferroptosis. However, the question of whether mitochondrial GPX4 inhibits ferroptosis remains a subject of debate [13, 120, 121], and there is currently no research establishing the role of nuclear GPX4 in ferroptosis.
Global Gpx4 knockout mice do not survive, but the phenotype of conditional Gpx4 knockout mice depends on the specific cell or tissue involved. For instance, the conditional knockout of Gpx4 in kidney cells, T cells, or B cells can induce ferroptotic damage, which can be reversed by supplementing with vitamin E or the ferroptosis inhibitor liproxstatin-1 [58, 59, 122, 123]. Conversely, the conditional knockout of Gpx4 in myeloid cells, blood progenitor cells, and sperm cells can increase pyroptosis, necroptosis, and apoptosis in response to specific stresses [120, 124, 125]. Therefore, the antioxidant function mediated by GPX4 plays a context-dependent role in both ferroptosis and non-ferroptotic cell death.
GPX4-independent antioxidant pathway
Although the ER (endoplasmic reticulum) has been suggested as the primary site for the initiation of ferroptosis [126], various cellular compartments contribute to the ferroptosis process [127]. Consequently, different antioxidant systems possess unique capabilities and operate within distinct cellular locales to inhibit ferroptosis. GPX4 has a prominent function within the cytosol, actively inhibiting ferroptosis. Simultaneously, various GPX4-independent enzymes contribute to ferroptosis resistance in distinct cellular compartments (Fig. 4), primarily by generating radical-trapping antioxidants, with a notable emphasis on COQ10 (coenzyme Q10).

GPX4-independent pathway in ferroptosis. Organisms possess intricate networks of antioxidant and enzyme systems that collaborate to prevent lipid peroxidation-mediated ferroptosis. Apart from GPX4, several GPX4-independent pathways and proteins are involved. Specifically, AIFM2 (also known as FSP1) and DHODH act as ferroptosis suppressors by reducing COQ10 to COQ10H2. This process is facilitated by STARD7. The PARL-mediated cleavage of STARD7 occurs during its import into mitochondria, leading to the release of a portion of mature STARD7 into the cytosol. This cytosolic fraction delivers mitochondria COQ10 to the plasma membrane. AIFM2 also inhibits ferroptosis by either generating reduced vitamin K or activating the ESCRT-III membrane repair pathway. Furthermore, ER stress can induce ATF4-dependent expression of TXNDC12, which serves to limit lipid peroxidation
AIFM2/FSP1 (apoptosis inducing factor mitochondria associated 2), a member of the apoptosis-inducing factor family located in mitochondria, has garnered attention as a potent suppressor of ferroptosis in diverse cancer cells, including HT1080 [12, 15]. It achieves this by translocating from mitochondria to the cell membrane and participating in the reduction of COQ10 to COQ10H2. STARD7 (StAR-related lipid transfer domain-containing 7), located in the intermembrane space of mitochondria and in the cytosol after cleavage by the rhomboid protease PARL (presenilin-associated rhomboid-like), is involved in transporting COQ10 to the plasma membrane AIFM2, thus suppressing ferroptosis in HeLa (human cervical cancer cell line) and HCT116 (human colorectal cancer cell line) cells [128]. Furthermore, AIFM2 contributes to the inhibition of ferroptosis by engaging in membrane repair in pancreatic cancer cells (PANC1) and liver cancer cells (HepG2) [129] and is also involved in the reduction of vitamin K in 786-O (human renal carcinoma cell line) and A375 (human melanoma cell line) cells, which includes warfarin-resistant vitamin K reduction [14, 130, 131]. The involvement of AIFM2 in ferroptosis is influenced by phase separation, a process in which a homogeneous mixture of substances spontaneously separates into distinct phases, and can be initiated through N-terminal myristoylation, which is facilitated by a compound called icFSP1 in a variety of human cancer cells [14].
DHODH (dihydroorotate dehydrogenase (quinone)), a flavin-dependent mitochondrial enzyme involved in de novo pyrimidine biosynthesis, has garnered attention as a potential therapeutic target for various diseases. DHODH inhibitors (e.g., BAY 2402234, PF-06726209, and brequinar) have been developed for potential use in cancer treatment and immunosuppression. The activity of DHODH influences the vulnerability of cancer cells with low GPX4 levels to ferroptosis, likely due to DHODH-mediated utilization of COQ10 as an electron acceptor in HT1080 and NCI-H226 (human lung cancer cell line) cells [13]. Inhibiting DHODH activity leads to a reduction in COQ10 levels, compromising antioxidant capacity and increasing susceptibility to lipid peroxidation and ferroptosis [13]. Nonetheless, there is an ongoing debate regarding the potential unintended effects of the DHODH inhibitor brequinar on AIFM2 in cancer cells [132, 133].
TXNDC12 (thioredoxin domain-containing protein 12), also known as ERp18 or ERp19, plays a role in inhibiting ferroptosis independently of GPX4 in human leukemia cells within the ER [134]. Its expression is increased in human leukemia cells during ferroptosis due to the activation of transcription factor ATF4 (activating transcription factor 4) [134]. Solid cancer cells with higher baseline TXNDC12 expression tend to exhibit resistance to ferroptosis [135]. Conversely, genetic knockdown of TXNDC12 enhances ferroptosis sensitivity, leading to increased lipid peroxidation both in vitro and in vivo [134, 135]. Hence, targeting TXNDC12 could prove to be a valuable strategy in cancer therapy.
Redundancy in antioxidant systems acts as a backup mechanism to safeguard cellular integrity and function in the face of oxidative stress, reducing the risk of damage. Other antioxidant enzymes, including GCH1 (GTP cyclohydrolase 1) [136], NOS2/iNOS (nitric oxide synthase 2) [137], MGST1 (microsomal glutathione S-transferase 1) [138], GSTZ1/maleylacetoacetate isomerase (glutathione S-transferase zeta 1), TXNRD1 (thioredoxin reductase 1) [139], PLA2G6/iPLA2β/PNPLA9 (phospholipase A2 group VI) [16, 17], and peroxiredoxins (PRDX) [140], also inhibit ferroptosis in a context-dependent manner, even in GPX4- or AIFM2-knockout cancer cells. Each of these pathways plays a unique and complementary role, collectively providing comprehensive protection against oxidative damage during ferroptosis.
Metal ions, particularly iron, play pivotal roles in initiating oxidative stress and triggering ferroptosis. To counteract these detrimental reactions, cells employ a range of metal-binding proteins, such as transferrin (TF) and ferritin, which efficiently sequester free iron [77, 78]. Intracellular metal equilibrium is meticulously upheld by specialized proteins, including metal chaperones that are tasked with delivering metals to their intended protein targets. Metallothioneins, a family of cysteine-rich proteins, also contribute significantly to regulating the availability of metal ions, thereby reducing their participation in oxidative damage and the ferroptosis process in human liver cancer cells (HepG2) [141]. Furthermore, certain clinical drugs with metal-chelating properties have shown promise in preventing ferroptosis by interfering with iron-related processes. Examples of these drugs include deferoxamine, deferiprone, deferasirox, and ciclopirox [142]. These compounds hold potential as therapeutic agents that can mitigate the impact of iron-mediated oxidative stress, thereby offering a new avenue for intervention in diseases where ferroptosis plays a significant role.
Membrane repair system
The membrane repair system operates to mitigate and reverse damage to the plasma membrane resulting from mechanical stress, injury, or disruption. When the membrane is compromised, cellular mechanisms are activated to restore its integrity, thereby reducing the risk of ferroptosis.
Ca2+ plays a central role in initiating membrane repair. Following plasma membrane damage, Ca2+ enters the cytoplasm from extracellular sources, serving as a signal to trigger downstream repair processes such as ESCRT (endosomal sorting complexes required for transport)-III and exocytosis. Ca2+ signaling from different organelles has a dual impact on ferroptosis sensitivity, highlighting the need for timely signal monitoring.
During cellular stress, which includes oxidative stress associated with ferroptosis, ESCRT-III components are mobilized to sites of membrane damage, where they play a critical role in facilitating repair and preventing irreversible damage that can lead to ferroptosis [143]. For instance, when there is an influx of Ca2+ in pancreatic cancer cells, specific subunits of ESCRT-III, referred to as CHMPs (charged multivesicular body proteins), particularly CHMP5 and CHMP6, are recruited and assembled at the damaged site, actively contributing to the restoration of membrane integrity in liver and pancreatic cancer cells [143]. In contrast, the knockout of either CHMP5 or CHMP6 increases the susceptibility of pancreatic cancer cells (PANC1) to ferroptosis [143].
Exocytosis constitutes another crucial aspect of the membrane repair system, where intracellular vesicles fuse with the plasma membrane to facilitate repair. This essential process, involved in the transport and secretion of diverse molecules, including proteins, lipids, neurotransmitters, and hormones, plays a protective role. It particularly safeguards neurons from ferroptosis by facilitating the release of lipids and iron from autolysosomes [144].
While repair mechanisms in the plasma membrane are well-studied, it remains unclear whether organelles employ similar mechanisms for repair. Further exploration of these mechanisms and their associated signaling pathways has the potential to pave the way for the development of targeted therapies aimed at addressing diseases associated with impaired ferroptosis. Additionally, the emerging field of understanding the role of membrane repair in immune responses, including inflammation and tissue repair, holds great promise and continues to garner interest.
Advantages of ferroptosis-based antitumor therapy
Extensive studies suggest that ferroptosis plays a pivotal role in tumor suppression, thus providing new opportunities for cancer therapy.
Personalized medicine
Identifying specific vulnerabilities in cancer cells that make them susceptible to ferroptosis allows for more personalized treatment, potentially improving outcomes. Below, we highlight three key gene mutation signals in the regulation of ferroptosis sensitivity.
RAS mutations, commonly found in various cancer types, promote abnormal cell growth and resistance to certain therapies. Ferroptosis, initially recognized as a targeted therapy for RAS-mutated cancer cells, offers selectivity in eliminating them while sparing normal cells [4]. Blocking the RAS pathway or its downstream RAF-MEK-ERK axis can reverse the specific cytotoxic effects induced by erastin [145]. Additionally, mutations in EGFR (epidermal growth factor receptor), upstream of RAS, enhance the susceptibility to ferroptosis induced by cystine depletion [146]. This susceptibility is likely attributed to the activation of the RAS-MAPK axis mediated by mutant EGFR in non-small cell lung cancer cells [146]. A recent study demonstrated that mutant KRAS elevates AIFM2 levels by activating MAPK and NFE2L2/NRF2 (NFE2 like BZIP transcription factor 2), thereby suppressing ferroptosis in pancreatic cancer cells [147]. Combining the induction of ferroptosis with the inhibition of defense mechanisms, such as AIFM2 or NFE2L2 inhibitors, holds promise as a potential treatment strategy for pancreatic cancers with KRAS mutations. In a similar fashion, mutant KRAS can activate NFE2L2-dependent expression of SLC7A11, resulting in elevated intracellular cysteine levels and increased GSH biosynthesis [148]. Inhibition of SLC7A11 by erastin can induce synthetic lethality in KRAS-mutated lung adenocarcinoma cells [148]. Taken together, these findings indicate that cancer cells harboring KRAS mutations are susceptible to ferroptosis induction.
Subsequent studies have revealed vulnerabilities associated with gene mutations, particularly TP53, impacting ferroptosis sensitivity. TP53, a tumor suppressor protein known as the 'guardian of the genome,' plays a crucial role in maintaining genomic stability and regulating cell cycle progression. For example, the acetylation-deficient TP53 variant, TP53[3KR], lacks the ability to induce apoptosis and cell cycle arrest but retains its tumor suppression function akin to wild-type TP53. It achieves this by suppressing SLC7A11 expression, leading to ferroptosis induction in various cancer cells [149, 150]. TP53 also influences ferroptosis sensitivity through other mechanisms, such as downregulating VKORC1L1 (vitamin K epoxide reductase complex subunit 1 like 1) in vitamin K metabolism [151] and inducing SAT1 (spermidine/spermine N1-acetyltransferase 1), an enzyme in polyamine catabolism that produces ROS [152]. Conversely, TP53 can inhibit ferroptosis in certain conditions. For example, TP53 deletion in colorectal cancer cells increases ferroptosis sensitivity by activating DPP4-mediated NOX1 pathway [70]. TP53 mutations, such as R175H, can lead to modified TP53 proteins that act as suppressors of ferroptosis by preventing BACH1 (BTB domain and CNC homolog 1)-mediated downregulation of SLC7A11, thus promoting tumor growth [153]. These findings underscore the varied roles of TP53 in regulating ferroptosis sensitivity, which can vary depending on the specific tumor types.
KEAP1 (kelch like ECH associated protein 1), an enzyme often mutated in lung cancer, interacts with NFE2L2, leading to NFE2L2 degradation [154, 155]. Mutations in KEAP1 or the augmentation of SQSTM1 (sequestosome 1)-mediated KEAP1 protein degradation can enhance the stability of NFE2L2 protein, enabling cancer cells to evade ferroptosis and acquire drug resistance [34]. NFE2L2 plays a pivotal role in regulating antioxidant genes involved in both GPX4-dependent (e.g., GPX4 and SLC7A11) and GPX4 independent pathways (e.g., AIFM2 and MT1G [metallothionein 1G]) [34, 83, 156]. In TNBC (triple-negative breast cancer), methylation of KEAP1 by PRMT5 (protein arginine methyltransferase 5) results in decreased levels of NFE2L2 and its downstream target gene HMOX1 (heme oxygenase 1), thereby influencing the sensitivity of the cancer cells to ferroptosis [157]. These findings also demonstrate an epigenetic strategy for controlling ferroptosis sensitivity in TNBC cells by modulating the KEAP1-NFE2L2 pathway. p14ARF, also known as ARF tumor suppressor, is a protein product encoded by the alternate reading frame of the CDKN2A (cyclin dependent kinase inhibitor 2A) locus. p14ARF enhances ferroptosis, both with and without TP53, by inhibiting NFE2L2, particularly in TP53-knockout cells. Loss of p14ARF activates NFE2L2, aiding cancer cell survival during oxidative stress and ferroptosis [158]. Apart from KEAP1, other oncogenes, such as KRASG12D [147], BRAFV619E [159], and MYCERT2 [159], can increase NFE2L2 expression or stability, maintaining cellular antioxidant defenses and potentially defending against ferroptosis. Overall, dysregulation of NFE2L2 or its downstream pathways can lead to increased susceptibility to ferroptosis, making it an attractive target for research and potential therapeutic interventions in certain types of cancer.
In addition to genetic mutations, recent reviews have highlighted the influence of multiple tumor-related signaling pathways on ferroptosis sensitivity. These pathways, including MTOR (mechanistic target of rapamycin kinase) signaling [31, 43, 160], hypoxia response [161,162,163], the Hippo pathway [164, 165], and autophagic degradation [116, 166,167,168,169,170,171,172], play context-dependent roles in determining ferroptosis susceptibility. Moreover, bioinformatic analyses have uncovered aberrant expression patterns of ferroptosis regulator genes in cancer patients. The development of a ferroptosis potential index for patients offers a valuable tool for predicting disease progression, evaluating immune responses, and characterizing metabolic alterations.
Overcoming drug resistance
The persistence of cancer cells, including drug-tolerant populations, presents a substantial challenge in both preclinical research and clinical practice. Some cancer cells resist conventional therapies due to apoptosis deficiencies, rendering them resistant to elimination. Ferroptosis-based therapies show promise in targeting vulnerabilities unique to these resistant cells, offering a potential solution.
Acquired drug resistance frequently hinders cancer treatments from achieving stable and complete responses. Recent evidence highlights the significance of non-mutational drug resistance mechanisms in the survival of residual cancer 'persister' cells [173]. These persister cells act as a reservoir from which drug-resistant tumors can potentially emerge, making targeting them an attractive therapeutic opportunity to prevent tumor relapse. Cancer cells in a high mesenchymal therapy-resistant state rely on the lipid hydroperoxidase GPX4 for survival [174, 175]. Preclinical studies have demonstrated the efficacy of GPX4 inhibitors, such as RSL3 or ML210, in eliminating persister cancer cells in vitro and in xenograft models [174, 175].
In addition, there is a scarcity of ferroptosis activators capable of targeting cancer stem cell niches and populations of cells that inherently tolerate therapy or acquire drug resistance [176,177,178]. These findings support the potential clinical utility of GPX4 inhibitors to prevent tumor relapse or to combat cancer cells that can adopt a therapy-resistant state.
Complementary to other therapies
Ferroptosis-based treatments can be employed in combination with existing cancer therapies (e.g., chemotherapy, radiotherapy, or immunotherapy) to potentially enhance overall treatment efficacy. Combining ferroptosis activators with traditional chemotherapeutic drugs (e.g., temozolomide, oxaliplatin, cisplatin, gemcitabine, and 5-fluorouracil) or targeted drugs (e.g., olaparib, cetuximab, and sunitinib) may help overcome drug resistance or enhance their effects. Radiotherapy itself can induce ferroptosis in tumor cells, and the addition of ferroptosis agonists can improve radiation therapy outcomes in tumor models. For instance, in vitro and in vivo studies using cancer cell lines (HT1080, HeLa, NCI-H1975 [human non-small cell lung cancer cell line], and B16F10 [human melanoma cell line]) demonstrated that combining ferroptosis activators with radiation therapy reduced tumor growth compared to radiation therapy alone [179,180,181,182]. Mechanistically, this combination therapy not only inhibits the expression of anti-ferroptotic proteins, such as GPX4 and SLC7A11, but also induces the expression of ACSL4, leading to increased lipid peroxidation and ferroptosis [183]. In cancer cells, PARP1 (poly(ADP-Ribose) polymerase 1) and PARP2 can activate DNA repair pathways to prevent DNA damage and cell death caused by radiotherapy. When radiotherapy is combined with PARP inhibitors (e.g., niraparib), it can induce DNA damage and activate the CGAS (cyclic GMP-AMP synthase)-STING1 (stimulator of interferon response cGAMP interactor 1) pathway. This activation, in turn, triggers tumor ferroptosis and subsequently stimulates CD8+ T cells-mediated antitumor immunity in colorectal cancer models [184]. In addition to its benefits in inhibiting tumor growth, ACSL4-dependent ferroptosis is implicated in radiation-induced intestinal injury, a common gastrointestinal complication resulting from radiotherapy [185]. This highlights that ferroptosis can also be a side effect of radiation-induced damage in normal tissues.
The sensitivity of ferroptotic cancer cell death is influenced by the release of cytokines from immune cells. IFNG/IFN-γ (interferon gamma), a versatile cytokine released by CD8+ T cells or NK (natural killer) cells, is a crucial effector molecule in immunotherapy. IFNG can enhance the cell death of tumor cells (HT1080, A375, and ID8 [murine ovarian surface epithelial cell line]) induced by ferroptosis activators, such as erastin, RSL3, or cysteine depletion [186]. This effect is partly achieved by downregulating SLC7A11 through its receptor and activating the JAK (Janus kinase)-STAT1 (signal transducer and activator of transcription 1) pathway in cancer cells [186] (Fig. 5A). In addition, IFNG stimulates ACSL4 expression through an IRF1 (interferon regulatory factor 1)-dependent mechanism, thereby augmenting arachidonic acid-related ferroptosis in animal models and impeding tumor growth [187] (Fig. 5A). Combining ferroptosis inducers with immune checkpoint inhibitors, such as anti-CTLA4 (cytotoxic T-lymphocyte associated protein 4), anti-PDCD1/PD-1 (programmed cell death 1), or anti-CD274/PD-L1 (programmed death ligand 1) antibodies, enhances tumor suppression compared to using immune checkpoint inhibitors alone. Conversely, tumor-associated macrophage-derived TGFB1/TGFβ1 (transforming growth factor beta 1) suppresses ferroptosis through the activation of the HLF (HLF transcription factor, PAR BZIP family member)-GGT1 (gamma-glutamyltransferase 1)-GSH-GPX4 pathway in TNBC cells [188] (Fig. 5B). Moreover, IL1B/IL-1β (interleukin 1 beta) stimulation leads to KAT2B (lysine acetyltransferase 2B)-dependent acetylation and activation of NNT (nicotinamide nucleotide transhydrogenase) in gastric cancer cells, resulting in increased production of NADPH and the preservation of Fe-S clusters within mitochondria [189] (Fig. 5C). This mechanism functions to shield tumor cells from ferroptosis and immune-based therapies. However, IL1B is also a mediator of immunogenic cell death driven by pyroptotic cell death [190], adding complexity to the feedback mechanisms involving different cell death and immune mediators.

Effects of cytokines produced by immune cells on ferroptosis in cancer cells. A IFNG production by CD8+ T cells or natural killer cells can promote ferroptosis in cancer cells through STAT1-dependent downregulation of SLC7A11 or IRF1-dependent upregulation of ACSL4. B TGFB1 production by macrophages can inhibit ferroptosis in cancer cells by enhancing HLF-dependent GSH synthesis through GGT1, ultimately leading to the activation of GPX4. C IL1B production by macrophages can inhibit ferroptotis in cancer cells through KAT2B-dependent acetylation. This acetylation enhances NNT's affinity for NADP+ and leads to increased production of NADPH, thus promoting the maintenance of Fe‐S clusters
Side effects of ferroptosis-based antitumor therapy
There is growing evidence that current ferroptosis activators can trigger cell death in normal cells, resulting in adverse effects during cancer therapy.
Immune cell death
Recent studies have shown that classical ferroptosis activators or oxidized lipids can inadvertently lead to the death of these crucial antitumor immune cells. Among these immune cells are cytotoxic CD8+ T cells, specialized in directly recognizing and eliminating cancer cells by targeting cancer-specific antigens on their surfaces. Inducing ferroptosis in CD8+ T cells has been found to dampen antitumor immunity in mouse models of clone cancer (using B16 or MC38 cell line) in B6 mice [191, 192] (Fig. 6A). This effect is linked to the expression of CD36, a cell surface protein known for its roles in lipid metabolism, including fatty acid and lipoprotein uptake, as well as its involvement in innate immunity and inflammation. The uptake of OxLDL (oxidized low-density lipoproteins) or PUFA by CD36 can trigger GPX4 downregulation and MAPK14/p38 (mitogen-activated protein kinase 14) phosphorylation, ultimately leading to lipid peroxidation and ferroptosis or functional exhaustion of cytotoxic CD8+ T cells in mouse models of clone cancer in B6 mice [191, 192]. These effects can be prevented by ferroptosis inhibitors or by CD36 deficiency [191, 192]. GPX4 is not essential for T cell development, but plays a crucial role in maintaining T cell homeostasis and supporting T cell-dependent immune responses during acute viral infections [59]. Moreover, overexpressing GPX4 or depleting CD36 can rescue effector functions of cytotoxic CD8+ T cells, enhancing their ability to control tumor growth deficiency [191, 192]. CD8+ T cells can be categorized into various functional subsets, including Tc1 cells that produce high amounts of IFNG and Tc9 cells that produce IL9 (interleukin 9) at lower levels of IFNG. Unlike conventional cytotoxic CD8+ Tc1 cells, CD8+ Tc9 cells activate the IL9-STAT3-fatty acid oxidation pathway in B16 tumor–bearing Thy1.2+ B6 mice [193] (Fig. 6B). This pathway provides protection against ROS-induced lipid peroxidation and ferroptosis in the tumor microenvironment [193]. Furthermore, Gpx4-deficient Treg (T regulatory) cells are vulnerable to ferroptosis and exhibit elevated IL1B production, which, in turn, enhances T Th17 (helper cell 17) responses in B6 mice inoculated with B16.F10 melanoma cells [194] (Fig. 6C). This impairment compromises Treg-mediated immunosuppression within the tumor microenvironment, ultimately restricting in vivo tumor growth [194].

Mechanisms of ferroptosis in immune cells and the effects of DAMP on ferroptosis-related tumor immunity. A-F Mechanisms of ferroptosis in immune cells within the tumor microenvironment. G The impact of ferroptotic cancer cells on tumor immunity, which depends on the stage of cell death, DAMP release, and the infiltration of immune cells within the tumor microenvironment
DCs (dendritic cells) are another type of immune cell that contributes to antitumor immunity by activating cytotoxic T cells. Interestingly, the GPX4 inhibitor RSL3 can impair the maturation and function of DCs in tumor suppression by activating PPARG/PPARγ (peroxisome oroliferator activated receptor gamma)-dependent ferroptosis in DC2.4 cell line [195] (Fig. 6D). Early-stage ferroptosis in the MCA205 mouse fibrosarcoma cell line treated by RSL3 for 1 hour exhibits immunogenicity when exposed to activated DCs, in contrast to late-stage ferroptosis induced by ML162 for 14 hours in the MCA205 mouse fibrosarcoma cell line [196, 197]. The potential mechanism responsible for these differences may involve a switch in the release of immunostimulatory danger/damage-associated molecular patterns (DAMPs) (e.g., ATP and reduced HMGB1 [high mobility group box 1]) to immune-suppressive DAMPs (e.g., oxidized HMGB1 or PGE2 [prostaglandin E2]) [198, 199]. PGE2 is an immunosuppressive lipid that hinders the anti-tumor activity of cDC1 (conventional type 1 DC), NK cells, and effector T cells, while also activating immunosuppressive cells, such as MDSCs (myeloid-derived suppressor cells) and Tregs, promoting immune escape.
NK cells represent yet another class of immune cells capable of targeting and eliminating cancer cells without prior sensitization or antigen recognition. However, tumor-associated NK cells within the tumor microenvironment of ovarian cancer patients exhibit increased expression of proteins related to lipid peroxidation, oxidative damage, ferroptosis, as well as senescence and autophagy pathways [200]. These alterations impair the cytotoxic activity of NK cells against ovarian cancer cell OVCAR8 in vitro [200]. L-Kynurenine is a naturally occurring molecule and a critical intermediate in the tryptophan metabolic pathway. In the context of cancer and immunology, L-kynurenine has garnered attention for its potential immunosuppressive effects. A recent study revealed that L-kynurenine can contribute to immune tolerance by inducing lipid peroxidation and ferroptosis in NK cells, ultimately promoting gastric cancer growth in in vivo experiments [201] (Fig. 6E). Thus, the modulation of the tryptophan metabolic pathway contributes to the development of potential immunotherapeutic strategies by influencing NK cell death through ferroptosis.
Furthermore, pathologically activated neutrophils, known as PMN-MDSCs (myeloid-derived suppressor cells), act as major negative regulators of antitumor immunity. While PMN-MDSCs in the tumor microenvironment undergo spontaneous ferroptosis, this process leads to the release of oxygenated lipids (e.g., oxidized AA-PEox) or PGE2, limiting the activity of both human and mouse T cells [202] (Fig. 6F). Inhibition of ferroptosis using liproxstatin-1 results in PMNs' depletion and enhances the effectiveness of anti-PDCD1/PD-1 antibody treatment on pancreatic cancer cell growth in immunocompetent mice [202]. These observations highlight the significant impact of both the particular cell undergoing ferroptosis and the release of DAMP within the tumor microenvironment on the functionality of cytotoxic CD8+ T cells (Fig. 6G).
To address this unintended consequence, it is crucial to develop strategies that can specifically target tumor cells while preserving the functionality of immune cells and their ability to surveil and combat cancer. In a recent study, a screening of a library containing more than 4000 small-molecule compounds identified N6F11 as the first cell-specific ferroptosis activator [36]. N6F11 selectively induces ferroptosis in cancer cells by targeting TRIM25 (tripartite motif containing 25)-mediated GPX4 degradation in pancreatic cells, both in vitro and in various mouse models, such as xenografts, orthotopic models, and transgenic models [36] (Fig. 7). TRIM25 is primarily expressed in cancer cells, rather than in immune cells (DC, T, NK, and neutrophil cells) [36]. This finding may represent a safe and effective approach to enhance ferroptosis-driven antitumor immunity. Additional research is needed to determine whether there are other TRIM25-dependent protein substrates involved in the selective induction of ferroptosis in cancer cells.

Strategy for GPX4 Inhibition. A Traditional GPX4 inhibitors, such as RSL3, lead to the loss of GPX4 activity. As GPX4 is widely expressed in both immune and cancer cells, they induce non-selective ferroptosis. B N6F11 can bind to TRIM25, subsequently triggering TRIM25-dependent GPX4 degradation to induce ferroptosis. Given that TRIM25 is mainly expressed in cancer cells, the use of N6F11 does not induce ferroptosis in non-cancer cells
Bone marrow impairment
Anticancer agents can disrupt the bone marrow's capacity to generate blood cells, potentially causing conditions, such as anemia, thrombocytopenia (low platelet count), or leukopenia (low white blood cell count). Inducing ferroptosis can lead to the death of stem cells and damage to the bone marrow, which may impact hematopoiesis and result in bone marrow suppression.
Recent research has shed light on the mechanisms involved. HSCs (hematopoietic stem cells) possess unique physiological adaptations that sustain lifelong blood cell production, including finely regulated protein synthesis rates. Disrupting MYSM1 (Myb like, SWIRM and MPN domains 1), a histone deubiquitinase, can compromise human HSC function, mimicking the bone marrow failure seen in patients, by triggering lipid peroxidation and ferroptosis [203]. Additionally, healthy HSCs are susceptible to ferroptosis induced by compounds such as erastin, FIN56, FINO2, and RSL3, primarily due to their low protein synthesis levels [203]. Therefore, inhibiting ferroptosis represents a potential strategy to augment HSC numbers, supporting the production of all blood cell types, including red blood cells (erythrocytes), white blood cells (leukocytes), and platelets (thrombocytes).
Furthermore, the activation of FANCD2 (fanconi anemia complementation group D2), a nuclear protein involved in DNA damage repair, plays a protective role against bone marrow injury caused by ferroptosis [204]. These discoveries suggest the existence of a nuclear protection pathway designed to alleviate ferroptotic damage in bone marrow dysfunction. Activators of FANCD2 have the potential to enhance its capacity to repair damaged DNA and shield cells from the detrimental consequences of DNA damage.
In light of these findings, monitoring the bone marrow and blood counts during ferroptosis-mediated therapy is crucial for assessing the impact of treatment on the hematological system.
Liver and kidney damage
Many cancer treatments, including chemotherapy and targeted therapies, go through hepatic metabolism and renal elimination processes, which can increase the exposure of these organs to potentially harmful substances. As a result, the liver and kidneys may be at risk of damage. Similar to some other anticancer agents, specific ferroptosis activators have the potential to induce hepatotoxicity (liver damage) or nephrotoxicity (kidney damage).
Research has shown that when Gpx4 is conditionally knocked out in the kidney or liver, it can lead to spontaneous ferroptotic damage [58, 205]. Fortunately, this damage can be reversed through treatment with vitamin E or liproxstatin-1. Therefore, there is a possibility that inhibition of GPX4, which promote ferroptosis, may have toxicity effects on the liver and kidneys [206].
To proactively manage this risk and ensure patient safety, it is crucial to closely monitor the function of the liver and kidneys throughout ferroptosis treatment. This can be achieved by conducting regular blood tests, including liver function tests and kidney function tests, such as measuring serum creatinine and blood urea nitrogen levels. These monitoring measures are indispensable for the early detection of any signs of damage or dysfunction in these vital organs.
Cachexia
Cachexia is a complex metabolic syndrome often associated with chronic illnesses, such as cancer. It is characterized by symptoms, such as weight loss and muscle wasting [207]. Ferroptosis-based treatments can contribute to the development of cachexia.
In a study involving mice fed a high-fat, low-carbohydrate ketogenic diet, the induction of ferroptosis effectively suppressed tumor growth [208]. However, it also resulted in systemic effects, including increased lipid peroxidation and decreased levels of NADPH, leading to early-onset cachexia and reduced survival rates [208]. To address these systemic side effects, researchers employed a pulsed administration of glucocorticoids. This approach demonstrated the potential for improving therapeutic outcomes by gaining a deeper understanding of the mechanisms behind these side effects.
Furthermore, GDF15 (growth differentiation factor 15), a member of the TGFB superfamily produced by stressed cells, plays a role in reducing food intake by binding to its receptor GFRAL (GDNF family receptor alpha like) in the area postrema [208]. While GDF15 levels are elevated in cachexia resulting from ferroptosis, specific laboratory markers to distinguish it from other forms of cachexia have yet to be identified.
Secondary tumorigenesis
Secondary tumorigenesis refers to the occurrence of new, unrelated tumors in individuals who have previously undergone cancer treatment. While cancer therapies are designed to target and eliminate cancer cells, they can inadvertently affect healthy cells and tissues, potentially leading to the development of new cancers. Although direct evidence is currently limited, several transgenic animal studies have suggested a potential role for ferroptotic damage in initiating tumorigenesis.
For instance, experiments involving high-iron diets or the depletion of Gpx4, which induces ferroptosis, promote pancreatitis and pancreatic tumorigenesis in mice [209, 210]. Conversely, inhibiting ferroptosis through the administration of liproxstatin-1 reduce spontaneous pancreatic cancer formation in a mouse model driven by KrasG12D mutations, suggesting that ferroptosis may play a role in pancreatic cancer development [209, 210]. This ferroptosis-induced pancreatic cancer development could be linked to macrophage infiltration and polarization, mediated by the release of 8-OHG (8-hydroxy-2′-deoxyguanosine) resulting from ferroptotic damage and subsequent activation of the STING1-dependent DNA sensor pathway [209, 210]. Moreover, the oncogenic KRAS protein can be released by ferroptotic pancreatic cells, promoting pro-tumor M2 macrophage polarization through uptake by the AGER/RAGE (advanced glycosylation end-product specific receptor) [211]. In addition, the process of mitophagy-mediated degradation of mitochondrial iron importers (SLC25A37 and SLC25A28) results in elevated mitochondrial iron accumulation [212]. This, in turn, triggers the HIF1A (hypoxia inducible factor 1 subunit alpha)-dependent Warburg effect and AIM2 (absent in melanoma 2)-dependent inflammasome activation, contributing to KrasG12D-induced pancreatic tumorigenesis [212]. Thus, mitochondrial iron dysfunction can contribute to tumorigenesis through hypoxia and inflammasome signals. Some studies suggest that M1 macrophages exhibit greater resistance to ferroptosis compared to the M2 phenotype, despite similar expression levels of GPX4, ACSL4, and LPCAT3 between M1 and M2 macrophages [137]. As a result, the increased presence of M1 macrophages not only contributes to immunotherapy, but also sustains a proinflammatory tumor microenvironment that supports tumor growth at early stage [137].
Recent research has also emphasized ferroptosis as a significant form of hepatocyte death. This process can lead to inflammation, which in turn promotes the development of HCC (hepatocellular carcinoma) and compensatory cell proliferation. The activation of ATF4 has a protective effect against the progression from steatohepatitis to HCC [213]. ATF4 accomplishes this by upregulating SLC7A11, a factor that inhibits stress-related ferroptosis, thereby slowing down the onset of HCC [213]. Furthermore, experiments involving the conditional knockout of Gpx4 in the liver have shown an acceleration of diethylnitrosamine-induced liver cancer. This acceleration is attributed to the release of HMGB1, which in turn recruits MDSC infiltration and is associated with the upregulation of CD274/PD-L1 [214].
These findings suggest that secondary tumors may arise as a potential side effect of pharmacological ferroptosis induction, underscoring the need for long-term experiments to assess this possibility in suitable animal models.
Conclusion and perspective
Ferroptosis, characterized by iron-dependent lipid peroxidation, presents a unique avenue for targeting cancer cells with specific vulnerabilities. While recent advancements have underscored its therapeutic potential in oncology, the intricate mechanisms of ferroptosis display variability and plasticity [215, 216]. However, like a coin with two sides, we must carefully weigh the reported side effects and potential risks, as discussed in this review, against its benefits during ferroptosis therapy.
Several future directions necessitate interdisciplinary collaboration to deepen our understanding of ferroptosis mechanisms and its application in tumor therapy:
Selective induction
Effective targeted therapy should precisely eradicate cancer cells while safeguarding normal tissues and maintaining immune surveillance. Many existing ferroptosis activators lack specificity for particular cells or tissues. While N6F11 has shown some cell-specific properties, it requires higher concentrations to achieve effectiveness compared to well-known non-selective ferroptosis activators [36]. Therefore, it is imperative to make improvements in its structure, metabolic stability, and activity.
Molecular mechanism
The specific molecular factors that set ferroptosis apart from other forms of oxidative cell death, such as mitochondrial apoptosis, are still not well understood [21]. Additionally, it is crucial to investigate whether varying levels of oxidative stress trigger distinct types of cell death. A deeper exploration is required to comprehend the functioning of different antioxidant systems, especially when critical pathways like GPX4 fail. The ongoing challenge lies in addressing acquired resistance mechanisms in ferroptosis. Furthermore, the transition between reduced and oxidized forms of non-enzymatic antioxidants also plays a role in determining the outcome of ferroptotic cell death [217, 218].
Biomarker identification
Monitoring ferroptosis responses necessitates diverse biomarkers to assess iron and lipid metabolism as well as associated immune responses. Several biomarkers, such as TFRC [219], ACSL4 [87], and PTGS2 (prostaglandin-endoperoxide synthase 2) [9], hyperoxidized PRDX3 [220], have been measured at the mRNA or protein levels to monitor ferroptosis responses. Blood-based biomarkers, particularly danger signals, such as HMGB1 [221], ATP [222], SQSTM1 [223], and DCN (decorin) [224], hold translational potential for clinical use. LC-MS-based redox lipidomics is invaluable for characterizing ferroptotic biomarkers in vivo. An outstanding question is whether an easily applicable and cost-effective biomarker or method can be developed for clinical trials.
Benefits and side effects
Despite being designed for specific elimination of drug-resistant cancer cells, ferroptosis therapies can exhibit side effects of varying unpredictability and severity, dependent on tumor types and treatments. Vigilant monitoring and management of these side effects are critical for therapy safety and continuity. In addition to developing selectively inductive drugs, the development of targeted drug delivery systems, such as nanoparticles, is essential for enhancing therapeutic effectiveness and reducing systemic side effects.
In summary, the assessment of the advantages and limitations of ferroptosis therapies in cancer is a challenging undertaking, owing to the intricate complexities of cancer signaling, the interplay among various cell death pathways, and the dynamic stress responses associated with these treatments. Ongoing comprehensive research, advanced experimental designs, and rigorous scientific methodologies are enhancing our comprehension of these intricacies. Nevertheless, these factors remain pivotal in informing decisions regarding cancer treatment strategies.
Availability of data and materials
No datasets were generated or analysed during the current study.
References
Bedoui S, Herold MJ, Strasser A. Emerging connectivity of programmed cell death pathways and its physiological implications. Nat Rev Mol Cell Biol. 2020;21:678–95.
Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29:347–64.
Peng F, Liao M, Qin R, Zhu S, Peng C, Fu L, Chen Y, Han B. Regulated cell death (RCD) in cancer: key pathways and targeted therapies. Signal Transduct Target Ther. 2022;7:286.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–79.
Chen X, Huang J, Yu C, Liu J, Gao W, Li J, Song X, Zhou Z, Li C, Xie Y, et al. A noncanonical function of EIF4E limits ALDH1B1 activity and increases susceptibility to ferroptosis. Nat Commun. 2022;13:6318.
Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113:E4966-4975.
Juan CA. Perez de la Lastra JM, Plou FJ, Perez-Lebena E: The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int J Mol Sci. 2021;22:4642.
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31.
Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, Roveri A, Peng X, Porto Freitas F, Seibt T, et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell. 2018;172(409–422):e421.
Liang D, Feng Y, Zandkarimi F, Wang H, Zhang Z, Kim J, Cai Y, Gu W, Stockwell BR, Jiang X. Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones. Cell. 2023;186(2748–2764):e2722.
Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, Goya Grocin A, Xavier da Silva TN, Silva TN, Panzilius E, Scheel CH, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–8.
Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, Koppula P, Wu S, Zhuang L, Fang B, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593:586–90.
Nakamura T, Hipp C, Santos Dias Mourao A, Borggrafe J, Aldrovandi M, Henkelmann B, Wanninger J, Mishima E, Lytton E, Emler D, et al. Phase separation of FSP1 promotes ferroptosis. Nature. 2023;619:371–7.
Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–92.
Sun WY, Tyurin VA, Mikulska-Ruminska K, Shrivastava IH, Anthonymuthu TS, Zhai YJ, Pan MH, Gong HB, Lu DH, Sun J, et al. Phospholipase iPLA(2)beta averts ferroptosis by eliminating a redox lipid death signal. Nat Chem Biol. 2021;17:465–76.
Chen D, Chu B, Yang X, Liu Z, Jin Y, Kon N, Rabadan R, Jiang X, Stockwell BR, Gu W. iPLA2beta-mediated lipid detoxification controls p53-driven ferroptosis independent of GPX4. Nat Commun. 2021;12:3644.
Tang L, Yu Y, Deng W, Liu J, Wang Y, Ye F, Kang R, Tang D, He Q. TXNDC12 inhibits lipid peroxidation and ferroptosis. iScience. 2023;26:108393.
Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021;18:280–96.
Zhang C, Liu X, Jin S, Chen Y, Guo R. Ferroptosis in cancer therapy: a novel approach to reversing drug resistance. Mol Cancer. 2022;21:47.
Dai E, Chen X, Linkermann A, Jiang X, Kang R, Kagan VE, Bayir H, Yang WS, Garcia-Saez AJ, Ioannou MS, et al. A guideline on the molecular ecosystem regulating ferroptosis. Nat Cell Biol. 2024. https://doi.org/10.1038/s41556-024-01360-8. Epub ahead of print.
Liu J, Kang R, Tang D. Adverse effects of ferroptotic therapy: mechanisms and management. Trends Cancer. 2024;S2405–8033(24):00002–5.
Riegman M, Sagie L, Galed C, Levin T, Steinberg N, Dixon SJ, Wiesner U, Bradbury MS, Niethammer P, Zaritsky A, Overholtzer M. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat Cell Biol. 2020;22:1042–8.
Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, Uchida K, O’Connor OA, Stockwell BR. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem Biol. 2019;26(623–633):e629.
Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, Xie Y, Liu J, Klionsky DJ, Kroemer G, et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System X(c)(-) Activity. Curr Biol. 2018;28(2388–2399):e2385.
Cheff DM, Huang C, Scholzen KC, Gencheva R, Ronzetti MH, Cheng Q, Hall MD, Arner ESJ. The ferroptosis inducing compounds RSL3 and ML162 are not direct inhibitors of GPX4 but of TXNRD1. Redox Biol. 2023;62:102703.
Eaton JK, Furst L, Ruberto RA, Moosmayer D, Hilpmann A, Ryan MJ, Zimmermann K, Cai LL, Niehues M, Badock V, et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat Chem Biol. 2020;16:497–506.
Moosmayer D, Hilpmann A, Hoffmann J, Schnirch L, Zimmermann K, Badock V, Furst L, Eaton JK, Viswanathan VS, Schreiber SL, et al. Crystal structures of the selenoprotein glutathione peroxidase 4 in its apo form and in complex with the covalently bound inhibitor ML162. Acta Crystallogr D Struct Biol. 2021;77:237–48.
Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, Brown LM, Valenzuela CA, Wolpaw AJ, Stockwell BR. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12:497–503.
Gaschler MM, Andia AA, Liu H, Csuka JM, Hurlocker B, Vaiana CA, Heindel DW, Zuckerman DS, Bos PH, Reznik E, et al. FINO(2) initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol. 2018;14:507–15.
Yi J, Zhu J, Wu J, Thompson CB, Jiang X. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc Natl Acad Sci U S A. 2020;117:31189–97.
Huang C, Santofimia-Castano P, Liu X, Xia Y, Peng L, Gotorbe C, Neira JL, Tang D, Pouyssegur J, Iovanna J. NUPR1 inhibitor ZZW-115 induces ferroptosis in a mitochondria-dependent manner. Cell Death Discov. 2021;7:269.
Liu J, Song X, Kuang F, Zhang Q, Xie Y, Kang R, Kroemer G, Tang D. NUPR1 is a critical repressor of ferroptosis. Nat Commun. 2021;12:647.
Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, Tang D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63:173–84.
Luo H, Wang X, Song S, Wang Y, Dan Q, Ge H. Targeting stearoyl-coa desaturase enhances radiation induced ferroptosis and immunogenic cell death in esophageal squamous cell carcinoma. Oncoimmunology. 2022;11:2101769.
Li J, Liu J, Zhou Z, Wu R, Chen X, Yu C, Stockwell B, Kroemer G, Kang R, Tang D. Tumor-specific GPX4 degradation enhances ferroptosis-initiated antitumor immune response in mouse models of pancreatic cancer. Sci Transl Med. 2023;15:eadg3049.
Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS, Stockwell BR. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523.
Zheng J, Sato M, Mishima E, Sato H, Proneth B, Conrad M. Sorafenib fails to trigger ferroptosis across a wide range of cancer cell lines. Cell Death Dis. 2021;12:698.
Yao X, Xie R, Cao Y, Tang J, Men Y, Peng H, Yang W. Simvastatin induced ferroptosis for triple-negative breast cancer therapy. J Nanobiotechnology. 2021;19:311.
Chen GQ, Benthani FA, Wu J, Liang D, Bian ZX, Jiang X. Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death Differ. 2020;27:242–54.
Jiang Z, Wang Z, Chen L, Zhang C, Liao F, Wang Y, Wang Y, Luo P, Luo M, Shi C. Artesunate induces ER-derived-ROS-mediated cell death by disrupting labile iron pool and iron redistribution in hepatocellular carcinoma cells. Am J Cancer Res. 2021;11:691–711.
Eling N, Reuter L, Hazin J, Hamacher-Brady A, Brady NR. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience. 2015;2:517–32.
Liu Y, Wang Y, Liu J, Kang R, Tang D. Interplay between MTOR and GPX4 signaling modulates autophagy-dependent ferroptotic cancer cell death. Cancer Gene Ther. 2021;28:55–63.
Chen Y, Li L, Lan J, Cui Y, Rao X, Zhao J, Xing T, Ju G, Song G, Lou J, Liang J. CRISPR screens uncover protective effect of PSTK as a regulator of chemotherapy-induced ferroptosis in hepatocellular carcinoma. Mol Cancer. 2022;21:11.
Ren X, Li Y, Zhou Y, Hu W, Yang C, Jing Q, Zhou C, Wang X, Hu J, Wang L, et al. Overcoming the compensatory elevation of NRF2 renders hepatocellular carcinoma cells more vulnerable to disulfiram/copper-induced ferroptosis. Redox Biol. 2021;46:102122.
Balgoma D, Kullenberg F, Calitz C, Kopsida M, Heindryckx F, Lennernas H, Hedeland M. Anthracyclins Increase PUFAs: Potential Implications in ER Stress and Cell Death. Cells. 2021;10:1163.
Kang X, Huo Y, Jia S, He F, Li H, Zhou Q, Chang N, Liu D, Li R, Hu Y, et al. Silenced LINC01134 Enhances Oxaliplatin Sensitivity by Facilitating Ferroptosis Through GPX4 in Hepatocarcinoma. Front Oncol. 2022;12:939605.
Guo J, Xu B, Han Q, Zhou H, Xia Y, Gong C, Dai X, Li Z, Wu G. Ferroptosis: A Novel Anti-tumor Action for Cisplatin. Cancer Res Treat. 2018;50:445–60.
Ma S, Henson ES, Chen Y, Gibson SB. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016;7:e2307.
Li C, Zhang Y, Liu J, Kang R, Klionsky DJ, Tang D. Mitochondrial DNA stress triggers autophagy-dependent ferroptotic death. Autophagy. 2021;17:948–60.
Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3:285–96.
Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15:234–45.
Parker JL, Deme JC, Kolokouris D, Kuteyi G, Biggin PC, Lea SM, Newstead S. Molecular basis for redox control by the human cystine/glutamate antiporter system xc(). Nat Commun. 2021;12:7147.
Godeas C, Tramer F, Micali F, Roveri A, Maiorino M, Nisii C, Sandri G, Panfili E. Phospholipid hydroperoxide glutathione peroxidase (PHGPx) in rat testis nuclei is bound to chromatin. Biochem Mol Med. 1996;59:118–24.
Tan S, Schubert D, Maher P. Oxytosis: A novel form of programmed cell death. Curr Top Med Chem. 2001;1:497–506.
Rossler OG, Bauer I, Chung HY, Thiel G. Glutamate-induced cell death of immortalized murine hippocampal neurons: neuroprotective activity of heme oxygenase-1, heat shock protein 70, and sodium selenite. Neurosci Lett. 2004;362:253–7.
Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, Jiang X. Role of Mitochondria in Ferroptosis. Mol Cell. 2019;73(354–363):e353.
Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180–91.
Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med. 2015;212:555–68.
Li C, Liu J, Hou W, Kang R, Tang D. STING1 Promotes Ferroptosis Through MFN1/2-Dependent Mitochondrial Fusion. Front Cell Dev Biol. 2021;9:698679.
Nakamura T, Ogawa M, Kojima K, Takayanagi S, Ishihara S, Hattori K, Naguro I, Ichijo H. The mitochondrial Ca(2+) uptake regulator, MICU1, is involved in cold stress-induced ferroptosis. EMBO Rep. 2021;22:e51532.
Qiu S, Zhong X, Meng X, Li S, Qian X, Lu H, Cai J, Zhang Y, Wang M, Ye Z, et al. Mitochondria-localized cGAS suppresses ferroptosis to promote cancer progression. Cell Res. 2023;33:299–311.
Ahola S, Rivera Mejias P, Hermans S, Chandragiri S, Giavalisco P, Nolte H, Langer T. OMA1-mediated integrated stress response protects against ferroptosis in mitochondrial cardiomyopathy. Cell Metab. 2022;34(1875–1891):e1877.
Yuan H, Li X, Zhang X, Kang R, Tang D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem Biophys Res Commun. 2016;478:838–44.
Wu S, Mao C, Kondiparthi L, Poyurovsky MV, Olszewski K, Gan B. A ferroptosis defense mechanism mediated by glycerol-3-phosphate dehydrogenase 2 in mitochondria. Proc Natl Acad Sci U S A. 2022;119:e2121987119.
Deshwal S, Onishi M, Tatsuta T, Bartsch T, Cors E, Ried K, Lemke K, Nolte H, Giavalisco P, Langer T. Mitochondria regulate intracellular coenzyme Q transport and ferroptotic resistance via STARD7. Nat Cell Biol. 2023;25:246–57.
Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell. 2015;59:298–308.
Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, Tyagi S, Ma L, Westbrook TF, Steinberg GR, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. 2020;22:225–34.
Yang C, Zhao Y, Wang L, Guo Z, Ma L, Yang R, Wu Y, Li X, Niu J, Chu Q, et al. De novo pyrimidine biosynthetic complexes support cancer cell proliferation and ferroptosis defence. Nat Cell Biol. 2023;25:836–47.
Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, Zhong M, Yuan H, Zhang L, Billiar TR, et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017;20:1692–704.
Wang C, Zhu L, Yuan W, Sun L, Xia Z, Zhang Z, Yao W. Diabetes aggravates myocardial ischaemia reperfusion injury via activating Nox2-related programmed cell death in an AMPK-dependent manner. J Cell Mol Med. 2020;24:6670–9.
Yang WH, Huang Z, Wu J, Ding CC, Murphy SK, Chi JT. A TAZ-ANGPTL4-NOX2 Axis Regulates Ferroptotic Cell Death and Chemoresistance in Epithelial Ovarian Cancer. Mol Cancer Res. 2020;18:79–90.
Shiose A, Sumimoto H. Arachidonic acid and phosphorylation synergistically induce a conformational change of p47phox to activate the phagocyte NADPH oxidase. J Biol Chem. 2000;275:13793–801.
Feng Z, Hom ME, Bearrood TE, Rosenthal ZC, Fernandez D, Ondrus AE, Gu Y, McCormick AK, Tomaske MG, Marshall CR, et al. Targeting colorectal cancer with small-molecule inhibitors of ALDH1B1. Nat Chem Biol. 2022;18:1065–75.
Mameishvili E, Serafimidis I, Iwaszkiewicz S, Lesche M, Reinhardt S, Bolicke N, Buttner M, Stellas D, Papadimitropoulou A, Szabolcs M, et al. Aldh1b1 expression defines progenitor cells in the adult pancreas and is required for Kras-induced pancreatic cancer. Proc Natl Acad Sci U S A. 2019;116:20679–88.
Abe C, Miyazawa T, Miyazawa T. Current Use of Fenton Reaction in Drugs and Food. Molecules. 2022;27:5451.
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, Kang R, Tang D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425–8.
Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26:1021–32.
Qin X, Zhang J, Wang B, Xu G, Yang X, Zou Z, Yu C. Ferritinophagy is involved in the zinc oxide nanoparticles-induced ferroptosis of vascular endothelial cells. Autophagy. 2021;17:4266–85.
Wu H, Liu Q, Shan X, Gao W, Chen Q. ATM orchestrates ferritinophagy and ferroptosis by phosphorylating NCOA4. Autophagy. 2023;19:2062–77.
Hong Y, Ren T, Wang X, Liu X, Fei Y, Meng S, Han X, Sun C, Shen H, Li L, et al. APR-246 triggers ferritinophagy and ferroptosis of diffuse large B-cell lymphoma cells with distinct TP53 mutations. Leukemia. 2022;36:2269–80.
Zhou H, Zhou YL, Mao JA, Tang LF, Xu J, Wang ZX, He Y, Li M. NCOA4-mediated ferritinophagy is involved in ionizing radiation-induced ferroptosis of intestinal epithelial cells. Redox Biol. 2022;55:102413.
Anandhan A, Dodson M, Shakya A, Chen J, Liu P, Wei Y, Tan H, Wang Q, Jiang Z, Yang K, et al. NRF2 controls iron homeostasis and ferroptosis through HERC2 and VAMP8. Sci Adv. 2023;9:eade9585.
Xin S, Schick JA. PUFAs dictate the balance of power in ferroptosis. Cell Calcium. 2023;110:102703.
Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81–90.
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91–8.
Yuan H, Li X, Zhang X, Kang R, Tang D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun. 2016;478:1338–43.
Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, Song S, Tavana O, Gu W. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. 2019;21:579–91.
Magtanong L, Mueller GD, Williams KJ, Billmann M, Chan K, Armenta DA, Pope LE, Moffat J, Boone C, Myers CL, et al. Context-dependent regulation of ferroptosis sensitivity. Cell Chem Biol. 2022;29(1409–1418):e1406.
Cui J, Wang Y, Tian X, Miao Y, Ma L, Zhang C, Xu X, Wang J, Fang W, Zhang X. LPCAT3 is transcriptionally regulated by YAP/ZEB/EP300 and collaborates with ACSL4 and YAP to determine ferroptosis sensitivity. Antioxid Redox Signal. 2023;39(7–9):491–511.
Lin Z, Liu J, Long F, Kang R, Kroemer G, Tang D, Yang M. The lipid flippase SLC47A1 blocks metabolic vulnerability to ferroptosis. Nat Commun. 2022;13:7965.
Zhang HL, Hu BX, Li ZL, Du T, Shan JL, Ye ZP, Peng XD, Li X, Huang Y, Zhu XY, et al. PKCbetaII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat Cell Biol. 2022;24:88–98.
Cui W, Liu D, Gu W, Chu B. Peroxisome-driven ether-linked phospholipids biosynthesis is essential for ferroptosis. Cell Death Differ. 2021;28:2536–51.
Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P, Paradkar S, Boehnke N, Deik AA, Reinhardt F, et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature. 2020;585:603–8.
Bai Y, Meng L, Han L, Jia Y, Zhao Y, Gao H, Kang R, Wang X, Tang D, Dai E. Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun. 2019;508:997–1003.
You JH, Lee J, Roh JL. PGRMC1-dependent lipophagy promotes ferroptosis in paclitaxel-tolerant persister cancer cells. J Exp Clin Cancer Res. 2021;40:350.
Reed A, Ware T, Li H, Fernando Bazan J, Cravatt BF. TMEM164 is an acyltransferase that forms ferroptotic C20:4 ether phospholipids. Nat Chem Biol. 2023;19:378–88.
Liu J, Liu Y, Wang Y, Li C, Xie Y, Klionsky DJ, Kang R, Tang D. TMEM164 is a new determinant of autophagy-dependent ferroptosis. Autophagy. 2023;19:945–56.
Klasson TD, LaGory EL, Zhao H, Huynh SK, Papandreou I, Moon EJ, Giaccia AJ. ACSL3 regulates lipid droplet biogenesis and ferroptosis sensitivity in clear cell renal cell carcinoma. Cancer Metab. 2022;10:14.
Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A, Ward CC, Cho K, Patti GJ, Nomura DK, et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem Biol. 2019;26(420–432):e429.
Ma M, Kong P, Huang Y, Wang J, Liu X, Hu Y, Chen X, Du C, Yang H. Activation of MAT2A-ACSL3 pathway protects cells from ferroptosis in gastric cancer. Free Radic Biol Med. 2022;181:288–99.
Liu Y, Wang Y, Lin Z, Kang R, Tang D, Liu J. SLC25A22 as a Key Mitochondrial Transporter Against Ferroptosis by Producing Glutathione and Monounsaturated Fatty Acids. Antioxid Redox Signal. 2023;39:166–85.
Wenzel SE, Tyurina YY, Zhao J, St Croix CM, Dar HH, Mao G, Tyurin VA, Anthonymuthu TS, Kapralov AA, Amoscato AA, et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell. 2017;171(628–641): e626.
Xie Y, Song X, Sun X, Huang J, Zhong M, Lotze MT, Zeh HJR, Kang R, Tang D. Identification of baicalein as a ferroptosis inhibitor by natural product library screening. Biochem Biophys Res Commun. 2016;473:775–80.
Yan B, Ai Y, Sun Q, Ma Y, Cao Y, Wang J, Zhang Z, Wang X. Membrane Damage during Ferroptosis Is Caused by Oxidation of Phospholipids Catalyzed by the Oxidoreductases POR and CYB5R1. Mol Cell. 2021;81:355–69.
Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, Sandoval-Gomez G, Clish CB, Doench JG, Schreiber SL. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16:302–9.
Amoscato AA, Anthonymuthu T, Kapralov O, Sparvero LJ, Shrivastava IH, Mikulska-Ruminska K, Tyurin VA, Shvedova AA, Tyurina YY, Bahar I, et al. Formation of protein adducts with Hydroperoxy-PE electrophilic cleavage products during ferroptosis. Redox Biol. 2023;63:102758.
Xie Y, Kang R, Klionsky DJ, Tang D. GPX4 in cell death, autophagy, and disease. Autophagy. 2023;19:2621–38.
Borchert A, Kalms J, Roth SR, Rademacher M, Schmidt A, Holzhutter HG, Kuhn H, Scheerer P. Crystal structure and functional characterization of selenocysteine-containing glutathione peroxidase 4 suggests an alternative mechanism of peroxide reduction. Biochim Biophys Acta Mol Cell Biol Lipids. 2018;1863:1095–107.
Scheerer P, Borchert A, Krauss N, Wessner H, Gerth C, Hohne W, Kuhn H. Structural basis for catalytic activity and enzyme polymerization of phospholipid hydroperoxide glutathione peroxidase-4 (GPx4). Biochemistry. 2007;46:9041–9.
Liu H, Forouhar F, Seibt T, Saneto R, Wigby K, Friedman J, Xia X, Shchepinov MS, Ramesh SK, Conrad M, Stockwell BR. Characterization of a patient-derived variant of GPX4 for precision therapy. Nat Chem Biol. 2022;18:91–100.
Roveri A, Di Giacinto F, Rossetto M, Cozza G, Cheng Q, Miotto G, Zennaro L, Di Paolo ML, Arner ESJ, De Spirito M, et al. Cardiolipin drives the catalytic activity of GPX4 on membranes: Insights from the R152H mutant. Redox Biol. 2023;64:102806.
Chen X, Tsvetkov AS, Shen HM, Isidoro C, Ktistakis NT, Linkermann A, Koopman WJH, Simon HU, Galluzzi L, Luo S, et al. International consensus guidelines for the definition, detection, and interpretation of autophagy-dependent ferroptosis. Autophagy. 2024:1–34. https://doi.org/10.1080/15548627.2024.2319901. Epub ahead of print.
Zhu S, Zhang Q, Sun X, Zeh HJ 3rd, Lotze MT, Kang R, Tang D. HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res. 2017;77:2064–77.
Wu Z, Geng Y, Lu X, Shi Y, Wu G, Zhang M, Shan B, Pan H, Yuan J. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci U S A. 2019;116:2996–3005.
Wu K, Yan M, Liu T, Wang Z, Duan Y, Xia Y, Ji G, Shen Y, Wang L, Li L, et al. Creatine kinase B suppresses ferroptosis by phosphorylating GPX4 through a moonlighting function. Nat Cell Biol. 2023;25:714–25.
Han L, Bai L, Fang X, Liu J, Kang R, Zhou D, Tang D, Dai E. SMG9 drives ferroptosis by directly inhibiting GPX4 degradation. Biochem Biophys Res Commun. 2021;567:92–8.
Yant LJ, Ran Q, Rao L, Van Remmen H, Shibatani T, Belter JG, Motta L, Richardson A, Prolla TA. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med. 2003;34:496–502.
Liang H, Yoo SE, Na R, Walter CA, Richardson A, Ran Q. Short form glutathione peroxidase 4 is the essential isoform required for survival and somatic mitochondrial functions. J Biol Chem. 2009;284:30836–44.
Azuma K, Koumura T, Iwamoto R, Matsuoka M, Terauchi R, Yasuda S, Shiraya T, Watanabe S, Aihara M, Imai H, Ueta T. Mitochondrial glutathione peroxidase 4 is indispensable for photoreceptor development and survival in mice. J Biol Chem. 2022;298:101824.
Tadokoro T, Ikeda M, Ide T, Deguchi H, Ikeda S, Okabe K, Ishikita A, Matsushima S, Koumura T, Yamada KI, et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight. 2020;5:e132747.
Muri J, Thut H, Bornkamm GW, Kopf M. B1 and Marginal Zone B Cells but Not Follicular B2 Cells Require Gpx4 to Prevent Lipid Peroxidation and Ferroptosis. Cell Rep. 2019;29(2731–2744):e2734.
Zheng H, Jiang L, Tsuduki T, Conrad M, Toyokuni S. Embryonal erythropoiesis and aging exploit ferroptosis. Redox Biol. 2021;48:102175.
Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, Cao L, Xie M, Ran Q, Kroemer G, et al. Lipid Peroxidation Drives Gasdermin D-Mediated Pyroptosis in Lethal Polymicrobial Sepsis. Cell Host Microbe. 2018;24(97–108):e104.
Canli O, Alankus YB, Grootjans S, Vegi N, Hultner L, Hoppe PS, Schroeder T, Vandenabeele P, Bornkamm GW, Greten FR. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood. 2016;127:139–48.
von Krusenstiern AN, Robson RN, Qian N, Qiu B, Hu F, Reznik E, Smith N, Zandkarimi F, Estes VM, Dupont M, et al. Identification of essential sites of lipid peroxidation in ferroptosis. Nat Chem Biol. 2023;19:719–30.
Chen X, Kang R, Kroemer G, Tang D. Organelle-specific regulation of ferroptosis. Cell Death Differ. 2021;28:2843–56.
Deshwal S, Onishi M, Tatsuta T, Bartsch T, Cors E, Ried K, Lemke K, Nolte H, Giavalisco P, Langer T. Mitochondria regulate intracellular coenzyme Q transport and ferroptotic resistance via STARD7. Nat Cell Biol. 2023;25:246–57.
Dai E, Zhang W, Cong D, Kang R, Wang J, Tang D. AIFM2 blocks ferroptosis independent of ubiquinol metabolism. Biochem Biophys Res Commun. 2020;523:966–71.
Mishima E, Ito J, Wu Z, Nakamura T, Wahida A, Doll S, Tonnus W, Nepachalovich P, Eggenhofer E, Aldrovandi M, et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature. 2022;608:778–83.
Jin DY, Chen X, Liu Y, Williams CM, Pedersen LC, Stafford DW, Tie JK. A genome-wide CRISPR-Cas9 knockout screen identifies FSP1 as the warfarin-resistant vitamin K reductase. Nat Commun. 2023;14:828.
Mishima E, Nakamura T, Zheng J, Zhang W, Mourao ASD, Sennhenn P, Conrad M. DHODH inhibitors sensitize to ferroptosis by FSP1 inhibition. Nature. 2023;619:E9–18.
Mao C, Liu X, Yan Y, Olszewski K, Gan B. Reply to: DHODH inhibitors sensitize to ferroptosis by FSP1 inhibition. Nature. 2023;619:E19–23.
Tang L, Yu Y, Deng W, Liu J, Wang Y, Ye F, Kang R, Tang D. Q H: TXNDC12 inhibits lipid peroxidation and ferroptosis. iSciense. 2023;132:108449.
Yu H, Zhu K, Wang M, Jiang X. TXNDC12 knockdown promotes ferroptosis by modulating SLC7A11 expression in glioma. Clin Transl Sci. 2023;16:1957–71.
Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Muller C, Zandkarimi F, Merl-Pham J, Bao X, Anastasov N, Kossl J, et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent Sci. 2020;6:41–53.
Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R, St Croix CM, Mikulska-Ruminska K, Liu B, Shrivastava IH, et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol. 2020;16:278–90.
Kuang F, Liu J, Xie Y, Tang D, Kang R. MGST1 is a redox-sensitive repressor of ferroptosis in pancreatic cancer cells. Cell Chem Biol. 2021;28(765–775):e765.
Wang Q, Bin C, Xue Q, Gao Q, Huang A, Wang K, Tang N. GSTZ1 sensitizes hepatocellular carcinoma cells to sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis. Cell Death Dis. 2021;12:426.
Lovatt M, Adnan K, Kocaba V, Dirisamer M, Peh GSL, Mehta JS. Peroxiredoxin-1 regulates lipid peroxidation in corneal endothelial cells. Redox Biol. 2020;30:101417.
Sun X, Niu X, Chen R, He W, Chen D, Kang R, Tang D. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology. 2016;64:488–500.
Entezari S, Haghi SM, Norouzkhani N, Sahebnazar B, Vosoughian F, Akbarzadeh D, Islampanah M, Naghsh N, Abbasalizadeh M, Deravi N. Iron Chelators in Treatment of Iron Overload. J Toxicol. 2022;2022:4911205.
Dai E, Meng L, Kang R, Wang X, Tang D. ESCRT-III-dependent membrane repair blocks ferroptosis. Biochem Biophys Res Commun. 2020;522:415–21.
Ralhan I, Chang J, Moulton MJ, Goodman LD, Lee NYJ, Plummer G, Pasolli HA, Matthies D, Bellen HJ, Ioannou MS. Autolysosomal exocytosis of lipids protect neurons from ferroptosis. J Cell Biol. 2023;222:e202207130.
Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM, Boniface JJ, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447:864–8.
Poursaitidis I, Wang X, Crighton T, Labuschagne C, Mason D, Cramer SL, Triplett K, Roy R, Pardo OE, Seckl MJ, et al. Oncogene-Selective Sensitivity to Synchronous Cell Death following Modulation of the Amino Acid Nutrient Cystine. Cell Rep. 2017;18:2547–56.
Muller F, Lim JKM, Bebber CM, Seidel E, Tishina S, Dahlhaus A, Stroh J, Beck J, Yapici FI, Nakayama K, et al. Elevated FSP1 protects KRAS-mutated cells from ferroptosis during tumor initiation. Cell Death Differ. 2023;30:442–56.
Hu K, Li K, Lv J, Feng J, Chen J, Wu H, Cheng F, Jiang W, Wang J, Pei H, et al. Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. J Clin Invest. 2020;130:1752–66.
Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62.
Wang SJ, Li D, Ou Y, Jiang L, Chen Y, Zhao Y, Gu W. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep. 2016;17:366–73.
Yang X, Wang Z, Zandkarimi F, Liu Y, Duan S, Li Z, Kon N, Zhang Z, Jiang X, Stockwell BR, Gu W. Regulation of VKORC1L1 is critical for p53-mediated tumor suppression through vitamin K metabolism. Cell Metab. 2023;35(8):1474-1490.e8.
Ou Y, Wang SJ, Li D, Chu B, Gu W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci U S A. 2016;113:E6806–12.
Su Z, Kon N, Yi J, Zhao H, Zhang W, Tang Q, Li H, Kobayashi H, Li Z, Duan S, et al. Specific regulation of BACH1 by the hotspot mutant p53(R175H) reveals a distinct gain-of-function mechanism. Nat Cancer. 2023;4:564–81.
Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. 2013;51:618–31.
Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–23.
Cahuzac KM, Lubin A, Bosch K, Stokes N, Shoenfeld SM, Zhou R, Lemon H, Asara J, Parsons RE. AKT activation because of PTEN loss upregulates xCT via GSK3beta/NRF2, leading to inhibition of ferroptosis in PTEN-mutant tumor cells. Cell Rep. 2023;42:112536.
Wang Z, Li R, Hou N, Zhang J, Wang T, Fan P, Ji C, Zhang B, Liu L, Wang Y, et al. PRMT5 reduces immunotherapy efficacy in triple-negative breast cancer by methylating KEAP1 and inhibiting ferroptosis. J Immunother Cancer. 2023;11:e006890.
Chen D, Tavana O, Chu B, Erber L, Chen Y, Baer R, Gu W. NRF2 Is a Major Target of ARF in p53-Independent Tumor Suppression. Mol Cell. 2017;68(224–232):e224.
DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–9.
Wang Y, Tian Q, Hao Y, Yao W, Lu J, Chen C, Chen X, Lin Y, Huang Q, Xu L, et al. The kinase complex mTORC2 promotes the longevity of virus-specific memory CD4(+) T cells by preventing ferroptosis. Nat Immunol. 2022;23:303–17.
Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, Tseng YY, Deasy R, Kost-Alimova M, Dancik V, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun. 2019;10:1617.
Yang Z, Su W, Wei X, Qu S, Zhao D, Zhou J, Wang Y, Guan Q, Qin C, Xiang J, et al. HIF-1alpha drives resistance to ferroptosis in solid tumors by promoting lactate production and activating SLC1A1. Cell Rep. 2023;42:112945.
Lin Z, Song J, Gao Y, Huang S, Dou R, Zhong P, Huang G, Han L, Zheng J, Zhang X, et al. Hypoxia-induced HIF-1alpha/lncRNA-PMAN inhibits ferroptosis by promoting the cytoplasmic translocation of ELAVL1 in peritoneal dissemination from gastric cancer. Redox Biol. 2022;52:102312.
Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, Chen ZN, Jiang X. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature. 2019;572:402–6.
Yang WH, Ding CC, Sun T, Rupprecht G, Lin CC, Hsu D, Chi JT. The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma. Cell Rep. 2019;28(2501–2508):e2504.
Yang M, Chen P, Liu J, Zhu S, Kroemer G, Klionsky DJ, Lotze MT, Zeh HJ, Kang R. Tang D Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci Adv. 2019;5:eaaw2238.
Kremer DM, Nelson BS, Lin L, Yarosz EL, Halbrook CJ, Kerk SA, Sajjakulnukit P, Myers A, Thurston G, Hou SW, et al. GOT1 inhibition promotes pancreatic cancer cell death by ferroptosis. Nat Commun. 2021;12:4860.
Xue Q, Yan D, Chen X, Li X, Kang R, Klionsky DJ, Kroemer G, Chen X, Tang D, Liu J. Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy. 2023;19:1982–96.
Chen X, Song X, Li J, Zhang R, Yu C, Zhou Z, Liu J, Liao S, Klionsky DJ, Kroemer G, et al. Identification of HPCAL1 as a specific autophagy receptor involved in ferroptosis. Autophagy. 2023;19:54–74.
Kang R, Zhu S, Zeh HJ, Klionsky DJ, Tang D. BECN1 is a new driver of ferroptosis. Autophagy. 2018;14:2173–5.
Chen F, Cai X, Kang R, Liu J, Tang D. Autophagy-Dependent Ferroptosis in Cancer. Antioxid Redox Signal. 2023;39:79–101.
Liu K, Huang J, Liu J, Klionsky DJ, Kang R, Tang D. Induction of autophagy-dependent ferroptosis to eliminate drug-tolerant human retinoblastoma cells. Cell Death Dis. 2022;13:521.
Mikubo M, Inoue Y, Liu G, Tsao MS. Mechanism of Drug Tolerant Persister Cancer Cells: The Landscape and Clinical Implication for Therapy. J Thorac Oncol. 2021;16:1798–809.
Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, Galeas J, Dhruv HD, Berens ME, Schreiber SL, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551:247–50.
Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547:453–7.
Wu M, Zhang X, Zhang W, Chiou YS, Qian W, Liu X, Zhang M, Yan H, Li S, Li T, et al. Cancer stem cell regulated phenotypic plasticity protects metastasized cancer cells from ferroptosis. Nat Commun. 2022;13:1371.
Zhao Y, Zhao W, Lim YC, Liu T. Salinomycin-Loaded Gold Nanoparticles for Treating Cancer Stem Cells by Ferroptosis-Induced Cell Death. Mol Pharm. 2019;16:2532–9.
Xiong Y, Liu T, Chen J. Anisomycin has the potential to induce human ovarian cancer stem cell ferroptosis by influencing glutathione metabolism and autophagy signal transduction pathways. J Cancer. 2023;14:1202–15.
Wan C, Sun Y, Tian Y, Lu L, Dai X, Meng J, Huang J, He Q, Wu B, Zhang Z, et al. Irradiated tumor cell-derived microparticles mediate tumor eradication via cell killing and immune reprogramming. Sci Adv. 2020;6:eaay9789.
Almahi WA, Yu KN, Mohammed F, Kong P, Han W. Hemin enhances radiosensitivity of lung cancer cells through ferroptosis. Exp Cell Res. 2022;410:112946.
Lang X, Green MD, Wang W, Yu J, Choi JE, Jiang L, Liao P, Zhou J, Zhang Q, Dow A, et al. Radiotherapy and Immunotherapy Promote Tumoral Lipid Oxidation and Ferroptosis via Synergistic Repression of SLC7A11. Cancer Discov. 2019;9:1673–85.
Ye LF, Chaudhary KR, Zandkarimi F, Harken AD, Kinslow CJ, Upadhyayula PS, Dovas A, Higgins DM, Tan H, Zhang Y, et al. Radiation-Induced Lipid Peroxidation Triggers Ferroptosis and Synergizes with Ferroptosis Inducers. ACS Chem Biol. 2020;15:469–84.
Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, Ajani JA, Xiao Q, Liao Z, Wang H, Gan B. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020;30:146–62.
Shen D, Luo J, Chen L, Ma W, Mao X, Zhang Y, Zheng J, Wang Y, Wan J, Wang S, et al. PARPi treatment enhances radiotherapy-induced ferroptosis and antitumor immune responses via the cGAS signaling pathway in colorectal cancer. Cancer Lett. 2022;550:215919.
Kong P, Yang M, Wang Y, Yu KN, Wu L, Han W. Ferroptosis triggered by STAT1- IRF1-ACSL4 pathway was involved in radiation-induced intestinal injury. Redox Biol. 2023;66:102857.
Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270–4.
Liao P, Wang W, Wang W, Kryczek I, Li X, Bian Y, Sell A, Wei S, Grove S, Johnson JK, et al. CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell. 2022;40(365–378):e366.
Li H, Yang P, Wang J, Zhang J, Ma Q, Jiang Y, Wu Y, Han T, Xiang D. HLF regulates ferroptosis, development and chemoresistance of triple-negative breast cancer by activating tumor cell-macrophage crosstalk. J Hematol Oncol. 2022;15:2.
Han Y, Zhang YY, Pan YQ, Zheng XJ, Liao K, Mo HY, Sheng H, Wu QN, Liu ZX, Zeng ZL, et al. IL-1beta-associated NNT acetylation orchestrates iron-sulfur cluster maintenance and cancer immunotherapy resistance. Mol Cell. 2023;83(1887–1902):e1888.
Galluzzi L, Vitale I, Warren S, Adjemian S, Agostinis P, Martinez AB, Chan TA, Coukos G, Demaria S, Deutsch E, et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J Immunother Cancer. 2020;8:e000337.
Xu S, Chaudhary O, Rodriguez-Morales P, Sun X, Chen D, Zappasodi R, Xu Z, Pinto AFM, Williams A, Schulze I, et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity. 2021;54(1561–1577):e1567.
Ma X, Xiao L, Liu L, Ye L, Su P, Bi E, Wang Q, Yang M, Qian J, Yi Q. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab. 2021;33(1001–1012):e1005.
Xiao L, Ma X, Ye L, Su P, Xiong W, Bi E, Wang Q, Xian M, Yang M, Qian J, Yi Q. IL-9/STAT3/fatty acid oxidation-mediated lipid peroxidation contributes to Tc9 cell longevity and enhanced antitumor activity. J Clin Invest. 2022;132:e153247.
Xu C, Sun S, Johnson T, Qi R, Zhang S, Zhang J, Yang K. The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain Treg cell activation and suppression of antitumor immunity. Cell Rep. 2021;35:109235.
Han L, Bai L, Qu C, Dai E, Liu J, Kang R, Zhou D, Tang D, Zhao Y. PPARG-mediated ferroptosis in dendritic cells limits antitumor immunity. Biochem Biophys Res Commun. 2021;576:33–9.
Wiernicki B, Maschalidi S, Pinney J, Adjemian S, Vanden Berghe T, Ravichandran KS, Vandenabeele P. Cancer cells dying from ferroptosis impede dendritic cell-mediated anti-tumor immunity. Nat Commun. 2022;13:3676.
Efimova I, Catanzaro E, Van der Meeren L, Turubanova VD, Hammad H, Mishchenko TA, Vedunova MV, Fimognari C, Bachert C, Coppieters F, et al. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. J Immunother Cancer. 2020;8:e001369.
Tang D, Kang R, Zeh HJ, Lotze MT. The multifunctional protein HMGB1: 50 years of discovery. Nat Rev Immunol. 2023;23(12):824–41.
Finetti F, Travelli C, Ercoli J, Colombo G, Buoso E, Trabalzini L. Prostaglandin E2 and Cancer: Insight into Tumor Progression and Immunity. Biology (Basel). 2020;9:434.
Poznanski SM, Singh K, Ritchie TM, Aguiar JA, Fan IY, Portillo AL, Rojas EA, Vahedi F, El-Sayes A, Xing S, et al. Metabolic flexibility determines human NK cell functional fate in the tumor microenvironment. Cell Metab. 2021;33(1205–1220): e1205.
Cui JX, Xu XH, He T, Liu JJ, Xie TY, Tian W, Liu JY. L-kynurenine induces NK cell loss in gastric cancer microenvironment via promoting ferroptosis. J Exp Clin Cancer Res. 2023;42:52.
Kim R, Hashimoto A, Markosyan N, Tyurin VA, Tyurina YY, Kar G, Fu S, Sehgal M, Garcia-Gerique L, Kossenkov A, et al. Ferroptosis of tumour neutrophils causes immune suppression in cancer. Nature. 2022;612:338–46.
Zhao J, Jia Y, Mahmut D, Deik AA, Jeanfavre S, Clish CB, Sankaran VG. Human hematopoietic stem cell vulnerability to ferroptosis. Cell. 2023;186(732–747):e716.
Song X, Xie Y, Kang R, Hou W, Sun X, Epperly MW, Greenberger JS, Tang D. FANCD2 protects against bone marrow injury from ferroptosis. Biochem Biophys Res Commun. 2016;480:443–9.
Carlson BA, Tobe R, Yefremova E, Tsuji PA, Hoffmann VJ, Schweizer U, Gladyshev VN, Hatfield DL, Conrad M. Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol. 2016;9:22–31.
Wu J, Wang Y, Jiang R, Xue R, Yin X, Wu M, Meng Q. Ferroptosis in liver disease: new insights into disease mechanisms. Cell Death Discov. 2021;7:276.
Argiles JM, Lopez-Soriano FJ, Stemmler B, Busquets S. Cancer-associated cachexia - understanding the tumour macroenvironment and microenvironment to improve management. Nat Rev Clin Oncol. 2023;20:250–64.
Ferrer M, Mourikis N, Davidson EE, Kleeman SO, Zaccaria M, Habel J, Rubino R, Gao Q, Flint TR, Young L, et al. Ketogenic diet promotes tumor ferroptosis but induces relative corticosterone deficiency that accelerates cachexia. Cell Metab. 2023;35(1147–1162):e1147.
Dai E, Han L, Liu J, Xie Y, Zeh HJ, Kang R, Bai L, Tang D. Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING-dependent DNA sensor pathway. Nat Commun. 2020;11:6339.
Liu K, Liu J, Zou B, Li C, Zeh HJ, Kang R, Kroemer G, Huang J, Tang D. Trypsin-Mediated Sensitization to Ferroptosis Increases the Severity of Pancreatitis in Mice. Cell Mol Gastroenterol Hepatol. 2022;13:483–500.
Dai E, Han L, Liu J, Xie Y, Kroemer G, Klionsky DJ, Zeh HJ, Kang R, Wang J, Tang D. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy. 2020;16:2069–83.
Li C, Zhang Y, Cheng X, Yuan H, Zhu S, Liu J, Wen Q, Xie Y, Liu J, Kroemer G, et al. PINK1 and PARK2 Suppress Pancreatic Tumorigenesis through Control of Mitochondrial Iron-Mediated Immunometabolism. Dev Cell. 2018;46(441–455):e448.
He F, Zhang P, Liu J, Wang R, Kaufman RJ, Yaden BC, Karin M. ATF4 suppresses hepatocarcinogenesis by inducing SLC7A11 (xCT) to block stress-related ferroptosis. J Hepatol. 2023;79:362–77.
Conche C, Finkelmeier F, Pesic M, Nicolas AM, Bottger TW, Kennel KB, Denk D, Ceteci F, Mohs K, Engel E, et al. Combining ferroptosis induction with MDSC blockade renders primary tumours and metastases in liver sensitive to immune checkpoint blockade. Gut. 2023;72(9):1774–82.
Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: machinery and regulation. Autophagy. 2021;17:2054–81.
Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31:107–25.
Wang D, Peng Y, Xie Y, Zhou B, Sun X, Kang R, Tang D. Antiferroptotic activity of non-oxidative dopamine. Biochem Biophys Res Commun. 2016;480:602–7.
Sun J, Lin XM, Lu DH, Wang M, Li K, Li SR, Li ZQ, Zhu CJ, Zhang ZM, Yan CY, et al. Midbrain dopamine oxidation links ubiquitination of glutathione peroxidase 4 to ferroptosis of dopaminergic neurons. J Clin Invest. 2023;133:e165228.
Feng H, Schorpp K, Jin J, Yozwiak CE, Hoffstrom BG, Decker AM, Rajbhandari P, Stokes ME, Bender HG, Csuka JM, et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep. 2020;30(3411–3423):e3417.
Cui S, Ghai A, Deng Y, Li S, Zhang R, Egbulefu C, Liang G, Achilefu S, Ye J. Identification of hyperoxidized PRDX3 as a ferroptosis marker reveals ferroptotic damage in chronic liver diseases. Mol Cell. 2023;83(21):3931-3939.e5.
Wen Q, Liu J, Kang R, Zhou B, Tang D. The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun. 2019;510:278–83.
Efimova I, Catanzaro E, Van der Meeren L, Turubanova VD, Hammad H, Mishchenko TA, Vedunova MV, Fimognari C, Bachert C, Coppieters F. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. Journal for immunotherapy of cancer. 2020;8:e001369.
Yang L, Ye F, Liu J, Klionsky DJ, Tang D, Kang R. Extracellular SQSTM1 exacerbates acute pancreatitis by activating autophagy-dependent ferroptosis. Autophagy. 2023;19:1733–44.
Li J, Liu J, Zhou Z, Wu R, Chen X, Yu C, Stockwell BR, Kroemer G, Kang R, Tang D. Tumor specific GPX4 degradation enhances ferroptosis-initiated antitumor immune response in mouse models of pancreatic cancer. Sci Transl Med. 2023;15:eadg3049.
Acknowledgements
The authors would like to express their gratitude to all the pioneers and researchers in the field of ferroptosis study. They apologize for any omissions of important references in this field due to space limitations.
Funding
J.L. is supported by grants from National Natural Science Foundation of China (32200594 and 82372152).
Author information
Authors and Affiliations
Contributions
J.D., D.T., L.H., Y.Z., and L.M. conceived of and wrote the manuscript. Y.J., F.D., J.L., and R.K. edited the manuscript. All authors participated in the final version preparation and submission.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Diao, J., Jia, Y., Dai, E. et al. Ferroptotic therapy in cancer: benefits, side effects, and risks. Mol Cancer 23, 89 (2024). https://doi.org/10.1186/s12943-024-01999-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-024-01999-9