
Abstract
Background
Escherichia coli (E. coli) is a common human pathogen, responsible for a broad spectrum of infections. Sites of infection can vary, but the hepato-biliary system is of particular concern due to the infection-associated formation of gallstones and the spread of pathogens from the bile ducts into the bloodstream.
Case presentation
The presented case is striking, as the detected isolate showed a positive string test. This hypermucoviscous phenotype is atypical for E. coli and a particular feature of hypervirulent Klebsiella pneumoniae (K. pneumoniae) variants.
Objectives
To provide new insights into the genomic background of an E. coli strain with an unusual hypermucoviscous phenotype using hybrid short- and long-read sequencing approaches.
Results
Complete hybrid assemblies of the E. coli genome and plasmids were done and used for genome based typing. Isolate 537–20 was assigned to the multilocus sequence type ST88 and serotype O8:H4. The strain showed a close relationship to avian pathogenic strains. Analysis of the chromosome and plasmids revealed the presence of several virulence factors, such as the Conserved Virulence Plasmidic (CVP) region on plasmid 537-20_1, including several iron acquisition genes (sitABCD, iroABCDEN, iucABCD, hbd) and the iutA gene encoding the receptor of the siderophore aerobactin. The hypermucoviscous phenotype could be caused by encapsulation of putative K. pneumoniae origin.
Conclusions
Hybrid sequencing enabled detailed genomic characterization of the hypermucoviscous E. coli strain, revealing virulence factors that have their putative origin in K. pneumoniae.
Similar content being viewed by others

Background
Escherichia coli is a Gram-negative, rod shaped, facultative anaerobic bacterium of the Enterobacteriaceae family. The species is well known to be a frequent and numerous intestinal colonizer and pathogen of animals and humans, while also being ubiquitously present in the environment [1, 2]. E. coli strains are commonly categorized by their ability to cause specific intestinal or extraintestinal infections. Extraintestinal E. coli (ExPEC) have a well-described repertoire of virulence factors, and distinct clonal lineages are spread out globally [3,4,5]. Apart from these properties, in few cases atypical observations of hypermucoviscous (hmv) E. coli strains have been described in the literature [6,7,8,9]. Hypermucoviscosity (usual mucoid appearance on agar plates) is usually a characteristic of certain strain types of the Klebsiella pneumoniae (K. pneumoniae) species and is typically detected using the “string-test” [9]. A strain is considered positive, if it presents a mucoid string (> 5 mm) when touched with a glass rod or inoculation loop [10, 11]. It is typically described for clinical isolates that are associated with severe and invasive infections of otherwise healthy and immunocompetent, non-risk patients [12, 13]. This phenotype seems to be conditioned by several genetic components, including distinct capsule types and virulence genes (iucA, iutA, rmpA, rmpA2) [13,14,15,16]. The hmv phenotype decreases the immunological host defenses and enhances the bacterial survival rates [17, 18].
In the presented case, a string-positive E. coli strain was isolated from a patient with recurrent bacteremia, conjecturally causative colligated with the patient’s cholestatic cholangitis. The occurrence of E. coli in the biliary tract is well known, for example as a cause of gallstones [19, 20]. Intestinal bacteria such as E. coli, especially ExPEC, are able to invade the biliary tract during bile stasis, resulting in an acute infection [21, 22]. Furthermore, these severe infections are able to overcome the biliary system and thus allow E. coli to invade the bloodstream, leading to acute bacteremia [23, 24]. Furthermore, Søgaard and colleagues stated that gastrointestinal, hepatobiliary, and urinary tract cancer may debut with E. coli community-acquired bacteremia [25].
In this study, we aimed to analyze the genetic background of an E. coli strain displaying an hmv phenotype, using state-of-the-art genome analyses, including short- and long-read sequencing techniques.
Importance
Description of an unusual hmv E. coli isolated from a patient suffering from biliary tract carcinoma and recurrent bacteremia.
Methods
Case presentation and bacterial isolation
A 71-year-old male German patient presented to the emergency department with acute cholangitis caused by a perihilar cholangiocarcinoma, also known as “Klatskin tumor” [26, 27], with hepatic and lymphogenic metastases. Comorbidities included chronic kidney insufficiency (KDIGO G2), type 2 diabetes mellitus and paroxysmal atrial fibrillation. At the time of admission to the hospital, the patient had fever (39.7 °C) and elevated inflammatory values (leukocytes: 16 × 109/L, C-reactive protein: 88 mg/L, interleukin-6: 493 pg/mL). A few weeks earlier, the patient received piperacillin/tazobactam for similar clinical symptoms in another hospital.
As part of the extended routine diagnostics, a string-test positive E. coli isolate was identified in 2/2 peripherally obtained blood culture pairs and subjected to a detailed microbiological analysis.
The patient underwent endoscopic retrograde cholangiography (ERC) and biliary drainage was re-established by exchange of two bile duct plastic stents. In addition, an antibiotic therapy was immediately initiated with piperacillin/tazobactam. Later, the patient received cefotaxime and metronidazole and—due to a lack of clinical improvement—imipenem/cilastatine. Despite the treatment, the patient experienced several septic episodes during the following two months. Blood cultures were intermittently positive for the string-test positive E. coli isolate despite various antibiotic therapies. Finally, the patient received palliative chemotherapy and died 12 months after the initial diagnosis.
In vitro characterization
E. coli 537-20 was isolated from patient blood cultures and identified as Escherichia coli via biochemical, phenotypical tests and matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF). The isolate displayed an hmv phenotype on agar plates. A string test, typically used for hmv K. pneumoniae, was conducted to verify this phenotype. The string test was rated as positive as a mucoid string of > 5 mm could be observed, when touching bacterial plate growth with a standard inoculation loop and gentle pulling away, as described in the literature [11].
Antibiotic susceptibility testing (AST) was done using a VITEK 2 (GN AST N248) and a broth microdilution method, as described before [28]. The obtained results were interpreted according to EUCAST (European Committee on Antimicrobial Susceptibility Testing) standards and breakpoints v12.0 (https://eucast.org/clinical_breakpoints/). The following substances were used for AST: ampicillin, trimethoprim, cefotaxime, ceftazidime, gentamycin, chloramphenicol, colistin, nalidixic acid, ciprofloxacin, meropenem, sulfamethoxazole-trimethoprim, piperacillin, piperacillin-tazobactam, aztreonam, cefepime, tobramycin, amikacin and fosfomycin.
To investigate the general plasmid content and plasmid size, an S1-nuclease restriction and pulsed-field gel electrophoresis (PFGE) was performed, as described elsewhere [29]. The transferability of resistance genes by conjugative plasmids and a possible co-transfer of other phenotypic properties was investigated in a broth mating experiment using the sodium azide-resistant E. coli strain J53 Azir as recipient.
Whole-genome sequencing, downstream data processing and assembly
DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen) and the MagAttract Kit (Qiagen) for high molecular weight DNA. The Qubit dsDNA HS Assay Kit (Invitrogen) was used for DNA quantification. DNeasy extracted DNA was sequenced on a NextSeq2000 benchtop device (Illumina) as described before [14]. The short-read whole-genome sequencing data analysis workflow was performed as described before, including several steps for quality control [14]. Long read sequencing was done similar as described before [30]. To this end high molecular weight DNA was size selected using SPRISelect beads (Beckman Coulter) and subjected to long-read sequencing with barcode 5 of the rapid barcoding kit (SQK-RBK004) on an R9.4 (FLO-MIN106) MinION flow cell and a Mk1c device for 21 h with live fast base-calling using guppy (v4.2.3) and auto de-multiplexing. This resulted in 137 k passed reads and 0.822 Gbp data for isolate 537–20. Reads were quality controlled using pycoqc (v2.5.0.23, https://github.com/a-slide/pycoQC) and kraken (v1.0) using an 8 GB mini kraken database. Adaptors were trimmed with porechop (v0.2.4, https://github.com/rrwick/Porechop) and the best 500 Mbp selected using filtlong (v0.2.0) and otherwise default parameters. Adaptor-clipped Illumina and filtered long-read data were hybrid assembled with Unicycler (v0.4.9b) [31] using default parameters, except for pinning SPAdes to version v3.13.0. The assembly was annotated with PGAP [32], first locally and later upon submission at and through NCBI again. One contig of 1760 bp was almost identical to a stretch of plasmid p537-20_1, had a reported depth of 0.4 × (chromosome was 1.04 ×), did not result in a circular contig and did not contain any plasmid replication genes. This contig was therefore removed from the assembly as it was thought to be an artifact.
In silico characterization and prediction of virulence and resistance
The downstream analyses included the SeqSphere+ software suite (v7.7.5) [33] and web tools provided by the center for genomic epidemiology, including ResFinder (v4.1) [34], VirulenceFinder (v2.0) [35] and the Mobile Element Finder (v1.0) [36]. Also, the EnteroBase (v1.1.2) [37] platform was used for typing and investigations on a population level, including wgMLST SNP analyses and phylogenetic investigation of comparative isolates from Europe. Further, the Kleborate (v2.0.0) tool was used to investigate potential Klebsiella-related virulence traits [38]. The PLSDB was used to identify plasmid replicon types [39] and BLASTN was used to identify closely related plasmids. To this end, the whole plasmid sequence was used as a query in a BLASTN (megablast) [40] search against NCBI nr/nr and the results sorted according to accession length (ascending), percent identity (descending) and finally query cover (descending). The top three results were then selected. CGView Server BETA v0.1 [41] was used for visualization purposes of plasmids. Whole plasmid alignments were done with LASTZ (v1.02.00) [42, 43] (http://www.bx.psu.edu/~rsharris/lastz/) in the Geneious software (v2021.2.2, https://www.geneious.com) using either p537-20_1 or the comparison plasmid as a reference.
Results and discussion
Microbiological investigation of of E. coli strain 537–20
Strain 537–20 was isolated from blood cultures of an elderly patient suffering from advanced perihilar cholangiocarcinoma and was identified as Escherichia coli via MALDI-TOF. AST revealed susceptibility of strain 537–20 to all tested antibiotics with exception of nalidixic acid and moxifloxacin. The in vitro transfer of these quinolone resistances in a broth mating experiment using the E. coli J53 recipient strain was not successful, incidicating a chromosomal, not plasmid based origin. Strain 537–20 showed a positive string test (Fig. 1), that has been typically described for K. pneumoniae strains and is caused by overproduction of mucus, an important feature of many hypervirulent K. pneumoniae strains [15]. The hypermucoviscosity characteristic in K. pneumoniae is discussed as general advantage for invasive infections, also considering the associated fitness costs [44] but the genetic cause in E. coli is unclear.

String test of E. coli isolate 537–20. A Strain was streaked out on Müller-Hinton sheep blood agar and incubated overnight at 37 °C. An inoculation loop (blue) was rubbed onto the colonies and pulled up vertically, forming a string (string test positive). B Close-up of string formed and indicated by arrows
Bacterial strain typing and chromosomal investigations
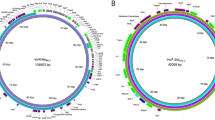
The complete, circular chromosome assembly of isolate 537–20 was 4,979,149 bp long (Fig. 2, Additional file 1: Table S1) and was used for in silico typing using Enterobase (Table 1) [37]. The isolate was assigned to Escherichia phylogroup C, serotype O8:H4, multilocus sequence type ST88 (Achtmann scheme) and cgMLST type ST173013. The SeqSphere cgMLST type was CT12213.

Chromosome of E. coli isolate 537–20 (CP091534). Coding sequences (CDS) are displayed separately for each of the six frames (rings with blue wedges). Prophages (as detected by Phaster [57, 58]), are shown in the outmost ring in green. tRNA: transfer RNA, rRNA: ribosomal RNA, GC skew ± : overabundance or lack of GC nucleotides. Image created with CGview Server v0.1 (http://cgview.ca/) [41]
E. coli of phylogenetic group C have been described as of commensal bacteria in humans and birds, but have been also reported in the clinical context [45, 46]. Serotype O8 has been commonly identified in clinical E. coli, but also in strains isolated from animals and sewage. [47,48,49,50]. Serotype O8 E. coli isolates are common EHEC variants; but the present isolate 537–20 did not contain a shiga toxin gene locus [48, 51]. The identified sequence type ST88, which is frequently associated with ExPEC strains in Europe, has been described as associated with colonization or urinary tract-infections, but not with bloodstream-infections [52]. Apart from that, a study by de Lastours et al. could show an association of increased mortality for bloodstream infections with the particular E. coli genotype combination ST88 and phylogenetic group C [53].
Analyzing the chromosomally encoded capsule of strain 537–20 using the Klebsiella-specific tool Kleborate resulted in the Klebsiella-capsule type KL54 (78.9% identity) and O5 (91.45% identity). The presence of Klebsiella-capsule genes in E. coli isolates seems to be uncommon, but not rare. Nanayakkara et al. [54] showed a wide diversity of Klebsiella-capsule types in E. coli isolates from Australia and were able to derive associations with E. coli subgroups. Their analyses revealed that the phylogenetic group C and serotype O8 are associated with emergence of Klebsiella-capsules. The detection of genes of a putative Klebsiella-capsule might be an explanation for the hmv phenotype of strain 537–20, especially since capsule types KL54have been found associated with positive string test results in previous studies [55]. Detailed functional studies are necessary to confirm the association between assessment of this capsule type and the hmv phenotype. Further, by applying the Kleborate tool, the operon genes for yersiniabactin and salmochelin were identified (Table2). This co-occurrence of the operons is a hint for the chromosomal integration of the K. pneumoniae integrative conjugative element (ICEKp) [56]. Furthermore, we identified several E. coli virulence factors in the chromosome of strain 537–20, including genes encoding adherence proteins (iha), siderophores (fyuA) and iron transporters (sitA) among others (Table 2).
We further hypothesized, that the hmv phenotype of strain 537–20 could be caused by genome- or mobilome- integrated phages, that induce bacterial cell lysis. Hence we investigated the occurrence of phages in chromosome using PHASTER [57, 58]. We identified four intact phages (PHAGE_Entero_lambda_NC_001416, PHAGE_Escher_HK639_NC_016158, PHAGE_Entero_mEp460_NC_019716, PHAGE_Entero_DE3_NC_042057) from the Siphoviridae family on the chromosome with sizes of 39–62 kbp and three incomplete phages with sizes of 16–25 kbp. Some of the identified phages (e.g. the Lambda phage) can exhibit lytic life cycles that could lead to bacterial lysis. However, because these phages are also present in other, non-hmv E. coli strains, we have no evidence for an involvement of these phages in the observed hmv phenotype.
The genetic background of quinolone resistance of strain 537–20 was identified subsequently by analysis of the gyrA gene sequence. The detected 1 bp mutation in gyrA resulting in amino acid substitution S38L has been described to cause quinolone resistance [59, 60]. This was in accordance to the MIC results, which indicated nalidixic acid resistance and the mating experiments which pointed to a chromosomal source of resistance. Interestingly, this gyrase modification bas been found to be implicated in reduced virulence by reducing the expression of fimA, papA, papB and the ompA genes resulting in decreased capacity to cause cystitis and pyelonephritis [61].
Plasmid analyses
S1-PFGE analysis indicated the presence of at least one large plasmid of approx. 120 kbp in isolate 537–20 (Fig. 3a). Several smaller plasmids of approximately 6000 bp, 2100 bp and 1500 bp (double band) were visible in a native plasmid preparation (Qiagen plasmid mini Kit) (Additional file 1: Fig. S1).

Analysis of plasmid p537-20_1 (CP091535). A Visible plasmids after S1 nuclease restriction and PFGE (right lane, molecular marker strain S. Braenderup H9812, restricted with XbaI). B Map of plasmid p537-20_1. Image created with CGview Server v0.1 (http://cgview.ca/) [41]
In accordance, five plasmids were present in the final short and long-read hybrid assembly (Table 3): one large plasmid of 126,021 bp (p537-20_1) (Fig. 3b) and four smaller plasmids of 6,647 bp (p537-20_2), 2,182 bp (p537-20_3), 1,552 bp (p537-20_4) and 1,433 bp (p537-20_5).
The plasmid p537-20_1 was of IncFIC(FII)_1/IncFIB(AP001918)_type, carried a vapBC toxin-antitoxin system, a potential hok/sok toxin-antitoxin system, several virulence genes, including the increased serum survival gene iss and, regulatory genes for iron metabolism (iroBCDEN, iucABCD, sitABCD, hbp [hemoglobin-binding protease autotransporter Hbp]). Many of these virulence genes and loci belong to the previously described Conserved Virulence Plasmidic (CVP) region. Lemaître et al. proposed the CVP region to be responsible for the virulence of an ExPEC strain of E. coli phylogroup C [46]. A LASTZ alignment using the original CVP sequence (HF922624) [46] as a reference, revealed that plasmid 537-20_1 contained most features of the CVP (Additional file 1: Fig. S2) with the main difference being an inversion of the sitABCDE-iucABCD-iutA region. When p537-20_1 served as the reference, the absence of a 40 kbp tra region was noted, but this could be attributed to sequence HF922624 possibly not including the full plasmid but only the CVP region (Additional file 1: Fig. S2). We further compared p537-20_1 to the sequence of the CVP containing plasmid pECOS88 (CU928146), which was proposed to be associated with meningitis in neonates and displaying high levels of bacteremia in a neonatal rat model [62]. The two plasmids showed a high degree of similarity in structure (Additional file 1: Fig. S3). Of note, p537-20_1 also contained the hbp gene which was absent in pECOS88. Hbp is a protease that is involved in host hemoglobin proteolysis and used to acquire iron from the host [63]. The hbp gene was shown to be associated with iron-limited infection-sites (e.g. intra-abdominal abscesses, [63] which might have served as a source of the blood stream infection). Interestingly, p537-20_1 also carried the iutA gene encoding the receptor of aerobactin, which is an important virulence factor for ExPEC and hypervirulent K. pneumoniae [52]. The carriage of iutA has been shown to be associated with increased mortality in E. coli, as well as K. pneumoniae bloodstream infection [23, 64]. The possession of the iutA gene but also many other iron acquisition genes likely represents a fitness advantage in the biliary tract, due to the general iron limitation in bile [23, 65]. The co-occurrence of iutA and iucA in APEC E. coli was described before, but as characteristic of Col (V) plasmids [66].
The other plasmids of strain 537-20 were considerably smaller and did not contain any notable features, except for two cea (Colicin E1) genes in p537-20_2. Plasmid p537-20_2 was identified as a ColRNAI plasmid and plasmids p537-20_4 and p537-20_5 as Col(MG828) plasmids. No replicon information could be identified for plasmid p537-20_3.
Global phylogenetic comparison
We further investigated the relationship of strain 537–20 to other E. coli isolates of the same sequence type. A total of 194 E. coli-ST88 isolates submitted to EnteroBase, were subjected for wgMLST SNP analyses. These originated from 21 European countries, including human origin and animal-associated origins (animals, livestock, food). The resulting phylogenetic tree (Fig. 4) visualizes the population structure of ST88 isolates.

Phylogenetic tree based on wgMLST SNP analyses of 194 European ST88 isolates, submitted to EnteroBase, and strain 537–20. A phylogenetic based on wgMLST SNP analyses of European ST88 isolates, submitted to EnteroBase. Visualization was realized with iTOL (v6.3) and information about isolate origin (country and isolation source) were added
The data analysis showed that E. coli ST88 is widely distributed in both human and animal-associated resources. Generally, ST88 is common for the European region [52]. Human- associated clusters can be observed, as well as animal-associated clusters. Other ST88 isolates of human origin, especially from Germany, were clearly separated from strain 537–20 in the phylogenetic tree (Fig. 4). Surprisingly, strain 537–20 clustered closely with isolates of animal origin (poultry/livestock) from Luxembourg and Denmark. This finding supports a hypothesis of a putative zoonotic origin, also because CVP containing plasmids were shown to be linked to extraintestinal avian pathogenic E. coli (APEC) [46, 62].
Conclusions
The hybrid sequencing approach allowed deep insights in the genome and plasmidome of the hypermucoviscous E. coli strain 537–20 causing recurrent bacteremia. The cause of the hypermucoviscous phenotype remains speculative, it might be due to the expression of a capsule of putative K. pneumoniae origin. As has been discussed for uropathogenic hypermucoviscous E. coli isolates [8], the direct linkage between the hmv phenotype and the clinical outcome is difficult to determine and raises the question of routine string-test screening.
The conducted typing and comparative phylogenetic analyses revealed a close relationship of this ST88 strain to ExPEC and APEC isolates. The virulence potential could be traced back to the acquisition of a conserved plasmid-located virulence island, the CVP region and ICEKp, that is a common virulence mediating element in K. pneumoniae. In addition, plasmid p537-20_1 contains several iron acquisition genes that enable growth under iron limiting conditions such as in the bile. Further in-depth studies are needed to investigate the interactions between this E. coli strain and human host its role in the processes of infection in bile duct and blood.
Availability of data and materials
Raw read data of both, Illumina and MinION were submitted to NCBI and are available under BioProject accession PRJNA800416, BioSample accession SAMN25247371 and Sequence Read Archive (SRA) accession numbers SRR17758648 (Illumina) and SRR17758649 (MinION). Hybrid assembly is available under accession numbers CP091534-CP091539 and the short-read assembly is available in EnteroBase (Uberstrain ESC_WA1159AA).
Abbreviations
- APEC:
-
Extraintestinal avian pathogenic E. coli
- AST:
-
Antibiotic susceptibility testing
- CVP:
-
Conserved virulence plasmidic region
- ERC:
-
Endoscopic retrograde cholangiography
- ExPEC:
-
Extraintestinal E. coli
- hmv:
-
Hypermucoviscous
- MALDI-TOF:
-
Matrix-assisted laser desorption ionization-time of flight mass spectrometry
- PFGE:
-
Pulsed-field gel electrophoresis
References
Jang J, Hur HG, Sadowsky MJ, Byappanahalli MN, Yan T, Ishii S. Environmental Escherichia coli: ecology and public health implications-a review. J Appl Microbiol. 2017;123(3):570–81.
Kolenda R, Burdukiewicz M, Schierack P. A systematic review and meta-analysis of the epidemiology of pathogenic Escherichia coli of calves and the role of calves as reservoirs for human pathogenic E. coli. Front Cell Infect Microbiol. 2015;5:23.
Riley LW. Pandemic lineages of extraintestinal pathogenic Escherichia coli. Clin Microbiol Infect. 2014;20(5):380–90.
Johnson TJ, Wannemuehler Y, Doetkott C, Johnson SJ, Rosenberger SC, Nolan LK. Identification of minimal predictors of avian pathogenic Escherichia coli virulence for use as a rapid diagnostic tool. J Clin Microbiol. 2008;46(12):3987–96.
Luthje P, Brauner A. Virulence factors of uropathogenic E. coli and their interaction with the host. Adv Microb Physiol. 2014;65:337–72.
Wacharotayankun R, Arakawa Y, Ohta M, Tanaka K, Akashi T, Mori M, Kato N. Enhancement of extracapsular polysaccharide synthesis in Klebsiella pneumoniae by RmpA2, which shows homology to NtrC and FixJ. Infect Immun. 1993;61(8):3164–74.
Harris S, Piotrowska MJ, Goldstone RJ, Qi R, Foster G, Dobrindt U, Madec JY, Valat C, Rao FV, Smith DG. Variant O89 O-antigen of E. coli is associated with group 1 capsule loci and multidrug resistance. Front Microbiol. 2018;9:2026.
Benham A, Davis J, Puzio C, Blakey G, Slobodov G. Renal abscess yields elusive hypermucoviscous phenotype of, uropathogenic Escherichia coli: a case report. J Okla State Med Assoc. 2013;106(11):435–8.
Bottone EJ. Hypermucoviscous phenotype expressed by an isolate of uropathogenic Escherichia coli: an overlooked and underappreciated virulence factor. Clin Microbiol Newsl. 2010;32(11):81–5.
Shon AS, Bajwa RP, Russo TA. Hypervirulent (hypermucoviscous) Klebsiella pneumoniae: a new and dangerous breed. Virulence. 2013;4(2):107–18.
Kumabe A, Kenzaka T. String test of hypervirulent Klebsiella pneumonia. QJM. 2014;107(12):1053.
Lam MMC, Wyres KL, Duchene S, Wick RR, Judd LM, Gan YH, Hoh CH, Archuleta S, Molton JS, Kalimuddin S, Koh TH, Passet V, Brisse S, Holt KE. Population genomics of hypervirulent Klebsiella pneumoniae clonal-group 23 reveals early emergence and rapid global dissemination. Nat Commun. 2018;9(1):2703.
Choby JE, Howard-Anderson J, Weiss DS. Hypervirulent Klebsiella pneumoniae - clinical and molecular perspectives. J Intern Med. 2020;287(3):283–300.
Klaper K, Wendt S, Lübbert C, Lippmann N, Pfeifer Y, Werner G. Hypervirulent Klebsiella pneumoniae of Lineage ST66-K2 Caused Tonsillopharyngitis in a German Patient. Microorganisms. 2021;9(1):133.
Catalan-Najera JC, Garza-Ramos U, Barrios-Camacho H. Hypervirulence and hypermucoviscosity: two different but complementary Klebsiella spp phenotypes? Virulence. 2017;8(7):1111–23.
Shu C, Zha H, Long H, Wang X, Yang F, Gao J, Hu C, Zhou L, Guo B, Zhu B. C3a–C3aR signaling promotes breast cancer lung metastasis via modulating carcinoma associated fibroblasts. J Exp Clin Cancer Res. 2020;39(1):11.
Lin YT, Jeng YY, Chen TL, Fung CP. Bacteremic community-acquired pneumonia due to Klebsiella pneumoniae: clinical and microbiological characteristics in Taiwan, 2001–2008. BMC Infect Dis. 2010;10:307.
Lin JC, Siu LK, Fung CP, Tsou HH, Wang JJ, Chen CT, Wang SC, Chang FY. Impaired phagocytosis of capsular serotypes K1 or K2 Klebsiella pneumoniae in type 2 diabetes mellitus patients with poor glycemic control. J Clin Endocrinol Metab. 2006;91(8):3084–7.
Blesl A, Stadlbauer V. The gut-liver axis in cholestatic liver diseases. Nutrients. 2021;13(3):1018.
Tajeddin E, Sherafat SJ, Majidi MR, Alebouyeh M, Alizadeh AH, Zali MR. Association of diverse bacterial communities in human bile samples with biliary tract disorders: a survey using culture and polymerase chain reaction-denaturing gradient gel electrophoresis methods. Eur J Clin Microbiol Infect Dis. 2016;35(8):1331–9.
Gu XX, Zhang MP, Zhao YF, Huang GM. Clinical and microbiological characteristics of patients with biliary disease. World J Gastroenterol. 2020;26(14):1638–46.
Weber A, Schneider J, Wagenpfeil S, Winkle P, Riedel J, Wantia N, Feihl S, Rommler F, Baur DM, Schmid RM, Algul H, Huber W. Spectrum of pathogens in acute cholangitis in patients with and without biliary endoprosthesis. J Infect. 2013;67(2):111–21.
Ikeda M, Kobayashi T, Fujimoto F, Okada Y, Higurashi Y, Tatsuno K, Okugawa S, Moriya K. The prevalence of the iutA and ibeA genes in Escherichia coli isolates from severe and non-severe patients with bacteremic acute biliary tract infection is significantly different. Gut Pathog. 2021;13(1):32.
Wang MC, Tseng CC, Chen CY, Wu JJ, Huang JJ. The role of bacterial virulence and host factors in patients with Escherichia coli bacteremia who have acute cholangitis or upper urinary tract infection. Clin Infect Dis. 2002;35(10):1161–6.
Søgaard KK, Veres K, Vandenbroucke-Grauls C, Vandenbroucke JP, Sørensen HT, Schønheyder HC. Community-acquired Escherichia coli bacteremia after age 50 and subsequent incidence of a cancer diagnosis: a danish population-based cohort study. Cancer Epidemiol Biomarkers Prev. 2020;29(12):2626–32.
Zhang X, Liu H. Klatskin tumor: a population-based study of incidence and survival. Med Sci Monit. 2019;25:4503–12.
Dangtakot R, Intuyod K, Ahooja A, Wongwiwatchai J, Hanpanich P, Lulitanond A, Chamgramol Y, Pinlaor S, Pinlaor P. Profiling of bile microbiome identifies district microbial population between choledocholithiasis and cholangiocarcinoma patients. Asian Pac J Cancer Prev. 2021;22(1):233–40.
Neumann B, Rackwitz W, Hunfeld KP, Fuchs S, Werner G, Pfeifer Y. Genome sequences of two clinical Escherichia coli isolates harboring the novel colistin-resistance gene variants mcr-1.26 and mcr-1.27. Gut Pathog. 2020;12:40.
Barton BM, Harding GP, Zuccarelli AJ. A general method for detecting and sizing large plasmids. Anal Biochem. 1995;226(2):235–40.
Schuster CF, Weber RE, Weig M, Werner G, Pfeifer Y. Ultra-deep long-read sequencing detects IS-mediated gene duplications as a potential trigger to generate arrays of resistance genes and a mechanism to induce novel gene variants such as blaCTX-M-243. J Antimicrob Chemother. 2021. https://doi.org/10.1093/jac/dkab407.
Wick RR, Judd LM, Gorrie CL, Holt KE. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. 2017;13(6): e1005595.
Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, Lomsadze A, Pruitt KD, Borodovsky M, Ostell J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016;44(14):6614–24.
Jünemann S, Sedlazeck FJ, Prior K, Albersmeier A, John U, Kalinowski J, Mellmann A, Goesmann A, von Haeseler A, Stoye J, Harmsen D. Updating benchtop sequencing performance comparison. Nat Biotechnol. 2013;31(4):294–6.
Bortolaia V, Kaas RS, Ruppe E, Roberts MC, Schwarz S, Cattoir V, Philippon A, Allesoe RL, Rebelo AR, Florensa AF, et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J Antimicrob Chemother. 2020;75(12):3491–500.
MalbergTetzschner AM, Johnson JR, Johnston BD, Lund O, Scheutz F. In silico genotyping of Escherichia coli isolates for extraintestinal virulence genes by use of whole-genome sequencing data. J Clin Microbiol. 2020. https://doi.org/10.1128/JCM.01269-20.
Johansson MHK, Bortolaia V, Tansirichaiya S, Aarestrup FM, Roberts AP, Petersen TN. Detection of mobile genetic elements associated with antibiotic resistance in Salmonella enterica using a newly developed web tool: MobileElementFinder. J Antimicrob Chemother. 2021;76(1):101–9.
Zhou Z, Alikhan NF, Mohamed K, Fan Y, Achtman M, Agama Study. The EnteroBase user’s guide, with case studies on Salmonella transmissions, Yersinia pestis phylogeny, and Escherichia core genomic diversity. Genome Res. 2020;30(1):138–52.
Lam MMC, Wick RR, Watts SC, Cerdeira LT, Wyres KL, Holt KE. A genomic surveillance framework and genotyping tool for Klebsiella pneumoniae and its related species complex. Nat Commun. 2021;12(1):4188.
Schmartz GP, Hartung A, Hirsch P, Kern F, Fehlmann T, Muller R, Keller A. PLSDB: advancing a comprehensive database of bacterial plasmids. Nucleic Acids Res. 2022;50(D1):D273–8.
Morgulis A, Coulouris G, Raytselis Y, Madden TL, Agarwala R, Schaffer AA. Database indexing for production MegaBLAST searches. Bioinformatics. 2008;24(16):1757–64.
Grant JR, Stothard P. The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 2008;36(2):W181-4.
Harris RS. Improved pairwise alignment of genomic DNA. In: Computer Science and Engineering. The Pennsylvania State University; 2007.
Schwartz S, Kent WJ, Smit A, Zhang Z, Baertsch R, Hardison RC, Haussler D, Miller W. Human-mouse alignments with BLASTZ. Genome Res. 2003;13(1):103–7.
Mike LA, Stark AJ, Forsyth VS, Vornhagen J, Smith SN, Bachman MA, Mobley HLT. A systematic analysis of hypermucoviscosity and capsule reveals distinct and overlapping genes that impact Klebsiella pneumoniae fitness. PLoS Pathog. 2021;17(3): e1009376.
Milenkov M, Rasoanandrasana S, Rahajamanana LV, Rakotomalala RS, Razafindrakoto CA, Rafalimanana C, Ravelomandranto E, Ravaoarisaina Z, Westeel E, Petitjean M, Mullaert J, Clermont O, Raskine L, Samison LH, Endtz H, Andremont A, Denamur E, Komurian-Pradel F, Armand-Lefevre L. Prevalence, risk factors, and genetic characterization of extended-spectrum beta-lactamase Escherichia coli isolated from healthy pregnant women in Madagascar. Front Microbiol. 2021;12: 786146.
Lemaître C, Mahjoub-Messai F, Dupont D, Caro V, Diancourt L, Bingen E, Bidet P, Bonacorsi S. A conserved virulence plasmidic region contributes to the virulence of the multiresistant Escherichia coli meningitis strain S286 belonging to phylogenetic group C. PLoS ONE. 2013;8(9): e74423.
Hayashi W, Tanaka H, Taniguchi Y, Iimura M, Soga E, Kubo R, Matsuo N, Kawamura K, Arakawa Y, Nagano Y, Nagano N. Acquisition of mcr-1 and cocarriage of virulence genes in avian pathogenic Escherichia coli isolates from municipal wastewater influents in Japan. Appl Environ Microbiol. 2019. https://doi.org/10.1128/AEM.01661-19.
Mora A, Blanco JE, Blanco M, Alonso MP, Dhabi G, Echeita A, Gonzalez EA, Bernardez MI, Blanco J. Antimicrobial resistance of Shiga toxin (verotoxin)-producing Escherichia coli O157:H7 and non-O157 strains isolated from humans, cattle, sheep and food in Spain. Res Microbiol. 2005;156(7):793–806.
Czirok E, Herpay M, Milch H. Computerized complex typing of Escherichia coli strains from different clinical materials. Acta Microbiol Hung. 1993;40(3):217–37.
Vu-Khac H, Holoda E, Pilipcinec E, Blanco M, Blanco JE, Dahbi G, Mora A, Lopez C, Gonzalez EA, Blanco J. Serotypes, virulence genes, intimin types and PFGE profiles of Escherichia coli isolated from piglets with diarrhoea in Slovakia. Vet J. 2007;174(1):176–87.
Saile N, Schuh E, Semmler T, Eichhorn I, Wieler LH, Bauwens A, Schmidt H. Determination of virulence and fitness genes associated with the pheU, pheV and selC integration sites of LEE-negative food-borne Shiga toxin-producing Escherichia coli strains. Gut Pathog. 2018;10:43.
Manges AR, Geum HM, Guo A, Edens TJ, Fibke CD, Pitout JDD. Global extraintestinal pathogenic Escherichia coli (ExPEC) lineages. Clin Microbiol Rev. 2019. https://doi.org/10.1128/CMR.00135-18.
de Lastours V, Laouenan C, Royer G, Carbonnelle E, Lepeule R, Esposito-Farese M, Clermont O, Duval X, Fantin B, Mentre F, Decousser JW, Denamur E, Lefort A. Mortality in Escherichia coli bloodstream infections: antibiotic resistance still does not make it. J Antimicrob Chemother. 2020;75(8):2334–43.
Nanayakkara BS, O’Brien CL, Gordon DM. Diversity and distribution of Klebsiella capsules in Escherichia coli. Environ Microbiol Rep. 2019;11(2):107–17.
Zhu J, Wang T, Chen L, Du H. Virulence factors in hypervirulent Klebsiella pneumoniae. Front Microbiol. 2021;12: 642484.
Lam MMC, Wick RR, Wyres KL, Gorrie CL, Judd LM, Jenney AWJ, Brisse S, Holt KE. Genetic diversity, mobilisation and spread of the yersiniabactin-encoding mobile element ICEKp in Klebsiella pneumoniae populations. Microb Genom. 2018. https://doi.org/10.1099/mgen.0.000196.
Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, Wishart DS. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016;44(W1):W16-21.
Arndt D, Marcu A, Liang Y, Wishart DS. PHAST, PHASTER and PHASTEST: Tools for finding prophage in bacterial genomes. Brief Bioinform. 2019;20(4):1560–7.
Bagel S, Hüllen V, Wiedemann B, Heisig P. Impact of gyrA and parC mutations on quinolone resistance, doubling time, and supercoiling degree of Escherichia coli. Antimicrob Agents Chemother. 1999;43(4):868–75.
Martin-Gutierrez G, Rodriguez-Beltran J, Rodriguez-Martinez JM, Costas C, Aznar J, Pascual A, Blazquez J. Urinary tract physiological conditions promote ciprofloxacin resistance in low-level-quinolone-resistant Escherichia coli. Antimicrob Agents Chemother. 2016;60(7):4252–8.
Sánchez-Céspedes J, Sáez-López E, Frimodt-Møller N, Vila J, Soto SM. Effects of a mutation in the gyrA gene on the virulence of uropathogenic Escherichia coli. Antimicrob Agents Chemother. 2015;59(8):4662–8.
Peigne C, Bidet P, Mahjoub-Messai F, Plainvert C, Barbe V, Medigue C, Frapy E, Nassif X, Denamur E, Bingen E, Bonacorsi S. The plasmid of Escherichia coli strain S88 (O45:K1:H7) that causes neonatal meningitis is closely related to avian pathogenic E. coli plasmids and is associated with high-level bacteremia in a neonatal rat meningitis model. Infect Immun. 2009;77(6):2272–84.
Otto BR, Sijbrandi R, Luirink J, Oudega B, Heddle JG, Mizutani K, Park SY, Tame JR. Crystal structure of hemoglobin protease, a heme binding autotransporter protein from pathogenic Escherichia coli. J Biol Chem. 2005;280(17):17339–45.
Wu X, Shi Q, Shen S, Huang C, Wu H. Clinical and bacterial characteristics of Klebsiella pneumoniae affecting 30-day mortality in patients with bloodstream infection. Front Cell Infect Microbiol. 2021;11: 688989.
Urdaneta V, Casadesus J. Interactions between bacteria and bile salts in the gastrointestinal and hepatobiliary tracts. Front Med (Lausanne). 2017;4:163.
Ling J, Pan H, Gao Q, Xiong L, Zhou Y, Zhang D, Gao S, Liu X. Aerobactin synthesis genes iucA and iucC contribute to the pathogenicity of avian pathogenic Escherichia coli O2 strain E058. PLoS ONE. 2013;8(2): e57794.
Acknowledgements
We thank Sibylle Müller-Bertling for excellent technical assistance.
Funding
Open Access funding enabled and organized by Projekt DEAL. Not applicable.
Author information
Authors and Affiliations
Contributions
NL, SW, TK and CL identified the case and provided clinical data. BN and CFS performed the WGS analyses. YP performed phenotypical characterization and PFGE analyses. BN, CFS, GW and YP wrote the manuscript and designed the figures and tables. All authors made a substantial, direct and intellectual contribution to the work, in interpreting results, providing critical feedback and finalizing the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The patient provided written informed consent for this case presentation.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Visualization of plasmids of strain 537-20 by native plasmid preparation (Plasmid Mini Kit, Qiagen, Hilden, Germany) and agarose gel electrophoresis. The plasmid containing E. coli strain V515 was used as a reference (lane M). Several plasmids were visible in strain 537-20 (lane 1).Plasmid sizes that were bioinformatically identified are indicated on the right. Figure S2. LASTZ alignment of p537-20_1 (CP091535) and Conserved Virulence Plasmidic (CVP) region (HF922624). In the top panel, plasmid p537-20_1 was aligned to the CVP region (HF922624, reference) using LASTZ. The LASTZ algorithm allows to identify regions of similarity as indicated in the “LASTZ Alignment Graph". Blue regions indicate identity, whereas red regions indicate inversions compared to the reference sequence. The X-axis in the graph describes the bp location. In the lower panel, reference and comparison sequences are switched to identify regions that are absent in the reference sequence. Figure S3. LASTZ alignment of p537-20_1 (CP091535) and pECOS88 (CU928146). Table S1. Sequencing and assembly statistics.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Neumann, B., Lippmann, N., Wendt, S. et al. Recurrent bacteremia with a hypermucoviscous Escherichia coli isolated from a patient with perihilar cholangiocarcinoma: insights from a comprehensive genome-based analysis. Ann Clin Microbiol Antimicrob 21, 28 (2022). https://doi.org/10.1186/s12941-022-00521-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12941-022-00521-7