
Abstract
Background
Epigenome-wide association studies of ambient fine particulate matter (PM2.5) have been reported. However, few have examined PM2.5 components (PMCs) and sources or included repeated measures. The lack of high-resolution exposure measurements is the key limitation. We hypothesized that significant changes in DNA methylation might vary by PMCs and the sources.
Methods
We predicted the annual average of 14 PMCs using novel high-resolution exposure models across the contiguous U.S., between 2000–2018. The resolution was 50 m × 50 m in the Greater Boston Area. We also identified PM2.5 sources using positive matrix factorization. We repeatedly collected blood samples and measured leukocyte DNAm with the Illumina HumanMethylation450K BeadChip in the Normative Aging Study. We then used median regression with subject-specific intercepts to estimate the associations between long-term (one-year) exposure to PMCs / PM2.5 sources and DNA methylation at individual cytosine-phosphate-guanine CpG sites. Significant probes were identified by the number of independent degrees of freedom approach, using the number of principal components explaining > 95% of the variation of the DNA methylation data. We also performed regional and pathway analyses to identify significant regions and pathways.
Results
We included 669 men with 1,178 visits between 2000–2013. The subjects had a mean age of 75 years. The identified probes, regions, and pathways varied by PMCs and their sources. For example, iron was associated with 6 probes and 6 regions, whereas nitrate was associated with 15 probes and 3 regions. The identified pathways from biomass burning, coal burning, and heavy fuel oil combustion sources were associated with cancer, inflammation, and cardiovascular diseases, whereas there were no pathways associated with all traffic.
Conclusions
Our findings showed that the effects of PM2.5 on DNAm varied by its PMCs and sources.
Similar content being viewed by others


Introduction
Ambient fine particulate matter (an aerodynamic diameter ≤ 2.5 µm; PM2.5) is a complex mixture of many components (PM2.5 components; PMCs) that differ in their physicochemical, and toxicological properties [1]. Studies have not only found that exposure to ambient PM2.5 was linked with death [2] and multi-systemic diseases [3,4,5,6,7] but have observed that PM2.5-related effects vary by its PMCs [8,9,10]. In addition, there are multiple sources of ambient PM2.5, including direct emissions (e.g., biomass burning, and inefficient fuel combustion) [11, 12] and secondary particles (chemical reactions of gas-phase pollutant precursors, e.g., nitrogen oxides). Studies also have suggested that PM2.5-related adverse health effects vary by different sources [13,14,15]. However, there are inconsistent results on which PMCs or PM2.5 sources are responsible for the adverse health effects [16,17,18,19]. Further, the underlying molecular changes caused by PMCs and the sources have not been adequately investigated.
DNA methylation (DNAm), a chemical modification of DNA with a methyl group addition predominantly at a cytosine-phosphate-guanine (CpG) site [20], has been associated with poor health outcomes, such as cardiovascular diseases (CVDs), cancer, aging, oxidative stress, and inflammation [20,21,22,23,24]. Meanwhile, DNAm has been linked with PM2.5 across different time windows [25,26,27,28]. For instance, our group have conducted an epigenome-wide association study (EWAS) of PM2.5 across a 28-day time window in the Normative Aging Study and found 2,717 statistically significant CpGs [29]. Since PM2.5 is a combination of multiple PMCs with different characteristics, these PMCs may be associated with DNAm at different sites. To date, only six studies have assessed the associations between PMCs and DNAm across different time windows [29,30,31,32,33,34], among which only one examined the long-term effects of PMCs on DNAm in an epigenome-wide scope [34] and two were conducted by our group previously [29, 34]. However, five [29,30,31, 33, 34] of the six studies obtained the PMCs’ concentrations from limited monitoring sites that do not reflect the spatial variation in the concentrations.
One vital limitation in studies using PMCs from fixed monitors is the lack of high-resolution exposure measurements. It leads to a low spatial resolution of PMCs in these studies despite evidence that some components (e.g., black carbon) can vary substantially over distances as small as 100 ~ 200 m [35, 36]. Emerging exposure prediction models which utilize methods such as chemical transport models [37, 38] and land use regression models [39, 40] provide higher exposure resolution. Nevertheless, these models have relatively moderate prediction accuracies and/or spatial resolutions. Our group recently developed novel prediction models with 50 m resolution of PMCs across the contiguous U.S, using machine learning and a mixture of land use, remote sensing, and other inputs [41]. These new models not only diminish the measurement errors for each PMC (out of sample R2 ~ 0.9) but minimize the extent of measurement errors across PMCs. We have applied the predicted PMCs in several epidemiological studies [42, 43]. To date, however, no EWAS of PMCs from high-resolution models nor EWAS of PM2.5 sources has been previously performed.
This study, therefore, sought to investigate the associations between long-term exposure to PMCs/sources and DNAm by conducting EWAS analyses, using whole-blood samples and exposure data from high-resolution models. We hypothesize that the changes in DNAm varied by PMCs and sources.
Materials and methods
Study population
The participants in this study included 669 elderly men in the Greater Boston Area who are part of the Normative Aging Study, a closed and ongoing cohort established by the U.S. Veterans Administration [44]. For the initial cohort, the participants were aged 21–82 years and were free of any known chronic diseases. They have physical examinations, including blood collection, and questionnaires in a clinical center every 3–5 years. In this study, we included subjects who had visits with their DNA samples collected in 2000 and later. To dimmish study heterogeneity that may be introduced by diverse genetic ancestry, we dropped non-white participants (~ 3%) [45]. The Harvard T.H. Chan School of Public Health and the Institutional Review Boards of the Department of Veterans Affairs approved the study proposal. All study participants provided their written informed consent before enrollment and at sample collection.
PM2.5 and its 14 components measures
We predicted annual average ambient PMCs based on a combination of machine learning algorithms in a geographically weighted regression. The resolution was 50 m × 50 m in the Greater Boston Area. The algorithms used ground monitoring data collected from 987 monitoring sites across the contiguous U.S., satellite-derived measurements (available through the Google Earth Engine), chemical transport model simulations, meteorological conditions, and land-use data (e.g., traffic counts, distance to OpenStreetMap features), between 2000–2018. The predicted PMCs included Bromine (Br), Calcium (Ca), Copper (Cu), elemental carbon (EC), Iron (Fe), Lead (Pb), Nickel (Ni), nitrate (NO3−), organic carbon (OC), Potassium (K), Silicon (Si), sulfate (SO42−), Vanadium (V), and Zinc (Zn). Excellent model performance was achieved with out of sample validation R2 for individual PMCs ranging from 0.821 (Br) to 0.975 (SO42−). We further matched the annual average PMCs data with each residential address based on the grid cell centroid closest to the address and year at the time of DNA samples’ collection in the study population. Additionally, we predicted daily PM2.5 mass concentrations between 2000–2016, using an ensemble model at a resolution of 1 km × 1 km (R2 = 0.86). We then calculated the annual PM2.5 concentrations based on the daily data across that year [46]. The National Human Activity Pattern Survey in the U.S. reported that U.S. adults spent 69% of their time at home and 8% of the time immediately outside their home [47]. Given the age range of our study population, the time that they stayed at or near their residence was probably even longer. It is reasonable to use residential air pollution to capture the exposure.
Source apportionment using positive matrix factorization
We used positive matrix factorization (PMF, version 5.0) analysis to apportion the measured PMCs to realizable sources [48]. PMF was developed by Paatero and Tapper [49] and has been widely used for sources apportionment studies [17, 50,51,52]. It requires two input files: the measured concentrations of the species and the estimated uncertainty of the concentration [48]. Similarly to cluster analysis, a correlation between species indicates a common factor which can represent a source category [48]. We tested for possible source numbers of 4,5, and 6. For each possible source number, 100 base runs were conducted to obtain the best factorization fit over all runs that achieved the minimization of weighted residual error for the linear fitting of a multivariate system of variables. We then selected the idea number of sources based on not only the minimization of weighted residual error but also the realistic scenario of component sources [50].
DNAm measures
DNA samples were extracted using the IQAamp DNA Blood Kit (Qiagen, CA, U.S.) from the buffy coat of the whole food collected between 1999 and 2013 (We dropped samples in 1999 in this study because there were no predicted PMCs in that year). We measured DNAm using Illumina Infinium Human Methylation450K BeadChip (450 K; Illumina Inc., San Diego, CA, U.S.), which includes ~ 485,000 CpG sites. Based on a two-stage age-stratified algorithm, we randomized the samples across plates and chips to minimize batch effects [34]. As described previously, we preprocessed DNAm data via the ewastools package in Github [45]. We dropped low-quality samples [53] and corrected dye-bias using a regression on the logarithm of internal control probes [54]. We elaborated on the steps for probes cleaning previously [45]. In total, we included 360,272 high-quality probes remote from SNPs in this study.
We normalized DNAm data by controlling for the normalization factors in the outcome regression instead of using other commonly used approaches, such as beta-mixture quantile normalization [55]. This normalization approach ensures a better adjustment for batch effects as their impact often varies across probes, and we have applied it previously [45, 56, 57]. The normalization factors included five experimental covariates (i.e., Non-polymorphic Red, Specificity I Red, Bisulfite Conversion I Red, Bisulfite Conversion II, Extension Red) [58]. DNAm level was expressed as the ratio of methylated cytosines over the sum of the methylated and unmethylated cytosines at each CpG location and then multiplied by 100 (mean %5-methylcytosine, i.e., %5-mC). Thus, the DNAm level ranged from 0- to 100%5-mC.
Statistical analyses
We examined EWAS of annual exposure to PMCs/sources at three levels: single CpG site, regional, and pathway analyses.
Single CpG analyses
We performed traditional EWAS at single CpG level to identify statistically significantly differentially methylated probes (DMPs) by PMCs/sources. As described previously [45], we used median regression to analyze associations between exposures and DNAm because median regression has no assumption on the distribution of dependent variables (i.e., DNAm in this study) [59]. In addition, we applied median regression for longitudinal data using the Koenker et al. method [60] because ~ 60% of the participants had repeated DNAm measures. It allowed us to model fixed-effects and correlated random-intercepts for each subject and use bootstrap for statistical inference. For PMCs, we investigated the effects of each PMC one a time with PM2.5 mass in the model to control for other particle components; for sources, we investigated the effects of each source with other three sources and PM2.5 mass in the model. In all regression analyses, we controlled for the following covariates a priori based on the relevant literature [34, 57]: chronological age (years), years of education, smoking status (ever/never), cigarette pack-years, alcohol consumption (< 2 or ≥ 2 drinks/day), body mass index (BMI, kg/m2), the estimated cell type compositions (CD4 + T lymphocytes, CD8 + T lymphocytes, natural killer cells, B cells, and monocytes) by the Houseman et al. method [61], technical factors such as batch effects and five normalization factors, and ambient annual mean temperature and relative humidity from gridMET [62]. The model for PMC is shown in Eq. (1) and the model for sources is shown in Eq. (2).
, where Mij is the median of DNAm level for subject i at visit j. PMCij in Eq. (1) is the annual average of its concentration. Sourcemij in Eq. (2) is the annual concentration of the mth (4 ≤ m ≤ 6) source for subject i at visit j. (We determined the value of m as described in 2.3 Source appointment using positive matrix factorization.) PM2.5ij are the annual average for subject i at visit j. Xij are the covariates that we listed above. \({\omega }_{i}\) in Eq. (1) and \({\theta }_{i}\) in Eq. (2) are the random intercepts for participant i. and \({\varepsilon }_{ij}\) in Eq. (1) and in Eq. (2) are the residuals. In order to account for the possible selection bias that healthier men were more likely to return for subsequent exams, we used inverse probability weighting [63]. It calculated the probability of having a subsequent visit given chronological age, education, BMI, blood pressure, smoking status, cigarette pack years, alcohol consumption, C-reactive protein, asthma, chronic bronchitis, and emphysema at previous visit, using logistic regression. We reported our results as the median difference in DNAm (%5-mC) per one interquartile range (IQR) increase in PMCs and its sources after annual exposure.
To account for the multiple testing in the context of the high correlation among CpG sites, we needed a method to consider that correlation, which reduces the effective number of independent tests. Following related work this area [64, 65], we used the “number of independent degrees of freedom” approach. Specifically, we used principal component analysis to project the probes to fewer dimensions. Fig. S1 showed the scree plot and cumulative scree plot that explained more than 95% of the variation of the DNAm data in this study. Thus, we set the number of independent degrees of freedom to be the number of components that explained 95% of the variation of the DNAm data (see Fig. S1). We then obtained the Bonferroni threshold for statistical significance of each estimate by dividing 0.05 by the independent degrees of freedom (i.e., PC-correction; p-value < 0.05/908/14 (3.93 × 10–6) for PMCs; p-value < 0.05/908/m for sources (4 ≤ m ≤ 6)).
Regional and pathway analyses
Single CpG may weakly associated with PMCs and its sources, and be difficult to identify. A region containing multiple DMPs that are functionally correlated may have more biological implications [66]. Thus, we investigated statistically significantly differentially methylated regions (DMRs) in relation to the exposures using the comb-p function from the ENmix package in R Bioconductor [67] because the comb-p tool has the best sensitivity and highest control of false-positive rate compared to the other DMR tools [68]. We defined a significant DMR as one with three or more probes within kilobase pair and its Sidak p-value < 0.05 [45].
In addition, we used the Ingenuity Pathway Analysis (IPA) database (QIAGEN Inc.) to identify significantly enriched gene pathways in the top ranked 100 CpGs that were associated with each PMC/source. We calculated permutation p-values based on the results of 10,000 random shuffles of association p-values for the CpGs on the 450 K array [69]. We defined significant pathways if p-value < 0.05 and gene set contains ≥ 3 genes with top ranked probes [45].
Sensitivity analyses
To check the robustness of our results, we conducted sensitivity analyses. In the main analysis, we only accounted for the selection bias due to healthier men being more likely to return for the subsequent exams. In the sensitivity analyses, we then further controlled for mortality that occurred prior to year 2000. We applied inverse probability weighting [63] via logistic regression to calculate the probability of death given the same factors that we mentioned above. We then multiplied this inverse probability weight with the one in the main analyses (for the probability of a subsequent visit). Thus, the visits in this study were representative of the original population. We compared the effects sizes and p-values of the top 5 probes for each source from the main analyses with the ones from the sensitivity analyses.
Results
Population description
We included 669 men with 1,178 visits. The summary characteristics of the study subjects are shown in Table 1. In this present study, almost sixty percent of the participants had more than one visit. The participants were older men with a mean age [standard deviation (SD)] of 74 (7) and 75 (7) at the first and all visits, respectively.
Concentrations of PM2.5 and 14 PMCs
Table 2 presents the summary statistics of annual PM2.5 and its PMCs during the study period (2000–2013). The mean (SD) concentration of annual PM2.5 mass concentration was 9.75 (1.80) µg/m3, with an IQR of 2.39 µg/m3. Among the investigated PMCs, SO42− accounted for the largest proportion of PM2.5 total mass (30.5%), followed by OC (19.4%). The annual average of metal components such as Pb and Fe is 3.22 (SD 1.09) ng/m3 and 50.84 (15.60) ng/m3, respectively. We reported the correlation coefficients among PM2.5 mass and 14 PMCs in Table S1 (see Table S1 in the supplementary material). The correlation coefficients ranged from 0.00 for Cu and K to 0.80 for Cu and Fe. K is the components that had the lowest correlation with other PMCs.
Sources from PMF analysis
Based on an evaluation of three PMF models with the number of sources equal to 4, 5, and 6, we chose 4 as it provided the most feasible source results. The source profiles and the distributions of 14 PMCs are presented in Fig. 1. The four sources included biomass burning (source 1), all traffic (source 2), secondary particles (source 3), and coal burning and heavy fuel oil combustion (source 4).

The proportion of PMCs in each source. Abbreviations: PMC, particulate matter components; Br, bromine; Ca, calcium; Cu, copper; EC, element carbon; Fe, iron; K, potassium; Ni, nickel; NO3-, nitrate; OC, organic carbon; Pb, lead; Si, silicon; SO42-, sulfate; V, vanadium; Zn, zinc
EWAS of PMCs and sources
Significant probes
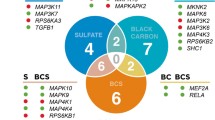
In the site-by-site analyses, we observed multiple significant DMPs for the 14 PMCs and 4 sources (see Table 3). For example, we found 7 significant DMPs associated with SO42−, 11 with K, 10 with source 1, and 8 with source 2. We presented the significant DMPs associated with PMCs ranked by p-values with their annotated genes in Tables S2; we showed the significant DMPs associated with sources ranked by p-values with their annotated genes in Tables S3. We also presented the Manhattan plots and the quantile–quantile plots with the estimated genomic inflation factor for each exposure (Fig. S2) and source (Fig. S3).We compared the significant DMPs by the 4 sources and found a few common probes across sources: source 1 had a common probe with source 2 (cg15911114), 6 common probes with source 3 (cg09852920, cg04660698, cg25277509, cg01252659, cg01733795, cg10692118), and 4 common probes with source 4 (cg09852920, cg25277509, cg01252659, cg10692118); source 3 and source 4 had 12 overlapping probes (cg09852920, cg16756998, cg21675770, cg25277509, cg25631650, cg10692118, cg05524450, cg21468420, cg01252659, cg17367077, cg08753391, cg05970846). Additionally, sources 1, 3, and 4 had 4 common probes (cg09852920, cg25277509, cg01252659, cg10692118). We also compared the significant DMPs by 14 PMCs (see supplementary material). There were many DMPs that were not similar across sources or PMCs.
Significant regions and pathways
We identified multiple significant DMRs for PMCs and sources (see Table 3). For example, we observed 6 DMRs due to EC: chr19: 37,825,307–37825680, chr4: 57,773,149–57,773,309, chr12: 14,720,834–14721289, chr3: 48,694,451–48,694,674; chr14: 24,779,959–24,780,405; chr17: 3,704,494–3704622 and 5 DMRs due to source 3 (i.e., secondary particles): chr6: 29,594,830–29595662, chr 16: 8,806,531–8807044, chr6: 33,048,254–33048486; chr10: 32,216,031–32216391, chr11: 6,291,879–6,292,312. We presented the significant regions ranked by p-values for PMCs in Table S4 and for sources in Tables S5. We compared the significant DMRs and the annotated genes with 4 sources and found that sources 3 and 4 had 5 common DMRs (chr16: 8,806,531–8807044, chr6: 29,594,830–29595662, chr10: 32,216,031–32216391, chr6: 33,048,254–33048286, chr11: 6,291,879–6,292,312) and 5 common annotated genes (ABAT, GABBR1, ARHGAP12, HLA-DPB1, CCKBR). We also compared the significant DMRs by 14 PMCs (see supplementary material).
In the pathway analyses, we found multiple significant pathways in relation to PMCs and sources (see Table 3). For example, we found 8 pathways for EC: role of NFAT in cardiac hypertrophy, telomerase signaling, tight junction signaling, cellular effects of sildenafil, osteoarthritis pathway, Wnt/ β-catenin signaling, cyclins and cell cycle regulation, axonal guidance signaling and 2 pathways for source 3: PTEN signaling and ILK signaling.
We showed the significant pathways ranked by p-values for PMCs in Tables S6 and for sources in Tables S7. We compared the significant pathways of the 4 sources: the two significant pathways of source 3 and the seven significant pathways of source 4 were all included in the eight pathways of source 1 (e.g., PTEN signaling).
Sensitivity analyses
We extracted the top 5 probes from the EWAS of 4 sources in the main analyses and compared their effect sizes and p-values with that from the sensitivity analyses. The effect sizes in the sensitivity analyses for all four sources were almost the same as in the main analyses (see Fig. S4).
Discussion
To our knowledge, this is the first EWAS of PMCs/sources using high-resolution air pollution models with 50 m × 50 m resolution. We identified multiple DMPs, DMRs, and pathways associated with both PMCs and PM2.5 sources. Moreover, the identified DMPs, DMRs, and pathways were different across PMCs and sources. For example, the significant pathways suggest that the source 1 (i.e., biomass burning) was related to CVD cancer, diabetes, inflammation, Alzheimer’s disease (AD), and oxidative stress, whereas the source 3 (i.e., secondary particles) was annotated to CVD and cancer.
In terms of the significant pathways associated with PM2.5 sources, source 1 (i.e., biomass burning) and source 4 (i.e., coal burning and heavy fuel oil combustion) were associated with almost the same pathways, such as cancer [70, 71], inflammation [72], CVD [73], AD [74, 75], and diabetes [76]. For example, the mTOR signaling pathway is related to cancer [70], and AD [74]; IL-8 signaling is associated with inflammation [72]; insulin receptor signaling is linked to diabetes [76]. The two pathways associated with the source 3 (i.e., secondary air pollution) were linked with cancer [71] and CVD [73]. For example, ILK signaling has been associated with the human heart [73]. There were no significant pathways that were associated with source 2 (i.e., all traffic). These findings suggest that biomass burning, coal burning, and heavy fuel oil combustion were the most impactful PM2.5 sources with respect to DNAm; all traffic had the least impact on DNAm in the Greater Boston Area.
We further discuss the significant pathways associated with individual PMCs based on their proportions across four sources. The highest proportion of OC was from the source 1 (i.e., biomass burning) and it was associated with pathways of cancer [77] and inflammation [72]. For example, RhoGDI signaling has been shown to mediate cancer progression [77]. Source 3 (i.e., secondary air pollution) had the highest percentage of NO3−. The pathways linked with NO3− were involved with CVD [78,79,80,81,82,83,84,85], cancer [86,87,88], inflammation [72, 89, 90], obesity [91], depression [92], and schizophrenia [93]. For example, cAMP-mediated signaling is typically involved in the regulation of heart function [78]; CXCR4 signaling contributes to tumor growth and invasion [86]. Source 4 (i.e., coal burning and heavy fuel oil combustion) accounted for the highest V, which was associated with pathways in immune system [94], cancer [77, 88, 95, 96], and AD [97]. Although source 2 (i.e., all traffic) was not associated with any pathways, its main components Ca, EC, and K were related with a few pathways, including cancer [70].
While studies of PM2.5 have been done, DMPs and pathways associated with only certain PMCs would be less likely to be detected in studies of that composite exposure. Hence this study, with repeated measures and high-resolution exposure to many components, is an important advance. Until recently, only a few epidemiological studies have linked long-term exposure to PMCs/sources with the pathways that we identified in this study. [98, 99]. For example, Ostro et al. found that long-term exposure to high-sulfur fuel combustion and the secondary particle NO3− was associated with CVD mortality in a longitudinal study [99]. This is consistent with our findings of pathways associated with secondary particles and coal burning.
We did not observe any significant associations with long-term exposure to source 2 (i.e., all traffic) and pathways. However, a few epidemiological studies found that exposure to traffic-related exposures were related cellular immunity, cardiovascular and neurological systems development, inflammation [28, 100, 101] among the DNA methylation features. For example, Eze et al. performed EWAS of transportation air pollution exposures. Their agnostic functional networks found cellular immunity, gene expression, cell growth/proliferation, cardiovascular, auditory, embryonic, and neurological systems pathways [28]. The inconsistency may be attributable to the agents in the traffic sources. In our study, the traffic source is mostly consistent of Cu, Fe, EC, and Ca, whereas other studies use nitrogen dioxide and PM2.5 as the main agents for traffic-related toxicity [102,103,104].
To date, only one study examined the associations between long-term exposure to PMCs and DNAm in an epigenome-wide scope [34], which was also conducted by our group (We did not perform an EWAS of sources in the previous study). This study used a central site for measurements of particle components. The number of significant DMPs (N = 29) and pathways (N = 9) was fewer in the previous study compared with this present one. We did not investigate the regions significantly associated with PMCs in our previous study, which observed a total of 29 DMPs (20 for Fe, 8 for Ni, and 1 for V) and 9 pathways (8 for Fe, 2 for Ni). In contrast, this present study totally identified 143 DMPs (among which 6 for Fe, 8 for Ni, and 5 for V) and 143 pathways (among which 2 for Fe, 4 for Ni, and 27 for V). We did not find any common DMPs, but a few overlapping pathways in the two studies, such as pathways in cancer by Ni. The different results in the two studies are mainly attributed to the data source of PMCs. The previous study estimated the concentrations of PMCs from monitors at a stationary site whereas the present study predicted the PMCs from high-resolution models with 50 m × 50 m.
In addition, we compared the significant DMPs in the present study with EWAS of long-term exposure to other air pollutants, such as PM2.5 mass [25, 27, 28, 105], smoking [106], coaling-burning [107], nitrogen oxides [27], sulfur oxide [108], and polycyclic aromatic hydrocarbons [109]. We found 8 overlapping DMPs for our components with DMPs previously identified for smoking [108], and 6 overlapping DMPs for sulfur oxide exposure [108], respectively. Specially, Joehanes et al. compared both current and past smokers with nonsmokers in DNAm using 16 cohorts and identified 18, 760 DMPs in current smokers and 2,623 DMPs in former smokers. Among the significant DMPs, 8 from current smokers (i.e., cg07450086, cg06644515, cg11436113, cg02324920, cg24807850, cg07197831, cg27134322, cg05661533) and 1 from former smokers (i.e., cg11436113, which was also identified in current smokers) were overlapped in this present study. These 8 DMPs were significantly associated with Fe, Zn, Ni, and K in our study. Among the mapped genes, the SELENOT gene is highly expressed in the cerebral globus pallidus and caudate nucleus in patients with Parkinson’s disease [110]. Choi et al. found a total of 6,733 DMPs were associated with prenatal exposure to sulfur oxide during the 3rd trimester at age 2 [108], among which 6 DMPs were also identified in our study (i.e., cg09835867, cg01747792, cg05871607, cg07143898, cg25142954, cg05661533). These 6 DMPs were significantly associated with Si, Zn, and Br in our study. Among the mapped genes, high expression of LPCAT1 gene plays an important role in breast cancer progression [111]. However, we did not find any common DMPs between our study and EWAS of PM2.5 [25, 27, 28, 107], nitrogen oxides [27], or polycyclic aromatic hydrocarbons [109]. This may reflect the heterogeneity in DMPs across different particle components.
This study has some limitations: 1) This study only included elderly white men, which limits the generalizability of the findings to other age groups, races, and sex. However, the studies that assess the modification effects on age, race, and sex in the associations between PMCs and DNAm are not well-established [112]. 2) We do not have data on gene expression; thus, we are not able to determine the regulation directions between DNAm and the coded protein. 3). We only measured DNAm in leukocytes, hence the identified pathways via IPA database (which is built based on multiple tissues) may not reflect all the relevant pathways.
On the other hand, our study has a number of important strengths. 1) This is the first EWAS to assess the associations between DNAm and PMCs/sources using high-resolution models (i.e., 50 m × 50 m). 2) The median regression that we used in this study does not require normally distributed residuals. 3) Repeated measurements of DNAm and PMCs provide a wide variation of the outcomes and exposures within-subject; thus, the statistical power is increased. 4) We analyzed EWAS of PMCs/sources at multiple dimensions: single CpG site, region, and pathway. It ensures us to fully elucidate the genes and pathways lined with the exposures.
Conclusions
In summary, this EWAS of long-term exposure to PMCs/sources from high-resolution models indicates that the associations between DNAm and particles varies by the components and sources. PMCs with emission sources of biomass burning, coal burning, and heavy fuel oil combustion are the most harmful. More similar studies with diverse study populations from different areas, using DNAm from other tissues are needed, to enrich the present findings in the future.
Availability of data and materials
The PM2.5 components concentration data are generated from the exposure models developed by Heresh Amini (heresh.amini@sund.ku.dk), and are available online at the SEDAC website. The methylation data are available at dbGAP, while the phenotype data are not available due to privacy restrictions. The meteorology data are available for download from the GridMET website.
Abbreviations
- Br:
-
Bromine
- Ca:
-
Calcium
- Cu:
-
Copper
- EC:
-
Element carbon
- EWAS:
-
Epigenome-wide association analysis
- Fe:
-
Iron
- IQR:
-
Interquartile range
- K:
-
Potassium
- Ni:
-
Nickel
- NO3 − :
-
Nitrate
- OC:
-
Organic carbon
- Pb:
-
Lead
- PMC:
-
Particulate matter components
- PM2.5 :
-
Fine particulate matter
- Si:
-
Silicon
- SO4 2 − :
-
Sulfate
- V:
-
Vanadium
- Zn:
-
Zinc
References
Council NR. Research priorities for airborne particulate matter: IV. Continuing research progress. 2004.
Kloog I, Ridgway B, Koutrakis P, Coull BA, Schwartz JD. Long- and short-term exposure to PM2.5 and mortality: using novel exposure models. Epidemiology. 2013;24(4):555–61.
Brook RD, Rajagopalan S, Pope CA 3rd, Brook JR, Bhatnagar A, Diez-Roux AV, et al. Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the American Heart Association. Circulation. 2010;121(21):2331–78.
Gu J, Shi Y, Zhu Y, Chen N, Wang H, Zhang Z, et al. Ambient air pollution and cause-specific risk of hospital admission in China: a nationwide time-series study. PLoS Med. 2020;17(8): e1003188.
Hunt A, Abraham JL, Judson B, Berry CL. Toxicologic and epidemiologic clues from the characterization of the 1952 London smog fine particulate matter in archival autopsy lung tissues. Environ Health Perspect. 2003;111(9):1209–14.
Nassan FL, Wang C, Kelly RS, Lasky-Su JA, Vokonas PS, Koutrakis P, et al. Ambient PM(2.5) species and ultrafine particle exposure and their differential metabolomic signatures. Environ Int. 2021;151:106447.
Turner MC, Krewski D, Pope CA 3rd, Chen Y, Gapstur SM, Thun MJ. Long-term ambient fine particulate matter air pollution and lung cancer in a large cohort of never-smokers. Am J Respir Crit Care Med. 2011;184(12):1374–81.
Dai L, Koutrakis P, Coull BA, Sparrow D, Vokonas PS, Schwartz JD. Use of the adaptive LASSO method to identify PM2. 5 components associated with blood pressure in elderly men: the Veterans Affairs Normative Aging Study. Environ Health Perspect. 2016;124(1):120–5.
Franklin M, Koutrakis P, Schwartz J. The role of particle composition on the association between PM2. 5 and mortality. Epidemiology (Cambridge, Mass). 2008;19(5):680.
White AJ, Keller JP, Zhao S, Carroll R, Kaufman JD, Sandler DP. Air pollution, clustering of particulate matter components, and breast cancer in the sister study: a US-wide cohort. Environ Health Perspect. 2019;127(10): 107002.
Yokelson RJ, Crounse J, DeCarlo P, Karl T, Urbanski S, Atlas E, et al. Emissions from biomass burning in the Yucatan. Atmos Chem Phys. 2009;9(15):5785–812.
Bond TC, Bhardwaj E, Dong R, Jogani R, Jung S, Roden C, et al. Historical emissions of black and organic carbon aerosol from energy‐related combustion, 1850–2000. Glob. Biogeochem Cycles. 2007;21(2).
Penttinen P, Vallius M, Tiittanen P, Ruuskanen J, Pekkanen J. Source-specific fine particles in urban air and respiratory function among adult asthmatics. Inhalation Toxicol. 2006;18(3):191–8.
Organization WH. Health relevance of particulate matter from various sources: report on a WHO workshop, Bonn, Germany 26–27 March 2007. Copenhagen: WHO Regional Office for Europe; 2007.
Laden F, Neas LM, Dockery DW, Schwartz J. Association of fine particulate matter from different sources with daily mortality in six U.S. cities. Environ Health Perspect. 2000;108(10):941–7.
Lelieveld J, Evans JS, Fnais M, Giannadaki D, Pozzer A. The contribution of outdoor air pollution sources to premature mortality on a global scale. Nature. 2015;525(7569):367–71.
Lall R, Ito K, Thurston GD. Distributed lag analyses of daily hospital admissions and source-apportioned fine particle air pollution. Environ Health Perspect. 2011;119(4):455–60.
Jacquemin B, Lanki T, Yli-Tuomi T, Vallius M, Hoek G, Heinrich J, et al. Source category-specific PM2.5 and urinary levels of Clara cell protein CC16. The ULTRA study. Inhal Toxicol. 2009;21(13):1068–76.
Pennington AF, Strickland MJ, Gass K, Klein M, Sarnat SE, Tolbert PE, et al. Source-apportioned PM2.5 and cardiorespiratory emergency department visits: accounting for source contribution uncertainty. Epidemiology. 2019;30(6):789–98.
Robertson KD, Jones PA. DNA methylation: past, present and future directions. Carcinogenesis. 2000;21(3):461–7.
Menezo YJ, Silvestris E, Dale B, Elder K. Oxidative stress and alterations in DNA methylation: two sides of the same coin in reproduction. Reprod Biomed Online. 2016;33(6):668–83.
Gonzalo S. Epigenetic alterations in aging. J Appl Physiol. 2010;109(2):586–97.
Pepin ME, Ha C-M, Crossman DK, Litovsky SH, Varambally S, Barchue JP, et al. Genome-wide DNA methylation encodes cardiac transcriptional reprogramming in human ischemic heart failure. Lab Invest. 2019;99(3):371–86.
Wang Y-P, Lei Q-Y. Metabolic recoding of epigenetics in cancer. Cancer Commun. 2018;38(1):1–8.
Gondalia R, Baldassari A, Holliday KM, Justice AE, Méndez-Giráldez R, Stewart JD, et al. Methylome-wide association study provides evidence of particulate matter air pollution-associated DNA methylation. Environ Int. 2019;132: 104723.
Panni T, Mehta AJ, Schwartz JD, Baccarelli AA, Just AC, Wolf K, et al. Genome-wide analysis of DNA methylation and fine particulate matter air pollution in three study populations: KORA F3, KORA F4, and the normative aging study. Environ Health Perspect. 2016;124(7):983–90.
Plusquin M, Guida F, Polidoro S, Vermeulen R, Raaschou-Nielsen O, Campanella G, et al. DNA methylation and exposure to ambient air pollution in two prospective cohorts. Environ Int. 2017;108:127–36.
Eze IC, Jeong A, Schaffner E, Rezwan FI, Ghantous A, Foraster M, et al. Genome-wide DNA methylation in peripheral blood and long-term exposure to source-specific transportation noise and air pollution: the SAPALDIA study. Environ Health Perspect. 2020;128(6): 067003.
Wang C, Cardenas A, Hutchinson JN, Just A, Heiss J, Hou L, et al. Short-and intermediate-term exposure to ambient fine particulate elements and leukocyte epigenome-wide DNA methylation in older men: the Normative Aging Study. Environ Int. 2022;158: 106955.
Baccarelli A, Wright RO, Bollati V, Tarantini L, Litonjua AA, Suh HH, et al. Rapid DNA methylation changes after exposure to traffic particles. Am J Respir Crit Care Med. 2009;179(7):572–8.
Chen R, Qiao L, Li H, Zhao Y, Zhang Y, Xu W, et al. Fine particulate matter constituents, nitric oxide synthase DNA methylation and exhaled nitric oxide. Environ Sci Technol. 2015;49(19):11859–65.
Hou L, Zhang X, Zheng Y, Wang S, Dou C, Guo L, et al. Altered methylation in tandem repeat element and elemental component levels in inhalable air particles. Environ Mol Mutagen. 2014;55(3):256–65.
Madrigano J, Baccarelli A, Mittleman MA, Wright RO, Sparrow D, Vokonas PS, et al. Prolonged exposure to particulate pollution, genes associated with glutathione pathways, and DNA methylation in a cohort of older men. Environ Health Perspect. 2011;119(7):977–82.
Dai L, Mehta A, Mordukhovich I, Just AC, Shen J, Hou L, et al. Differential DNA methylation and PM2. 5 species in a 450K epigenome-wide association study. Epigenetics. 2017;12(2):139–48.
Karner AA, Eisinger DS, Niemeier DA. Near-roadway air quality: synthesizing the findings from real-world data. Environ Sci Technol. 2010;44(14):5334–44.
Patton AP, Perkins J, Zamore W, Levy JI, Brugge D, Durant JL. Spatial and temporal differences in traffic-related air pollution in three urban neighborhoods near an interstate highway. Atmos Environ. 2014;99:309–21.
Sommar JN, Hvidtfeldt UA, Geels C, Frohn LM, Brandt J, Christensen JH, et al. Long-term residential exposure to particulate matter and its components, nitrogen dioxide and ozone—a Northern Sweden cohort study on mortality. Int J Environ Res Public Health. 2021;18(16):8476.
Hvidtfeldt UA, Geels C, Sørensen M, Ketzel M, Khan J, Tjønneland A, et al. Long-term residential exposure to PM2. 5 constituents and mortality in a Danish cohort. Environ Int. 2019;133:105268.
Lavigne A, Sterrantino AF, Liverani S, Blangiardo M, De Hoogh K, Molitor J, et al. Associations between metal constituents of ambient particulate matter and mortality in England: an ecological study. BMJ Open. 2019;9(12): e030140.
Chen J, Rodopoulou S, de Hoogh K, Strak M, Andersen ZJ, Atkinson R, et al. Long-term exposure to fine particle elemental components and natural and cause-specific mortality—a pooled analysis of eight European cohorts within the ELAPSE project. Environ Health Perspect. 2021;129(4): 047009.
Amini H, Danesh-Yazdi M, Di Q, Requia W, Wei Y, Abu-Awad Y, et al. Hyperlocal super-learned PM2. 5 components across the contiguous US. 2022.
Jin T, Amini H, Kosheleva A, Danesh Yazdi M, Wei Y, Castro E, et al. Associations between long-term exposures to airborne PM2. 5 components and mortality in Massachusetts: mixture analysis exploration. Environ Health. 2022;21(1):1–13.
Qiu X, Wei Y, Amini H, Wang C, Weisskopf M, Koutrakis P, et al. Fine particle components and risk of psychiatric hospitalization in the US. Sci Total Environ. 2022;849: 157934.
Bell B, Rose CL, Damon A. The Veterans Administration longitudinal study of healthy aging. Gerontologist. 1966;6(4):179–84.
Wang C, Cardenas A, Hutchinson JN, Just A, Heiss J, Hou L, et al. Short- and intermediate-term exposure to ambient fine particulate elements and leukocyte epigenome-wide DNA methylation in older men: the Normative Aging Study. Environ Int. 2022;158: 106955.
Di Q, Amini H, Shi L, Kloog I, Silvern R, Kelly J, et al. An ensemble-based model of PM2. 5 concentration across the contiguous United States with high spatiotemporal resolution. Environment Int. 2019;130:104909.
Klepeis NE, Nelson WC, Ott WR, Robinson JP, Tsang AM, Switzer P, et al. The National Human Activity Pattern Survey (NHAPS): a resource for assessing exposure to environmental pollutants. J Expo Anal Environ Epidemiol. 2001;11(3):231–52.
USEPA.
Paatero P, Tapper U. Positive matrix factorization: A non-negative factor model with optimal utilization of error estimates of data values. Environmetrics. 1994;5(2):111–26.
Manchanda C, Kumar M, Singh V, Faisal M, Hazarika N, Shukla A, et al. Variation in chemical composition and sources of PM2. 5 during the COVID-19 lockdown in Delhi. Environment Int. 2021;153:106541.
Mohammed MOA, Song WW, Ma YL, Liu LY, Ma WL, Li WL, et al. Distribution patterns, infiltration and health risk assessment of PM2.5-bound PAHs in indoor and outdoor air in cold zone. Chemosphere. 2016;155:70–85.
Sharma SK, Mandal TK, Jain S, Saraswati, Sharma A, Saxena M. Source Apportionment of PM2.5 in Delhi, India Using PMF Model. Bull Environ Contam Toxicol. 2016;97(2):286–93.
Heiss JA, Just AC. Identifying mislabeled and contaminated DNA methylation microarray data: an extended quality control toolset with examples from GEO. Clin Epigenetics. 2018;10:73.
Xu Z, Langie SA, De Boever P, Taylor JA, Niu L. RELIC: a novel dye-bias correction method for Illumina Methylation BeadChip. BMC Genomics. 2017;18(1):4.
Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29(2):189–96.
Wang C, Just A, Heiss J, Coull BA, Hou L, Zheng Y, et al. Biomarkers of aging and lung function in the normative aging study. Aging (Albany NY). 2020;12(12):11942–66.
Wang C, Koutrakis P, Gao X, Baccarelli A, Schwartz J. Associations of annual ambient PM(2.5) components with DNAm PhenoAge acceleration in elderly men: The Normative Aging Study. Environ Pollut. 2020;258:113690.
BeadArray controls reporter.
Koenker R, Hallock KF. Quantile regression. Journal of economic perspectives. 2001;15(4):143–56.
Koenker R. Quantile regression for longitudinal data. J Multivar Anal. 2004;91(1):74–89.
Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86.
Abatzoglou JT. Development of gridded surface meteorological data for ecological applications and modelling. Int J Climatol. 2013;33(1):121–31.
Hogan JW, Lancaster T. Instrumental variables and inverse probability weighting for causal inference from longitudinal observational studies. Stat Methods Med Res. 2004;13(1):17–48.
Li MX, Yeung JM, Cherny SS, Sham PC. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum Genet. 2012;131(5):747–56.
Nyholt DR. A simple correction for multiple testing for single-nucleotide polymorphisms in linkage disequilibrium with each other. Am J Hum Genet. 2004;74(4):765–9.
Jaffe AE, Murakami P, Lee H, Leek JT, Fallin MD, Feinberg AP, et al. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int J Epidemiol. 2012;41(1):200–9.
Xu Z, Xie C, Taylor JA, Niu L. ipDMR: identification of differentially methylated regions with interval P-values. Bioinformatics. 2021;37(5):711–3.
Mallik S, Odom GJ, Gao Z, Gomez L, Chen X, Wang L. An evaluation of supervised methods for identifying differentially methylated regions in Illumina methylation arrays. Brief Bioinform. 2019;20(6):2224–35.
Xu Z, Sandler DP, Taylor JA. Blood DNA methylation and breast cancer: a prospective case-cohort analysis in the sister study. J Natl Cancer Inst. 2020;112(1):87–94.
Xu K, Liu P, Wei W. mTOR signaling in tumorigenesis. Biochim Biophys Acta. 2014;1846(2):638–54.
Keniry M, Parsons R. The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene. 2008;27(41):5477–85.
Harada A, Sekido N, Akahoshi T, Wada T, Mukaida N, Matsushima K. Essential involvement of interleukin-8 (IL-8) in acute inflammation. J Leukoc Biol. 1994;56(5):559–64.
Traister A, Aafaqi S, Masse S, Dai X, Li M, Hinek A, et al. ILK induces cardiomyogenesis in the human heart. PLoS ONE. 2012;7(5): e37802.
Chano T, Okabe H, Hulette CM. RB1CC1 insufficiency causes neuronal atrophy through mTOR signaling alteration and involved in the pathology of Alzheimer’s diseases. Brain Res. 2007;1168:97–105.
Rickle A, Bogdanovic N, Volkmann I, Zhou X, Pei JJ, Winblad B, et al. PTEN levels in Alzheimer’s disease medial temporal cortex. Neurochem Int. 2006;48(2):114–23.
Hall C, Yu H, Choi E. Insulin receptor endocytosis in the pathophysiology of insulin resistance. Exp Mol Med. 2020;52(6):911–20.
Harding MA, Theodorescu D. RhoGDI signaling provides targets for cancer therapy. Eur J Cancer. 2010;46(7):1252–9.
Perera R, Nikolaev V. Compartmentation of c AMP signalling in cardiomyocytes in health and disease. Acta Physiol. 2013;207(4):650–62.
Orekhov AN, Mukhamedova N, Ivanova EA, Rizzo M. PPAR in cardiovascular disorders. Hindawi; 2016.
Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of beta-adrenergic signaling in heart failure? Circ Res. 2003;93(10):896–906.
St-Louis J, Massicotte G. Chronic decrease of blood pressure by rat relaxin in spontaneously hypertensive rats. Life Sci. 1985;37(14):1351–7.
Lo CW. Role of gap junctions in cardiac conduction and development: insights from the connexin knockout mice. Circ Res. 2000;87(5):346–8.
Tran N, Garcia T, Aniqa M, Ali S, Ally A, Nauli SM. Endothelial Nitric Oxide Synthase (eNOS) and the Cardiovascular System: in Physiology and in Disease States. Am J Biomed Sci Res. 2022;15(2):153–77.
Nehme A, Zouein FA, Zayeri ZD, Zibara K. An update on the tissue renin angiotensin system and its role in physiology and pathology. J Cardiovasc Dev Dis. 2019;6(2):14.
O’Neal WT, Griffin WF, Dries-Devlin JL, Kent SD, Chen J, Willis MS, et al. Ephrin-Eph signaling as a potential therapeutic target for the treatment of myocardial infarction. Med Hypotheses. 2013;80(6):738–44.
Chatterjee S, Behnam Azad B, Nimmagadda S. The intricate role of CXCR4 in cancer. Adv Cancer Res. 2014;124:31–82.
Jeon SM. Regulation and function of AMPK in physiology and diseases. Exp Mol Med. 2016;48(7): e245.
Surawska H, Ma PC, Salgia R. The role of ephrins and Eph receptors in cancer. Cytokine Growth Factor Rev. 2004;15(6):419–33.
Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117(14):3720–32.
Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. 2014;6(10): a016295.
Obradovic M, Sudar-Milovanovic E, Soskic S, Essack M, Arya S, Stewart AJ, et al. Leptin and Obesity: Role and Clinical Implication. Front Endocrinol (Lausanne). 2021;12: 585887.
Paez-Pereda M, Hausch F, Holsboer F. Corticotropin releasing factor receptor antagonists for major depressive disorder. Expert Opin Investig Drugs. 2011;20(4):519–35.
Wang H, Xu J, Lazarovici P, Quirion R, Zheng W. cAMP Response Element-Binding Protein (CREB): A Possible Signaling Molecule Link in the Pathophysiology of Schizophrenia. Front Mol Neurosci. 2018;11:255.
Marshall JS, Warrington R, Watson W, Kim HL. An introduction to immunology and immunopathology. Allergy Asthma Clin Immunol. 2018;14(Suppl 2):49.
Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y, Hu LL. ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med. 2020;19(3):1997–2007.
Caretta A, Mucignat-Caretta C. Protein kinase a in cancer. Cancers. 2011;3(1):913–26.
Rowan MJ, Klyubin I, Cullen WK, Anwyl R. Synaptic plasticity in animal models of early Alzheimer’s disease. Philos Trans R Soc Lond B Biol Sci. 2003;358(1432):821–8.
Pope CA, 3rd, Thun MJ, Namboodiri MM, Dockery DW, Evans JS, Speizer FE, et al. Particulate air pollution as a predictor of mortality in a prospective study of U.S. adults. Am J Respir Crit Care Med. 1995;151(3 Pt 1):669–74.
Ostro B, Hu J, Goldberg D, Reynolds P, Hertz A, Bernstein L, et al. Associations of mortality with long-term exposures to fine and ultrafine particles, species and sources: results from the California Teachers Study Cohort. Environ Health Perspect. 2015;123(6):549–56.
Jiang Y, Niu Y, Xia Y, Liu C, Li Z, Wang W, et al. Effects of personal nitrogen dioxide exposure on airway inflammation and lung function. Environ Res. 2019;177: 108620.
Carmona J, Sofer T, Hutchinson J, Cantone L, Coull B, et al. Short-term airborne particulate matter exposure alters the epigenetic landscape of human genes associated with the mitogen-activated protein kinase network: a cross-sectional study. Environ Health. 2014;13:94.
Oftedal B, Nystad W, Brunekreef B, Nafstad P. Long-term traffic-related exposures and asthma onset in schoolchildren in oslo, norway. Environ Health Perspect. 2009;117(5):839–44.
Dijkema MB, Mallant SF, Gehring U, van den Hurk K, Alssema M, van Strien RT, et al. Long-term exposure to traffic-related air pollution and type 2 diabetes prevalence in a cross-sectional screening-study in the Netherlands. Environ Health. 2011;10:76.
Haddad P, Kutlar Joss M, Weuve J, Vienneau D, Atkinson R, Brook J, et al. Long-term exposure to traffic-related air pollution and stroke: a systematic review and meta-analysis. Int J Hyg Environ Health. 2023;247: 114079.
Sayols-Baixeras S, Fernández-Sanlés A, Prats-Uribe A, Subirana I, Plusquin M, Künzli N, et al. Association between long-term air pollution exposure and DNA methylation: the REGICOR study. Environ Res. 2019;176: 108550.
Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR, et al. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet. 2016;9(5):436–47.
Wei S, Wang W, Liu S, Sun B, Zeng Q, Wang G, et al. Genome-wide DNA methylation pattern in whole blood of patients with coal-burning arsenic poisoning. Ecotoxicol Environ Saf. 2022;248: 114323.
Choi YJ, Cho J, Hong YC, Lee DW, Moon S, Park SJ, et al. DNA methylation is associated with prenatal exposure to sulfur dioxide and childhood attention-deficit hyperactivity disorder symptoms. Sci Rep. 2023;13(1):3501.
Liu K, Jiang J, Lin Y, Liu W, Zhu X, Zhang Y, et al. Exposure to polycyclic aromatic hydrocarbons, DNA methylation and heart rate variability among non-current smokers. Environ Pollut. 2021;288: 117777.
Shao ZQ, Zhang X, Fan HH, Wang XS, Wu HM, Zhang L, et al. Selenoprotein T Promotes Proliferation and G1-to-S Transition in SK-N-SH Cells: Implications in Parkinson’s Disease. J Nutr. 2019;149(12):2110–9.
Mehta V, Suman P, Chander H. High levels of unfolded protein response component CHAC1 associates with cancer progression signatures in malignant breast cancer tissues. Clin Transl Oncol. 2022;24(12):2351–65.
Wu Y, Qie R, Cheng M, Zeng Y, Huang S, Guo C, et al. Air pollution and DNA methylation in adults: a systematic review and meta-analysis of observational studies. Environ Pollut. 2021;284: 117152.
Acknowledgements
This work was supported by the National Institute of Environmental Health Sciences (R01ES027747, R01ES025225, P30ES009089), HSPH-NIEHS Center for Environmental Health (ES000002), and Novo Nordisk Foundation Challenge Program (NNF17OC0027812). Its contents are solely the responsibility of the grantee and do not necessarily represent the official views of the National Institute of Environmental Health Sciences or the U.S. EPA. Further, Neither the National Institute of Environmental Health Sciences nor the U.S. EPA endorses the purchase of any commercial products or services mentioned in the publication.
Author information
Authors and Affiliations
Contributions
J.S. designed research and directed its implementation; C.W., H.A., Z.X., A.P., M.Y., X.Q., Y.W., and A.K. prepared datasets; C.W. analyzed data, made the figures and tables, and wrote the paper; and all authors contributed to the revision of the manuscript.
Corresponding author
Ethics declarations
Competing interests
There are no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, C., Amini, H., Xu, Z. et al. Long-term exposure to ambient fine particulate components and leukocyte epigenome-wide DNA Methylation in older men: the Normative Aging Study. Environ Health 22, 54 (2023). https://doi.org/10.1186/s12940-023-01007-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12940-023-01007-5