
Abstract
Background
Melioidosis, caused by the category B biothreat agent Burkholderia pseudomallei, is a disease with a high mortality rate and requires an immediate culture-independent diagnosis for effective disease management. In this study, we developed a highly sensitive qPCR assay for specific detection of Burkholderia pseudomallei and melioidosis disease diagnosis based on a novel target sequence.
Methods
An extensive in-silico analysis was done to identify a novel and highly conserved sequence for developing a qPCR assay. The specificity of the developed assay was analyzed with 65 different bacterial cultures, and the analytical sensitivity of the assay was determined with the purified genomic DNA of B. pseudomallei. The applicability of the assay for B. pseudomallei detection in clinical and environmental matrices was evaluated by spiking B. pseudomallei cells in the blood, urine, soil, and water along with suitable internal controls.
Results
A novel 85-nucleotide-long sequence was identified using in-silico tools and employed for the development of the highly sensitive and specific quantitative real-time PCR assay S664. The assay S664 was found to be highly specific when evaluated with 65 different bacterial cultures related and non-related to B. pseudomallei. The assay was found to be highly sensitive, with a detection limit of 3 B. pseudomallei genome equivalent copies per qPCR reaction. The detection limit in clinical matrices was found to be 5 × 102 CFU/mL for both human blood and urine. In environmental matrices, the detection limit was found to be 5 × 101 CFU/mL of river water and 2 × 103 CFU/gm of paddy field soil.
Conclusions
The findings of the present study suggest that the developed assay S664 along with suitable internal controls has a huge diagnostic potential and can be successfully employed for specific, sensitive, and rapid molecular detection of B. pseudomallei in various clinical and environmental matrices.
Similar content being viewed by others


Introduction
Burkholderia pseudomallei is a category B biothreat agent responsible for causing melioidosis, a serious invasive disease of humans with a high case fatality rate [1]. B. pseudomallei is a saprophytic environmental pathogen predominantly found in rhizospheric soil, paddy fields, and standing streams [2]. The human population living in rural areas is at high risk of acquiring this infection mostly in the rainy season through direct contact with contaminated soil and surface water [3]. The most common symptoms of melioidosis are associated with respiratory and cardiovascular systems and its non-specific clinical presentation hampers diagnosis and also delays early treatment which characteristically leads to high fatality rate [4].
Currently, the culturing method used for the isolation and identification of bacteria from clinical and environmental samples is the gold standard for the diagnosis and detection of B. pseudomallei. However, it requires expertise, special selective media, and a long incubation period (4–5 days) which delays diagnosis. Furthermore, isolated cultures are usually misidentified as Bacillus or Pseudomonas species [5]. Serological tests such as Indirect Haemagglutination Assay, Enzyme-Linked Immunosorbent Assay, and Lateral Flow Immuno-assays are used for the detection of B. pseudomallei-specific antibodies. These methods are not reliable for accurate disease diagnosis, especially in highly endemic areas where a high rate of background seropositivity in healthy populations is observed [6]. So, to conquer the established boundaries of microbiological and serological test methods, direct nucleic acid amplification-based specific molecular detection methods have been developed. Real-time quantitative polymerase chain reaction (qPCR) assays have been identified to have high degrees of specificity and sensitivity for organism detection in the samples [7]. At present, the most promising assay for the detection of B. pseudomallei generally targets gene clusters of type III secretion system (T3SS). The orf2 within the T3SS1 is considered the gold standard for molecular identification of B. pseudomallei [8]. The other gene targets such as 16S rRNA, TTSS1-orf11, mprA, YLF/BTFC, BPSL1664, phaC, lpxO, and Bp loci 8653 and 9438 have also been evaluated so far for their efficiency in the identification of B. pseudomallei [8, 9]. However, all the above-mentioned assays lack internal controls to monitor proper nucleic acid extraction and adequate nucleic acid amplification.
The genomic heterogeneity and high rate of genetic recombination are the few most striking features of B. pseudomallei [10]. The natural competency of B. pseudomallei for DNA uptake and catabolism adds to its genetic diversity [11]. Furthermore, efficient and simple gears have been developed for the compliant genetic manipulations in the genome of this category B biothreat agent [12]. Misuse of genetically manipulated or naturally occurring B. pseudomallei strains that lacks the specific target sequence will pose a serious threat to human life due to their high lethality. Hence, the existing assays are insufficient to counter and detect the altered pathogen in case of public health and biothreat emergencies. Therefore, there is an ever-increasing need to identify novel targets for specific detection and identification of B. pseudomallei in clinical and environmental settings. Keeping the above features of B. pseudomallei in mind, the present study was focused to develop a multiplex hydrolysis probe-based real-time qPCR assay targeting in-silico identified novel gene target. To the best of our knowledge, this is the first report describing the development of a novel multiplex qPCR assay employing suitable internal controls for melioidosis disease diagnosis and detection of B. pseudomallei in different environmental matrices.
Materials and methods
In-silico identification of specific target and primer-probe designing
For the identification of B. pseudomallei-specific novel candidate sequence, the genomic regions of B. pseudomallei absent in the genome of Burkholderia mallei were initially shortlisted [13]. The basis of such an analysis was that the B. mallei evolved as a deletion clone of B. pseudomallei [13, 14]. The obtained gene sequences of these genomic regions were then analyzed in-silico to derive unanimously unique sequences of B. pseudomallei. The nucleotide BLAST (BLASTn, https://blast.ncbi.nlm.nih.gov/blast.cgi) was done for the shortlisted genes against the RefSeq Genome Database (refseq genomes) of B. pseudomallei. A novel gene was finally selected based on its specificity and higher B. pseudomallei strain coverage in comparison to orf2. The complete sequence of the novel gene from all the available strains was subsequently obtained and aligned employing ClustalW in MEGA X software in order to identify the highly conserved region within the gene. Further, the BLASTn server was used to retrieve the orf2 sequences from available strains, and strain-wise comparative analysis for both the sequences was performed. Three sets of the primers and respective probes for the identified gene segment were initially designed by the PrimerQuest tool (https://sg.idtdna.com/pages/tools/primerquest) and then individually screened, analyzed, and sorted using the BLASTn server to reduce the possibility of cross-reactivity with other organisms [15].
Bacterial culture condition and DNA preparation
The cultures of B. pseudomallei were grown on Ashdown’s agar medium containing 4% Glycerol (Fisher Scientific, #CAS 56-81-5), 1% Tryptone Soya Broth (HiMedia, #M011), 0.5 mg/L Crystal Violet (HiMedia, #GRM961), 5 mg/L Neutral Red (HiMedia, #RM122), and 1.5% Bacteriological Agar (HiMedia, #GRM026) supplemented with 5 mg/L of Gentamicin (HiMedia, #RM461) at 37 °C for 48–72 h [16]. The other cultures used in the study were grown on Brain Heart Infusion (BHI) agar medium (HiMedia, #M211) at 37 °C for 16 to 48 h. Obtained single colonies were inoculated into BHI broth (HiMedia, #M210) for DNA extraction. The genomic DNA from bacterial cells was extracted using DNeasy Blood and Tissue kit (Qiagen, #69504) in accordance with the manufacturer’s protocol. The purity and quantity of DNA were measured using NanoDrop (Thermo). Isolated purified bacterial genomic DNA was aliquoted and stored at -20 °C till further use.
Hydrolysis probe-based qPCR assay S664
The qPCR assay S664 was performed using GoTaq Probe qPCR master mix (Promega, #A6102) on StepOne Real-Time PCR System (Applied Biosystems) and CFX96 Touch Real-Time PCR detection systems (Bio-Rad). The qPCR reactions were prepared in a total volume of 20 µL containing 1× master mix, 1000 nM of each forward and reverse primer, 250 nM hydrolysis probe (Eurofins genomics) and 2 µL of DNA. The list of primer and probes used in the study is mentioned in Table 1. The thermal profile of the assay consisted of 10 min of initial denaturation and polymerase activation at 95 °C followed by 40 cycles of denaturation at 95 °C for 15 s and annealing/extension at 60 °C for 60 s.
Analytical sensitivity and specificity of assay S664
To determine the analytical sensitivity of assay S664, a 10-fold serially diluted B. pseudomallei genomic DNA ranging from 3 × 106 to 3 × 10− 1 genome equivalent (GE) copies/reaction was used. The amount of DNA was converted to GE copies based on the size of B. pseudomallei genome (7.25 × 106 bp) [13, 16]. All the qPCRs were carried out in triplicate, and at least two separate experiments were performed. The obtained cycle threshold (Ct) values were used to generate standard curve. The efficiency of the assay was calculated by the formula E = (-1 + 10− 1/slope) × 100 using the slope of the standard curve [17]. The coefficient of variation for inter-assay and intra-assay were calculated. The specificity of the developed assay S664 was tested three times by incorporating genomic DNA (∼ 10 ng) from B. pseudomallei-related and non-related bacterial cultures (Table 2). B. pseudomallei (NCTC 13392) served as a positive control and nuclease-free water served as no template control (NTC) in the specificity analysis.
Detection of B. pseudomallei in clinical matrices
For the feasibility of assay S664 to detect B. pseudomallei in clinical samples, a multiplex assay S664 was developed using the novel target BPSS0664 and the human RNaseP gene as endogenous control (Table 1) [18]. The multiplex qPCR reaction was prepared in total 20 µL volume containing 1× master mix, 250 nM of forward primer, reverse primer, and probe of gene BPSS0664, 750 nM of forward and reverse primer, and 250 nM hydrolysis probe of gene RNaseP and 2 µL of DNA. To determine the limit of detection in clinical matrices, 10-fold serial dilutions of B. pseudomallei cells were spiked in healthy human blood collected in EDTA-coated vials [19] and urine ranging from 5 × 107 to 5 × 100 cells/mL. The total DNA was extracted from spiked blood and urine using DNeasy Blood and Tissue kit according to the manufacturer’s instructions. The qPCR was performed in triplicates for each dilution along with non-spiked control and NTC to determine the detection limit.
Detection of B. pseudomallei in environmental matrices
B. pseudomallei prefer moist, nutrient-rich rhizospheric soil and also found in water bodies [2]. For the applicability of the developed assay S664 to detect B. pseudomallei in water and soil, a multiplex assay S664 has been developed using the novel target BPSS0664 and the cry1 gene of Bacillus thuringiensis (Table 1) [20]. The multiplex qPCR reaction was prepared in total 20 µL volume containing 1× master mix, 250 nM of forward primer, reverse primer, and probe of gene BPSS0664, 750 nM of forward and reverse primer and 250 nM of hydrolysis probe of gene cry1 and 2 µL of DNA. The river water was collected from the Narmada River, Khandwa, Madhya Pradesh, India (GPS coordinates: N 22° 14’ 36.58”, E 76° 9’ 39.79”) and paddy field soil from Bharatpur village in Lucknow, Uttar Pradesh, India (GPS coordinates: N 27° 2’ 43.05”, E 80° 53’ 39.74”) [16]. The river water was spiked with B. pseudomallei cells at a concentration of 5 × 107 to 5 × 100 CFU/mL of water. The B. thuringiensis cells (105) were chosen as an internal control for DNA extraction and PCR amplification for detecting B. pseudomallei in water samples [21]. The concentration of B. thuringiensis cells was empirically determined to yield cycle threshold (Ct) values between 28 and 30 along with B. pseudomallei-specific amplification. The DNA was extracted from spiked water (B. pseudomallei and B. thuringiensis cells) using the DNeasy Blood and Tissue kit according to the manufacturer’s instructions. The paddy field soil was spiked with B. pseudomallei cells at a concentration of 2 × 107 to 2 × 100 CFU/gm of soil. The B. thuringiensis spores (105) were chosen as an internal control for DNA extraction and PCR amplification for detecting B. pseudomallei in soil samples [20]. The concentration of B. thuringiensis spores was empirically determined to yield Ct values between 28 and 30 along with B. pseudomallei-specific amplification. The total DNA from spiked soil (B. pseudomallei and B. thuringiensis cells) was extracted using the NucleoSpin Soil kit (Macherey-Nagel, #REF740780.50) according to the manufacturer’s instructions. All the qPCR reactions for spiked water as well as spiked soil were performed in triplicates for each dilution along with the non-spiked control and NTC to determine the detection limit.
Results
Identification of B. pseudomallei-specific target and primer-probe designing
The results of the in-silico studies showed that the 85-bp region within the gene BPSS0664 was unique and had no significant similarity with the sequences of related or non-related organisms. The presence of in-silico identified novel gene sequence in 1794 out of 1796 strains of B. pseudomallei indicates enhanced strain coverage in contrast to orf2 of T3SS1 which is present in only 1791 strains. BPSS0664 is exclusively present in five strains (1258a, NRF80Bp1, SBCT-RF80-BP1, NAU14B-9, and MSHR1879) that are devoid of orf2 sequence (Table S1). Multiple sequence alignment analysis revealed that the 85-bp region of the BPSS0664 gene was highly conserved (Fig. S1). The forward and reverse primers amplifying an 85-bp long amplicon along with a labelled hydrolysis probe were designed and used for the real-time PCR assay S664 development (Table 1).
Analytical sensitivity and specificity of the assay S664
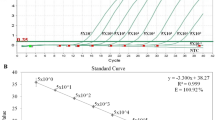
The limit of detection of the developed novel sequence-based assay S664 was found to be 3 GE copies of B. pseudomallei genome per qPCR reaction (Fig. 1A). A linear calibration line was obtained in the standard curve plotted using mean Ct values against the log concentration of a 10-fold serially diluted B. pseudomallei genomic DNA with a linear model equation of y = -3.227x + 37.434. A strong linear inverse relationship was observed between log10GE copies of B. pseudomallei and Ct values with a linear regression coefficient value (R2) of 0.997. The efficiency of assay S664 was found to be 104.12% (Fig. 1B). The intra-assay variations were estimated between 0.4% and 2.7% while inter-assay variations were between 0.3% and 1.9%. The developed assay S664 has specifically detected standard strains, soil isolates, and clinical isolates of B. pseudomallei, and no cross-reactivity was observed with bacterial species within the genus Burkholderia or other closely related organisms. Moreover, no cross-reactivity was also observed with other non B. pseudomallei related organisms (Table 2).

Analytical sensitivity of the assay S664 (A) Amplification plot showing sensitivity of 10-fold serially diluted B. pseudomallei GE copies from 3 × 106 to 3 × 100 per qPCR reaction (B) Graph plot showing straight calibration line for 10-fold serially diluted log B. pseudomallei GE copies from 3 × 106 to 3 × 100 per qPCR reaction
The feasibility of assay S664 to detect B. pseudomallei in clinical samples
The feasibility of the developed multiplex assay S664 for the clinical diagnosis of melioidosis was assessed by spiking healthy human blood and urine with B. pseudomallei cells and employing suitable internal control (RNaseP). The developed multiplex assay S664 was found to be highly sensitive with a detection limit of 5 × 102 CFU/mL for both human blood and urine. The efficiency of multiplex assay S664 was found to be 99.2% and 89.5% for the detection of B. pseudomallei in human blood and urine respectively (Fig. 2A and B). The RNaseP gene used as a control for nucleic acid extraction and amplification was readily detected in all the spiked clinical samples with a mean Ct value (± SD) of 23.9 ± 0.8 and 29.0 ± 0.8 for human blood and urine respectively.

The feasibility of multiplex assay S664 for detection of B. pseudomallei in clinical and environmental matrices (A) Graph plot showing straight calibration line for detection of B. pseudomallei cells spiked in human blood from 5 × 107 to 5 × 102 CFU/mL (B) Graph plot showing straight calibration line for detection of B. pseudomallei cells spiked in human urine from 5 × 107 to 5 × 102 CFU/mL (C) Graph plot showing straight calibration line for detection of B. pseudomallei cells spiked in river water from 5 × 107 to 5 × 101 CFU/mL (D) Graph plot showing straight calibration line for detection of B. pseudomallei cells spiked in paddy field soil in concentration of 2 × 107 to 2 × 103 CFU/gm of soil
The feasibility of assay S664 to detect B. pseudomallei in environmental samples
The feasibility of the developed multiplex assay S664 in detecting B. pseudomallei from the environmental matrices namely, water and soil was assessed by spiking B. pseudomallei cells along with suitable internal controls. The detection limit of assay S664 in B. pseudomallei-spiked river water was found to be 5 × 101 CFU/mL and no amplification was observed in non-spiked control river water (Fig. 2C). The B. thuringiensis vegetative cells used as a control for nucleic acid extraction and amplification was readily detected in all spiked river water samples with a mean Ct value of 29.9 ± 1.0. The detection limit of assay S664 in spiked paddy field soil was found to be 2 × 103 CFU/gm of soil and no amplification for B. pseudomallei was observed in non-spiked control soil (Fig. 2D). The B. thuringiensis spores used as a control for nucleic acid extraction and amplification was readily detected in all spiked paddy field soil samples with a mean Ct value of 28.3 ± 1.1. The efficiency of the multiplex assay S664 was 103.6% and 89.3% in B. pseudomallei-spiked river water and paddy field soil respectively.
Discussion
B. pseudomallei is an emerging pathogen as well as a potential biothreat agent owing to its remarkable capability to survive in extreme environmental conditions [22,23,24,25]. The disease melioidosis is acquired through direct contact with a pathogen from a contaminated environment [4]. The non-specific clinical manifestation leads to an inaccurate diagnosis on clinical grounds. The special culture method, which is a mainstay for diagnosis is confounded by its slow growth, requirement of special selective media, and expertise in identifying B. pseudomallei culture. All these limiting factors are collectively accountable for the high case fatality rate [6]. Therefore, specific and rapid identification of the pathogen is essential for the early control and prevention of melioidosis. Molecular detection techniques such as PCRs and other isothermal assays offer several advantages over conventional serological methods in terms of sensitivity and specificity. The reported molecular assays are mostly based on orf2 of T3SS1 of B. pseudomallei [26,27,28,29]. Orf2 is available in 1791 genomic assemblies of B. pseudomallei out of 1796. Hence, the assays based on orf2 can detect only 1791 strains of B. pseudomallei out of 1796 due to the lost target sequence. Furthermore, the highly plastic genome of B. pseudomallei is exceptionally vulnerable to natural genetic recombination and artificial genetic manipulations which can alter the outcome of molecular assays based on the orf2 sequence [10,11,12,13]. Additionally, the molecular assays developed in the past lacked suitable internal controls for appropriate monitoring of nucleic acid extraction and amplification from the clinical and environmental samples [9]. Therefore, there is an indispensable need to develop molecular assays based on novel gene targets accompanied by internal controls for the specific, sensitive, and reliable detection of B. pseudomallei in clinical and environmental samples.
In the present study, we have identified a novel and highly specific 85-bp-long nucleotide sequence within the BPSS0664 gene using extensive bioinformatic analysis. The identified sequence is highly conserved in the genomes of 1794 B. pseudomallei strains out of 1796. The comparative analysis of orf2 and BPSS0664 suggests the presence of both targets in 1789 strains of B. pseudomallei, whereas BPSS0664 is exclusively present in 5 strains i.e. 1258a (human isolate, Thailand), NRF80Bp1 (environmental isolate, Thailand), SBCT-RF80-BP1 (environmental isolate, Thailand), NAU14B-9 (environmental isolate, Australia), and MSHR1879 (human isolate, Australia) which are lacking the orf2 sequence and hence, the in-silico identified novel gene BPSS0664 has an advantage over orf2 for specific and sensitive assay development. This newly identified gene sequence was used for the development of the hydrolysis probe-based qPCR assay S664. The analytical sensitivity of the developed qPCR assay S664 was evaluated with freshly isolated genomic DNA of B. pseudomallei (NCTC 13392). The assay S664 could detect 3 GE copies of the genome per reaction which is more sensitive than reported real-time PCR assays [16, 29,30,31,32]. The specificity of assay S664 was further evaluated with 65 different B. pseudomallei-related and non-related bacterial cultures. The assay S664 was found to be highly specific for the identification of B. pseudomallei as no cross-reactivity was observed with other species of the genus Burkholderia (B. thailandensis, B. mallei, B. cepacia, and B. gladioli). Further, no cross-reactivity of the newly developed assay was also observed with related bacterial pathogens classified in group proteobacteria including Brucella, Coxiella, Francisella, Pseudomonas, Klebsiella, Salmonella and Shigella as well as other bacteria used in the present study.
The diagnostic and detection applicability of the assay S664 to detect B. pseudomallei in clinical and environmental samples, respectively, was evaluated by spiking B. pseudomallei cells in human blood, urine, river water, and paddy field soil. To ensure the proper nucleic acid extraction from different clinical and environmental matrices and to differentiate a true from a false negative result, the singleplex assay S664 was translated into a multiplex assay by incorporating suitable internal controls. For the clinical diagnosis of melioidosis in humans, a multiplex assay incorporating BPSS0664 and the RNaseP gene as an extraction and amplification control was developed. The basis for the selection of the RNaseP is its presence in every human cell, and hence it can be readily detectable in all human clinical samples [33]. The developed multiplex assay S664 was found to be highly sensitive in the detection of B. pseudomallei in clinical matrices with a detection limit of 5 × 102 CFU/mL for both human blood and urine. The assay S664 has higher sensitivity as compared to orf2-based real-time qPCR assay in clinical matrices [29, 32, 34]. The amplification of the RNaseP gene used as an internal control was also observed in all the B. pseudomallei spiked and non-spiked human blood and urine samples and no cross-reactivity was observed with human DNA. The lower assay efficiency in human urine (89.5%) was observed as compared to human blood (99.2%), which could be due to the presence of PCR inhibitors in urine samples such as urea [35, 36]. Together with the application of RNaseP as the internal control for both nucleic acid extraction and amplification, the developed multiplex assay assures a highly reliable and specific diagnosis of melioidosis in human clinical samples.
For the detection of B. pseudomallei in environmental samples such as water and soil which are the primary sources of infection, a multiplex assay incorporating BPSS0664 and the cry1 gene as an extraction and amplification control was developed [20]. The B. thuringiensis vegetative cells and spores were spiked to water and soil samples, respectively, before the nucleic acid extraction. The multiplex assay S664 could detect 5 × 101 cells of B. pseudomallei per mL of water and 2 × 103 cells of B. pseudomallei per gm of soil which is higher than the detection limit reported by Saxena et al. [37] and similar to the detection limit reported by Peng et al. [32]. The amplification of the cry1 gene was observed in all B. thuringiensis spiked water and soil samples with Ct values ranging from 28 to 31. The lower assay efficiency in paddy field soil (89.3%) was observed as compared to the river water (103.6%) which could be due to the presence of PCR inhibitors in soil samples such as humic substances [36, 38]. Moreover, no amplification was observed with total DNA isolated from unspiked water and soil which are the primary habitats of many micro and macroorganisms showing the high degree of specificity of developed multiplex assay S664 [39, 40]. These results indicate the potential usefulness of the developed multiplex assay using the cry1 gene as an internal control for the detection of B. pseudomallei in environmental samples.
In conclusion, the developed multiplex qPCR assay targeting a novel gene with suitable internal controls has the potential for both sensitive and specific melioidosis disease diagnosis and it can provide an early and specific detection of B. pseudomallei in environmental samples in an outbreak or in a biothreat scenario. Altogether, the novel assay S664 can be a potential substitute for orf2-based molecular assays for detecting B. pseudomallei in diverse clinical and environmental matrices.
Data availability
All data generated or analysed during this study are included in this article [and its supplementary information files].
References
Cheng AC, Currie BJ. Melioidosis: epidemiology, pathophysiology, and management. Clin Microbiol Rev. 2005;18(2):383–416. https://doi.org/10.1128/CMR.18.2.383-416.2005.
Kaestli M, Schmid M, Mayo M, Rothballer M, Harrington G, Richardson L, Hill A, Hill J, Tuanyok A, Keim P, Hartmann A, Currie BJ. Out of the ground: aerial and exotic habitats of the melioidosis bacterium Burkholderia pseudomallei in grasses in Australia. Environ Microbiol. 2012;14(8):2058–70. https://doi.org/10.1111/j.1462-2920.2011.02671.x.
Currie BJ. Melioidosis: evolving concepts in epidemiology, pathogenesis, and treatment. Semin Respir Crit Care Med. 2015;36(1):111–25. https://doi.org/10.1055/s-0034-1398389.
Wiersinga WJ, Virk HS, Torres AG, Currie BJ, Peacock SJ, Dance DAB, Limmathurotsakul D. Melioidosis Nat Rev Dis Primers. 2018;4:17107. https://doi.org/10.1038/nrdp.2017.107.
Hoffmaster AR, AuCoin D, Baccam P, Baggett HC, Baird R, Bhengsri S, Blaney DD, Brett PJ, Brooks TJ, Brown KA, et al. Melioidosis diagnostic workshop, 2013. Emerg Infect Dis. 2015;21(2):e141045. https://doi.org/10.3201/eid2102.141045.
Wagner GE, Föderl-Höbenreich E, Assig K, Lipp M, Berner A, Kohler C, Lichtenegger S, Stiehler J, Karoonboonyanan W, Thanapattarapairoj N, et al. Melioidosis DS rapid test: a standardized serological dipstick assay with increased sensitivity and reliability due to multiplex detection. PLoS Negl Trop Dis. 2020;14(7):e0008452. https://doi.org/10.1371/journal.pntd.0008452.
Richardson LJ, Kaestli M, Mayo M, Bowers JR, Tuanyok A, Schupp J, Engelthaler D, Wagner DM, Keim PS, Currie BJ. Towards a rapid molecular diagnostic for melioidosis: comparison of DNA extraction methods from clinical specimens. J Microbiol Methods. 2012;88(1):179–81. https://doi.org/10.1016/j.mimet.2011.10.023.
Price EP, Dale JL, Cook JM, Sarovich DS, Seymour ML, Ginther JL, Kaufman EL, Beckstrom-Sternberg SM, Mayo M, Kaestli M, et al. Development and validation of Burkholderia pseudomallei-specific real-time PCR assays for clinical, environmental or forensic detection applications. PLoS ONE. 2012;7(5):e37723. https://doi.org/10.1371/journal.pone.0037723.
Lowe W, March JK, Bunnell AJ, O’Neill KL, Robison RA. PCR-based methodologies used to detect and differentiate the Burkholderia pseudomallei complex: B. Pseudomallei, B. mallei, and B. Thailandensis. Curr Issues Mol Biol. 2014;16:23–54. https://doi.org/10.21775/cimb.016.023.
Pearson T, Giffard P, Beckstrom-Sternberg S, Auerbach R, Hornstra H, Tuanyok A, Price EP, Glass MB, Leadem B, Beckstrom-Sternberg JS, et al. Phylogeographic reconstruction of a bacterial species with high levels of lateral gene transfer. BMC Biol. 2009;7:78. https://doi.org/10.1186/1741-7007-7-78.
Norris MH, et al. Burkholderia pseudomallei natural competency and DNA catabolism: identification and characterization of relevant genes from a constructed fosmid library. PLoS ONE. 2017;12(12):e0189018. https://doi.org/10.1371/journal.pone.0189018.
Choi KH, Mima T, Casart Y, Rholl D, Kumar A, Beacham IR, Schweizer HP. Genetic tools for select-agent-compliant manipulation of Burkholderia pseudomallei. Appl Environ Microbiol. 2008;74(4):1064–75. https://doi.org/10.1128/AEM.02430-07.
Holden MT, Titball RW, Peacock SJ, Cerdeño-Tárraga AM, Atkins T, Crossman LC, Pitt T, Churcher C, Mungall K, Bentley SD, et al. Genomic plasticity of the causative agent of melioidosis, Burkholderia pseudomallei. Proc Natl Acad Sci U S A. 2004;101(39):14240–5. https://doi.org/10.1073/pnas.0403302101.
Galyov EE, Brett PJ, DeShazer D. Molecular insights into Burkholderia pseudomallei and Burkholderia mallei pathogenesis. Annu Rev Microbiol. 2010;64:495–517. https://doi.org/10.1146/annurev.micro.112408.134030.
Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. J Comput Biol. 2000;7(1–2):203–14. https://doi.org/10.1089/10665270050081478.
Trung TT, Hetzer A, Göhler A, Topfstedt E, Wuthiekanun V, Limmathurotsakul D, Peacock SJ, Steinmetz I. Highly sensitive direct detection and quantification of Burkholderia pseudomallei bacteria in environmental soil samples by using real-time PCR. Appl Environ Microbiol. 2011;77(18):6486–94. https://doi.org/10.1128/AEM.00735-11.
Rutledge RG, Côté C. Mathematics of quantitative kinetic PCR and the application of standard curves. Nucleic Acids Res. 2003;31(16):e93. https://doi.org/10.1093/nar/gng093.
Waggoner JJ, Abeynayake J, Sahoo MK, Gresh L, Tellez Y, Gonzalez K, Ballesteros G, Balmaseda A, Karunaratne K, Harris E, Pinsky BA. Development of an internally controlled real-time reverse transcriptase PCR assay for pan-dengue virus detection and comparison of four molecular dengue virus detection assays. J Clin Microbiol. 2013;51(7):2172–81. https://doi.org/10.1128/JCM.00548-13.
Dong M, Fisher C, Añez G, Rios M, Nakhasi HL, Hobson JP, Beanan M, Hockman D, Grigorenko E, Duncan R. Standardized methods to generate mock (spiked) clinical specimens by spiking blood or plasma with cultured pathogens. J Appl Microbiol. 2016;120(4):1119–29. https://doi.org/10.1111/jam.13082.
de Bruin A, Janse I, Koning M, de Heer L, van der Plaats RQ, van Leuken JP, van Rotterdam BJ. Detection of Coxiella burnetii DNA in the environment during and after a large Q fever epidemic in the Netherlands. J Appl Microbiol. 2013;114(5):1395–404. https://doi.org/10.1111/jam.12163.
Stoeckel DM, Stelzer EA, Dick LK. Evaluation of two spike-and-recovery controls for assessment of extraction efficiency in microbial source tracking studies. Water Res. 2009;43(19):4820–7. https://doi.org/10.1016/j.watres.2009.06.028.
Pumpuang A, Chantratita N, Wikraiphat C, Saiprom N, Day NP, Peacock SJ, Wuthiekanun V. Survival of Burkholderia pseudomallei in distilled water for 16 years. Trans R Soc Trop Med Hyg. 2011;105(10):598–600. https://doi.org/10.1016/j.trstmh.2011.06.004.
Hantrakun V, Rongkard P, Oyuchua M, Amornchai P, Lim C, Wuthiekanun V, Day NP, Peacock SJ, Limmathurotsakul D. Soil nutrient depletion is Associated with the Presence of Burkholderia pseudomallei. Appl Environ Microbiol. 2016;82(24):7086–92. https://doi.org/10.1128/AEM.02538-16.
Yip TW, Hewagama S, Mayo M, Price EP, Sarovich DS, Bastian I, Baird RW, Spratt BG, Currie BJ. Endemic melioidosis in residents of desert region after atypically intense rainfall in central Australia, 2011. Emerg Infect Dis. 2015;21(6):1038–40. https://doi.org/10.3201/eid2106.141908.
Pumirat P, Saetun P, Sinchaikul S, Chen ST, Korbsrisate S, Thongboonkerd V. Altered secretome of Burkholderia pseudomallei induced by salt stress. Biochim Biophys Acta. 2009;1794(6):898–904. https://doi.org/10.1016/j.bbapap.2009.01.011.
Chantratita N, Meumann E, Thanwisai A, Limmathurotsakul D, Wuthiekanun V, Wannapasni S, Tumapa S, Day NP, Peacock SJ. Loop-mediated isothermal amplification method targeting the TTS1 gene cluster for detection of Burkholderia pseudomallei and diagnosis of melioidosis. J Clin Microbiol. 2008;46(2):568–73. https://doi.org/10.1128/JCM.01817-07.
Wong Tzeling JM, Engku Nur Syafirah EAR, Irekeola AA, Yusof W, Aminuddin Baki NN, Zueter A, Harun A, Chan YY. One-step, multiplex, dual-function oligonucleotide of loop-mediated isothermal amplification assay for the detection of pathogenic Burkholderia pseudomallei. Anal Chim Acta. 2021;1171:338682. https://doi.org/10.1016/j.aca.2021.338682.
Li J, Zhong Q, Shang MY, Li M, Jiang YS, Zou JJ, Ma SS, Huang Q, Lu WP. Preliminary Evaluation of Rapid Visual Identification of Burkholderia pseudomallei using a newly developed lateral Flow Strip-based recombinase polymerase amplification (LF-RPA) system. Front Cell Infect Microbiol. 2022;11:804737. https://doi.org/10.3389/fcimb.2021.804737.
Novak RT, Glass MB, Gee JE, Gal D, Mayo MJ, Currie BJ, Wilkins PP. Development and evaluation of a real-time PCR assay targeting the type III secretion system of Burkholderia pseudomallei. J Clin Microbiol. 2006;44(1):85–90. https://doi.org/10.1128/JCM.44.1.85-90.2006.
Supaprom C, Wang D, Leelayuwat C, Thaewpia W, Susaengrat W, Koh V, Ooi EE, Lertmemongkolchai G, Liu Y. Development of real-time PCR assays and evaluation of their potential use for rapid detection of Burkholderia pseudomallei in clinical blood specimens. J Clin Microbiol. 2007;45(9):2894–901. https://doi.org/10.1128/JCM.00291-07.
Kaestli M, Richardson LJ, Colman RE, Tuanyok A, Price EP, Bowers JR, Mayo M, Kelley E, Seymour ML, Sarovich DS, Pearson T, Engelthaler DM, Wagner DM, Keim PS, Schupp JM, Currie BJ. Comparison of TaqMan PCR assays for detection of the melioidosis agent Burkholderia pseudomallei in clinical specimens. J Clin Microbiol. 2012;50(6):2059–62. https://doi.org/10.1128/JCM.06737-1.
Peng Y, Zheng X, Kan B, Li W, Zhang W, Jiang T, Lu J, Qin A. Rapid detection of Burkholderia pseudomallei with a lateral flow recombinase polymerase amplification assay. PLoS ONE. 2019;14(7):e0213416. https://doi.org/10.1371/journal.pone.0213416.
Wozniak A, Cerda A, Ibarra-Henríquez C, Sebastian V, Armijo G, Lamig L, Miranda C, Lagos M, Solari S, Guzmán AM, et al. A simple RNA preparation method for SARS-CoV-2 detection by RT-qPCR. Sci Rep. 2020;10(1):16608. https://doi.org/10.1038/s41598-020-73616-w.
Podnecky NL, Elrod MG, Newton BR, Dauphin LA, Shi J, Chawalchitiporn S, Baggett HC, Hoffmaster AR, Gee JE. Comparison of DNA extraction kits for detection of Burkholderia pseudomallei in spiked human whole blood using real-time PCR. PLoS ONE. 2013;8(2):e58032. https://doi.org/10.1371/journal.pone.0058032.
Munch MM, Chambers LC, Manhart LE, Domogala D, Lopez A, Fredricks DN, Srinivasan S. Optimizing bacterial DNA extraction in urine. PLoS ONE. 2019;14(9):e0222962. https://doi.org/10.1371/journal.pone.0222962.
Schrader C, Schielke A, Ellerbroek L, Johne R. PCR inhibitors - occurrence, properties and removal. J Appl Microbiol. 2012;113(5):1014–26. https://doi.org/10.1111/j.1365-2672.2012.05384.x.
Saxena A, Pal V, Tripathi NK, Goel AK. A recombinase polymerase amplification lateral flow assay for rapid detection of Burkholderia pseudomallei, the causative agent of melioidosis. Braz J Microbiol. 2022;53(1):185–93. https://doi.org/10.1007/s42770-021-00669-y.
Ding C, Xu X, Liu Y, Huang X, Xi M, Liu H, Deyett E, Dumont MG, Di H, Hernández M, Xu J, Li Y. Diversity and assembly of active bacteria and their potential function along soil aggregates in a paddy field. Sci Total Environ. 2023;866:161360. https://doi.org/10.1016/j.scitotenv.2022.161360.
Liu S, Sun Y, Shi F, Liu Y, Wang F, Dong S, Li M. Composition and diversity of Soil Microbial Community Associated with Land Use types in the Agro-pastoral Area in the Upper Yellow River Basin. Front Plant Sci. 2022;13:819661. https://doi.org/10.3389/fpls.2022.819661.
Pernthaler J. Freshwater Microbial communities. In: Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F, editors. The prokaryotes. Berlin, Heidelberg: Springer; 2013. pp. 97–112. https://doi.org/10.1007/978-3-642-30123-0_40.
Acknowledgements
The authors are thankful to the Director, Defence Research and Development Establishment, Gwalior for his motivation and continuous support for this study. The authors are also thankful to the Defence Research and Development Organization, Ministry of Defence, India, for providing the necessary facilities and instrumentation. The authors would also like to thank Dr. Gitanjali Javir, DRDO-Research Associate for providing help in the statistical analysis.
Funding
This research was supported by Defence Research and Development Organization (DRDO) research funds. Pranjal Kumar Yadav is the recipient of DRDO research fellowships.
Author information
Authors and Affiliations
Contributions
DT, SK, and PKY have designed the experiments. PKY has performed the experiments and written the manuscript. SS and MP helped in bacterial culture maintenance, DNA extraction, spore production, purification, and quantification. DT, SK, and PKY have analyzed the results. PKY, SS, MP, SK, SP, and DT have reviewed, revised, and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethical approval
The study was reviewed and approved by Institutional Research and Ethics Committee (IREC). This manuscript has been allotted DRDE accession number DRDE-IREC-30-28032023.
Competing interests
The authors declare that the research was conducted in the absence of any financial or non-financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yadav, P.K., Singh, S., Paul, M. et al. Development of a novel sequence based real-time PCR assay for specific and sensitive detection of Burkholderia pseudomallei in clinical and environmental matrices. Ann Clin Microbiol Antimicrob 23, 30 (2024). https://doi.org/10.1186/s12941-024-00693-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12941-024-00693-4