
Abstract
Background
Biology is experiencing a gradual but fast transformation from a laboratory-centred science towards a data-centred one. As such, it requires robust data engineering and the use of quantitative data analysis methods as part of database curation. This paper focuses on G protein-coupled receptors, a large and heterogeneous super-family of cell membrane proteins of interest to biology in general. One of its families, Class C, is of particular interest to pharmacology and drug design. This family is quite heterogeneous on its own, and the discrimination of its several sub-families is a challenging problem. In the absence of known crystal structure, such discrimination must rely on their primary amino acid sequences.
Methods
We are interested not as much in achieving maximum sub-family discrimination accuracy using quantitative methods, but in exploring sequence misclassification behavior. Specifically, we are interested in isolating those sequences showing consistent misclassification, that is, sequences that are very often misclassified and almost always to the same wrong sub-family. Random forests are used for this analysis due to their ensemble nature, which makes them naturally suited to gauge the consistency of misclassification. This consistency is here defined through the voting scheme of their base tree classifiers.
Results
Detailed consistency results for the random forest ensemble classification were obtained for all receptors and for all data transformations of their unaligned primary sequences. Shortlists of the most consistently misclassified receptors for each subfamily and transformation, as well as an overall shortlist including those cases that were consistently misclassified across transformations, were obtained. The latter should be referred to experts for further investigation as a data curation task.
Conclusion
The automatic discrimination of the Class C sub-families of G protein-coupled receptors from their unaligned primary sequences shows clear limits. This study has investigated in some detail the consistency of their misclassification using random forest ensemble classifiers. Different sub-families have been shown to display very different discrimination consistency behaviors. The individual identification of consistently misclassified sequences should provide a tool for quality control to GPCR database curators.
Similar content being viewed by others


Background
Biology in general is experiencing a gradual but nevertheless fast transformation from a laboratory-centred science towards a data-centred one [1]. Such transformation can be seen as the result of the coalescence of at least two drivers: the quick progression of information technology systems capabilities and the not much slower advances on data acquisition methods in the different sub-fields of biology.
All of this is particularly relevant to the omics sciences and to bioinformatics. Consistent and concerted efforts are being made in these fields to guarantee the scientific community reliable access to large, heterogeneous and ever-increasing databases. A central concept to the tasks of maintaining and managing such complex databases is that of data curation, which, in the context of biology, is sometimes referred to as biocuration [2].
The objective of the analyses reported in this paper is the large super-family of G protein-coupled receptors (GPCRs). These are eukaryotic cell membrane proteins that are considered of interest to biology in general. Within GPCRs, our specific interest is in one of its families, namely Class C. This family is of particular relevance to current pharmacology, as its members have shown to be therapeutic drug targets that could be involved in the treatment of specific neuro-degenerative diseases such as Alzheimer’s disease, schizophrenia and Parkinson’s disease, amongst others [3], given that most of them are expressed in the central nervous system.
Little is known of the complete crystal (expressing the 3-D) structure of GPCRs and it is only recently that some partial GPCR structures have been solved. They are mostly from Class A [4]. For Class C, instead, no full crystal structure has yet been solved; in fact, only two transmembrane domains and several extracellular domains have been described over the last few years in [5, 6]. This lack of knowledge about their tertiary and quaternary structures means that, faced with the challenge of investigating the functionality of these receptors, we are bound to rely on the analysis of their primary structure, that is, of their amino acid symbolic sequences. This information, fortunately, is publicly available from existing curated databases.
The Class C family is by no means homogeneous and its members have been tagged as belonging to a rich taxonomy of sub-families. Note that many of these are even further sub-divided into types at several levels. This means that the labeling of all these sub-families becomes quite a challenge for the expert and, consequently, any process attempting the automatic sub-family classification unavoidably becomes itself a challenging problem [7]. Again, and as stated earlier in this introduction, such sub-family discrimination becomes a data analysis problem, for which statistics and machine learning approaches can provide well-founded and robust solutions [8].
It must be stressed that, in this study, we are interested less in the problem of achieving maximum sub-family discrimination accuracy (as, for instance, in [7]) than in the exploration of sequence misclassification behavior. We assume that the majority of sequences are adequately characterized (that is, that they are correctly labeled in the database according to sub-family), but we are keen on investigating which sequences are commonly misclassified by computer-based methods and what type of misclassification they suffer. The starting point for these concerns are previously reported results indicating that the discriminatory classification of Class C GPCRs from transformations of their primary sequences shows clear limits [9, 10].
More specifically, we aim to isolate those sequences that show consistent misclassification, in the sense that they are very often misclassified and almost always assigned to the same sub-family (this sub-family being other than the one described by their database label). This is not to be mistaken with sequences that might be misclassified for their partial similarity with different sub-families (what we could call class borderline cases) and which could show far less consistent behavior.
According to this goal, the current study, which is an extension of [11], and its reported experiments aim at assisting the task of database curation by providing data-based evidence of potential GPCR quality control issues. This should be accomplished by identifying and shortlisting cases whose original sub-family assignment is highly questionable from the data modelling results, thus motivating further expert intervention.
Random forests (RF) are the machine learning method of choice in this work for the task of assessing the consistency of (mis)classification. This choice is justified by their ensemble nature, in which the many base classifiers they consist of coalesce in the decision of assigning each given sequence to a class (GPCR sub-family). This collective decision process makes them naturally suited to assess the consistency of the classification decisions.
Given that our analyses are based on GPCR primary sequences, a data pre-processing problem obviously arises, which is the choice of transformation of the varying-length sequential symbolic data into formats that are suitable for multivariate data analysis. Such transformations might use the complete unaligned sequences, or methods of multiple sequence alignment. Here, we use a number of unaligned sequence transformations.
Methods
This section first introduces some basics about the task of curation of protein databases. This is followed by a description of the data used in the reported experiments and an introduction to the RF algorithm as applied to the analysis of these data.
Curation of protein databases
As mentioned in the introduction, this paper presents a data-based analytical method for the assessment of the consistency of discriminatory classification of sub-families of GPCRs. It is suggested here that such method could be used to assist protein database curators in tasks of data quality control.
Biocuration has become a need in biology due to the exponential growth in data availability in all its many sub-fields and particularly in bioinformatics. It has been described as “the activity of organizing, representing and making biological information accessible” [2] to biologists. Therefore, it should at least partially concern data engineering tasks. Nevertheless, it is far from being an established and well-defined activity and failure to establish and standardize biocuration procedures and to fund these efforts properly would risk the possibility of channeling the data deluge for scientific knowledge extraction.
One of the challenges of curation is the unambiguous identification of biological entities (proteins in the case of the current study) from existing studies and literature. In the end, data trustworthiness can only be ensured through costly data management [12]. This task is uncertain and error-prone, so that the development of computational procedures to assist human experts in it is worth pursuing. Note that GPCRs have been categorized into the Classes A–F (as described in the next sub-section) based on sequence homology procedures [13]. That is, receptor labeling is itself homology model-based and, therefore, uncertain to a degree and at least debatable.
The background for our study is the preliminary evidence suggesting that there seems to exist an upper bound to Class C GPCR discriminability according to the existing sub-family labels [9, 10]. We aim to establish if certain receptors show clear patterns of consistent misclassification and whether this might be the cause for the existence of such upper bound.
Class C data from the GPCRdb
This study is based on the analysis of data extracted from GPCRdb, a publicly accessible molecular-class information repository for GPCRs [13]. This endeavour was started in 1993 and it is now in its fifth release, with stewardship by David Gloriam’s group at the University of Copenhagen from 2013 and part of the GLISTEN EU COST Action [14] for the creation of “a pan-European multidisciplinary network of researchers investigating G protein-coupled receptor signalling”.
In this repository, the GPCR super-family is divided into major classes following the IUPHAR [15] system, including: A (rhodopsin like), B (secretin and adhesion), C (glutamate), F (Frizzled) and others, based on the ligand types, functions and sequence similarities.
As previously stated, the current study focuses on Class C GPCRs, a quite heterogeneous family that includes seven main sub-families: metabotropic glutamate (mG) receptors, calcium sensing (CS), GABA\(_B\) (GB), vomeronasal (VN), pheromone (Ph), odorant (Od) and taste (Ta).
mG receptors are activated by glutamate, a major excitatory neurotransmitter in the brain. These receptors are involved in neurological disorders including Alzheimer’s and Parkinson’s diseases, Fragile X syndrome, depression, schizophrenia, anxiety, and pain. Some preliminary information about the binding sites and behavior of two subtypes (1 and 5) of mG receptors has been described in [5, 6] from their crystal structures, as summarily reviewed in [16]. The CS receptor is activated by the calcium ion and it is known to play a key role in extra-cellular calcium homeostasis regulation. GB is a neurotransmitter that mediates most inhibitory actions in the central nervous system; it is involved in chronic pain, anxiety, depression and addiction pathologies. VN, Ph, Od and Ta are all involved in physiological roles related to the senses of smell and taste.
A total of 1510 Class C GPCR sequences (from version 11.3.4, March 2011 of the database), belonging to the previously mentioned seven sub-families, were analyzed using the RF models described in the following sub-section. Their distribution of cases by sub-family is displayed in Table 1.
Given that the primary sequences cannot be analysed as symbolic arrays using standard statistical, pattern recognition and machine learning methods, they have to be transformed for subsequent investigation. Several transformations were considered in our experiments.
The first one uses directly the 20 amino acids of the receptor sequence alphabet. An example of this type of transformations is amino acid composition (AAC) [17], in which each sequence is described by the frequencies of appearance of the amino acids. By using only frequencies, AAC ignores the order embedded into the sequential information itself (i.e., the relative position of the amino acids in the sequence). Despite such simplicity, its use has previously yielded surprisingly solid sub-family discrimination results [17, 18].
Subsets of amino acids may share similar physico-chemical properties, which makes them equivalent at a functional level [19]. This equivalence would make them somehow redundant and the use of amino acid groupings based on physicochemical similarity becomes advisable. Amino acid grouping also helps computations by reducing the dimensionality of the analysed data set. For this study, two alternative groupings were used, in the form of sub-sequence frequencies (see Table 2): the Sezerman (SEZ) alphabet (11 groups) [20] and the Davies Random (DAV) alphabet (9 groups) [19]. We select these groupings for analysis because they are the outcome of a selection process and their performance in the classification of GPCRs into their major classes was positively evaluated in [19, 20].
Amino acids and their groupings were not just used as such in this study, but in the form of n-grams, which are subsequences of length n. The concept of n-grams is well-known in protein analysis [21, 22]. Here, we used the relative frequencies of the n-grams. Therefore, the n-gram representation consists of the relative frequency of each n-gram in a sequence (note that for Sezerman and Davies, the length of the n-gram is not taken in number of amino acids, but in number of groupings). Due to the exponential growth of the size of n-grams, experiments were limited to n-grams of size 1, 2 and 3.
Random forests
Since their definition in the first years of the century, RFs [23] have become a popular and widely-used machine learning tool for classification and regression tasks. This is particularly true in the areas of computational biology and bioinformatics [24]. In these fields, RFs have de facto become standard methods, especially adequate in settings with poor observations-to-variables ratios (of which the current study is a mild case). They are also capable of coping well with highly correlated variables and scale nicely to multi-class problems such as the one investigated here, avoiding the more complex one-vs-one or one-vs-all classification schemes that are common in the field [9].
The general graphical scheme of the RF algorithm is sketched in Fig. 1. At each split of the observed sample data, a random subset of variables is selected (in what is called a random subspace method) and the process is repeated until a specified number of base decision tree classifiers is generated. Each tree is built from a bootstrap sample drawn with replacement from the observed data (the available sample), and the predictions of all trees are finally aggregated through majority voting.

RF scheme. General graphical scheme of the RF algorithm
A feature of RFs is the definition of an out-of-bag (OOB) error, which is calculated from observations that were not used to build a particular base decision tree; it can thus be considered as an internal cross-validation error measure [24, 25]. This is an important feature for the type of experiments carried out in this study, because it simplifies the otherwise cumbersome cross-validation procedures that would be required if alternative classification methods such as, for instance, support vector machines or artificial neural networks were used [23].
The fact that RFs are defined as ensemble-of-trees classifiers also means that these models are naturally suited to the task of analysing protein sequence misclassification behavior. The reason for that is that we are interested in methods that naturally work according to a voting scheme in order to assign a sequence observation to a given class (sub-family). These votes from each individual decision tree allow us to more closely inspect the performance of each Class C GPCR sequence. Furthermore, pooling the votes also allows us to gauge the consistency of the (mis)classification results for the sequences of any given sub-family.
Results and discussion
In this section we first provide details of the experimental settings. This is followed by a presentation of all results and their discussion.
Experimental settings
As mentioned in the previous section, the subset of sequences belonging to Class C acquired from the GPCRdb database were transformed in three ways: AAC, Sezerman (11 groupings) and Davies (9 groupings). All possible n-grams of sizes 1 to 3 were built for each of them and the relative frequencies of the n-grams of size 1, 2 and 3 (for Sezerman and Davies, the length of the n-gram is not taken in number of amino acids, but in number of groupings) were calculated.
In previous research [26], feature selection was performed on these transformed datasets using statistical t test filtering to establish a ranking of relevance of the available features. This led to the choice of subsets of features whose test was significant for different numbers of binary classifiers (note that, taking into account that we analyze 7 Class C sub-families, we would have 21 different one-vs-one binary classifiers and, at best, a subset of features that was significant for all 21 of them might be obtained).
The subsets of features that achieved the best classification performance in [26] become the starting point for our RF models. Their number of features is 585 for n-grams using AAC, 386 for n-grams using Sezerman groupings and 238 for n-grams using Davies groupings. This makes the discrimination problem a mild case of poor observations-to-variables ratio, as previously indicated.
The RF model was trained using the randomForest and matrixStatsR packages of the R programming language. To ensure the reproducibility of the results, the random number generator (RNG) state was set to a value of 42. The model was stratified to ensure that the difference in the number of cases between sub-families did not significantly affect the results. It included 500 trees, yielding sufficient performance while keeping computational costs relatively small. On an Apple MacBook Pro with 2.3 GHz Quad-core Intel i7 CPU and 8 Gb RAM, the CPU times for the execution of the processes were 0.028, 0.024 and 0.026 s for selected subsets of n-grams generated using AAC, Sezerman and Davies transformations, respectively.
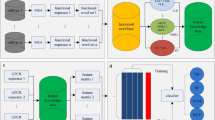
The whole experimental workflow is graphically summarized in Fig. 2.

Experimental workflow. Step-by-step graphical representation of the experimental workflow
Experimental results
In order to start from a basic reference assessment of performance of the models, overall accuracies (understood as the ratio of correctly classified sequences to all sequences) for all the models were calculated. They are as follows: 0.91 for the selected subset of n-grams using AAC; 0.90 for the subset selected using Sezerman transformation and 0.88 for the subset selected using Davies transformation.
Given that the main target is the investigation of the details of the Class C GPCR misclassification behavior, we first calculated the confusion matrices for our RFs. The results are presented in Table 3 for the selected subset of n-grams using AAC; in Table 4 for the selected subset of n-grams using Sezerman groupings; and in Table 5 for the selected subset of n-grams using Davies groupings. All these confusion matrices also include sub-family-specific classification errors (which are equivalent to 1-sensitivity for each sub-family). Confusion matrices provide us with an intuitive overall assessment of how scattered the classification results are.
The overall accuracy and the confusion matrices are aggregated measures that do not inform about the consistency of individual sequence classifications. We now recall the fact that the sub-family assignments made by the RF model are the result of the individual voting of 500 base trees.
On a first and still sub-family centered approximation, the votes of these 500 trees for each sub-family were extracted and mean values of their voting ratios were calculated as a measure of sub-family classification consistency. For better illustration, only the results for mG, GB and Ph are presented in the main text, in Table 6. These three sub-families are selected as opposite examples of rather well discriminated sub-families (mG and GB) vs. a comparatively poorly discriminated one (Ph). Additionally, the spread of the votes is shown in Table 7 using the standard deviation of the voting ratios.
On a second level of detail, the consistency of RF voting for each sequence in the analyzed receptors was calculated. A consistency of 100% can only be reached when the 500 RF trees agree on the sub-family assignment. Note that this does not necessarily mean that the sequence has been correctly classified (according to its database label); in fact, if the assignment was always to same wrong sub-family (in the sense that differs from the database label) the consistency would still be 100%.
The detailed consistencies for all mG and GB sequences for the subset generated using AAC are, in turn, displayed in Figs. 3 and 4; for the subset using the Sezerman transformation in Figs. 5 and 6; and for the subset using the Davies transformation in Figs. 7 and 8. As an illustration of a sub-family with poor discrimination behavior, similar figures are shown for Ph (see, in turn, Figs. 9, 10, 11). Results for the rest of sub-families can be found in Additional file 1.

mG consistency values per sequence (AAC). Individual consistency values for mG sequences described by the selected subset of n-grams using AAC. The horizontal axis only describes the position of the sequence in the GPCRdb extracted dataset

GB consistency values per sequence (AAC). Individual consistency values for GB sequences described by the selected subset of n-grams using AAC. The horizontal axis only describes the position of the sequence in the GPCRdb extracted dataset

mG consistency values per sequence (SEZ). Individual consistency values for mG sequences described by the Sezerman transformation. The horizontal axis only describes the position of the sequence in the GPCRdb extracted dataset

GB consistency values per sequence (SEZ). Individual consistency values for GB sequences described by the Sezerman transformation. The horizontal axis only describes the position of the sequence in the GPCRdb extracted dataset

mG consistency values per sequence (DAV). Individual consistency values for mG sequences described by the Davies transformation. The horizontal axis only describes the position of the sequence in the GPCRdb extracted dataset

GB consistency values per sequence (DAV). Individual consistency values for GB sequences described by the Davies transformation. The horizontal axis only describes the position of the sequence in the GPCRdb extracted dataset

Ph consistency values per sequence (AAC). Individual consistency values for Ph sequences described by the selected subset of n-grams using AAC. The horizontal axis only describes the position of the sequence in the GPCRdb extracted dataset

Ph consistency values per sequence (SEZ). Individual consistency values for Ph sequences described by the Sezerman transformation. The horizontal axis only describes the position of the sequence in the GPCRdb extracted dataset

Ph consistency values per sequence (DAV). Individual consistency values for Ph sequences described by the Davies transformation. The horizontal axis only describes the position of the sequence in the GPCRdb extracted dataset
From the point of view of database quality assessment and control, we are most interested in those Class C sequences that are not just very frequently misclassified from the point of view of their true (according to the database) class, but also very consistently misclassified to a given wrong sub-family by the majority of trees in the RF.
Shortlists of this type of sequences and values of the consistencies of their misclassification were obtained for all Class C sub-families. The criterion for inclusion is that the consistency for the true class (according to database label) is lower than 1/7, meaning that the consistency for some other class must be higher than 1/7. More restrictive criteria might be applied should the expert decide to restrict the data quality control procedure.
Again, we first illustrate this selection using mG, GB and Ph for each of the three transformations. Results can be found in Tables 8, 9 and 10. It would also be important to discount the possible impact of the type of data transformation on the misclassification consistency behavior. For that, we show in Tables 11 and 12, in turn, the sequences shortlisted in at least two out of three transformations and in all three transformations. Similar results for the rest of sub-families can be found in Additional file 2.
Discussion
As stated in the introduction, the target of the study is not the assessment of the overall classification accuracy that could be obtained for the different sub-families and for each type of data transformation. Nevertheless, such global accuracy was calculated for the three data transformations and reported in the previous section. Accuracies are fairly similar and all in the area of 90%, a result that is consistent with those reported in previous studies [26]. These results suggest that no transformation provides a clear advantage over the others in terms of overall Class C sub-family discrimination.
The confusion matrices in Tables 3, 4 and 5 yield several clear and more specific messages. First, the RF model is shown to have adequate overall discrimination capabilities for all data transformations. This classification, though, is not homogeneous across sub-families; mG, CS and GB are very well discriminated in all data transformations, while Ta is extremely well-discriminated in all data transformations and slightly less so when using Davies’. On the opposite extreme, Od is very poorly classified in all cases. Despite similarities for all data transformations, the selection of n-grams using AAC shows some advantage over the more parsimonious Sezerman and Davies transformations.
Inspecting these matrices in more detail, some other interesting patterns emerge: some sub-families do not show clear “preference” in their misclassification, namely mG, CS, GB and Ta; whereas VN, Ph and Od seem to mostly restrict misclassifications to happen between them. VN is mostly misclassified as Ph, while Ph is mostly misclassified as VN (and to a lesser extent to mG); in turn, Od is mostly misclassified as Ph and, to a lesser extent, as VN. Overall, the Ph sub-family seems to take a central role in this misclassification pattern, overlapping the other two sub-families.
The latter results are corroborated by the per-sub-family consistency means reported in Table 6 (and by the means corresponding to the rest of sub-families, not reported here), which show a sizeable overlapping between the Ph and VN sub-families.
Yet again, even if the overall results for each of the sub-families are relevant on their own right, the detailed consistency values per sequence for these sub-families are the key objective of the current study. Corroborating the overall findings, a sizeable proportion of the consistencies of the individual sequences belonging to mG have either 100% or near 100% consistency values, as seen in Figs. 3, 5 and 7. Nonetheless, the consistencies of many sequences with indices roughly between 50 and 150 fall sharply to values under 50% for all transformations. Note that the horizontal axis in these figures only reflects the position of the specific receptor sequence in the GPCRdb extracted data set and, therefore, these results could perhaps indicate the existence of a differentiated sub-group within this sub-family that would require specific attention. A first inspection of GPCRdb identifiers reveals that only a few of the sequences in positions between 50 and 150 are clearly tagged in the database as belonging to a given mG subtype and they are mostly from subtype 3. Many of them are actually uncharacterized. A similar but more attenuated pattern can be seen in Figs. 4, 6 and 8 for the GB sub-family, which also has two known sub-types (GABA\(_{B1}\) and GABA\(_{B2}\)).
Most of the individual consistencies of the sequences for the rest of sub-families (see Additional file 1) show a more homogeneous distribution, mostly with values between 60 and 90%. Sequences of very low consistencies seem to be quite evenly distributed.
Turning now to the final objective of this paper, which is the creation of shortlists of Class C GPCR sequences that are very consistently misclassified, the results reported in Tables 8, 9 and 10 turn out to be quite revealing. First, and unsurprisingly, a poorly discriminated sub-family such as Ph has many more consistent misclassifications than easier to discriminate sub-families such as mG and GB. These results are corroborated by those of the rest of sub-families, compiled in Additional file 2, where, as extreme opposite cases, many Od sequences are shortlisted, in stark contrast to the almost complete absence of Ta sequences. Also, and interestingly, most of the GB and Ph consistently misclassified sequences were assigned to the mG sub-family regardless of data transformation, whereas the shortlisted mG have been unevenly assigned to GB, VN, Ph and Od. A hypothesis to explain this behavior is that, due to the heterogeneity of the mG sub-family, known to include up to 8 different subtypes, some of these subtypes could be relatively close to GB, VN, Ph, or Od in the space spanned by their sequence transformations. For the rest of sub-families, it is also interesting to discover that most of the consistently misclassified Od sequences are assigned to Ph, corroborating the previous results that indicate the strong overlapping between both families.
For the analysis of the influence of data tranformations on the consistency of misclassifications, the results reported in Tables 11 and 12 for mG, GB and Ph are again revealing. Five sequences were consistently misclassified using two out of the three transformations and as much as another eight appeared in all three shortlists. These are sizeable numbers that reveal that, although some of the misclassifications could be data transformation-related, most are transformation-independent. Focusing only on Table 12, we see that it includes three mG sequences, one GB and four Ph.
The same results for the rest of sub-families, found in Additional file 2, indicate that the consistency of misclassification is most transformation-dependent for the most overlapping sub-families (Od and VN). Ten sequences of those sub-families were included in the refined shortlist of all transformations: two CS, two VN and six Od.
Although the investigation of each of these individual sequences is beyond the scope of this paper, some of them will be discussed here. Sequences a8dz71_danre and a8dz72_danre, reported in the refined list of Table 12, were, uncharacterized proteins derived from an Ensembl automatic analysis pipeline according to the UniProt database; they have since been deleted from the database. Ensembl characterized them as Class C olfactory receptors and note that our RF model predicted them to belong to the Ph sub-family. Also according to UniProt [27], q5i5c3_9tele, in the refined list of Table 12, is the unreviewed putative pheromone receptor CPpr14 (note that the RF assigns it consistently to Od and to Ph as a second choice).
Sequence q8c0m6_mouse, a CS in the refined shortlist in Additional file 2, is also an unreviewed sequence that should be considered only as preliminary data, according to the UniProt database [28]. Also in Additional file 2, XP_002740613 and XP_002940324, which are, in turn, a CS predicted to be a GB and an Od predicted to be a VN, would require further investigation as their sub-family assignment was automatically derived by computational analysis from an annotated genomic sequence by means of a gene prediction mode from the RefSeq databank [29].
Finally, let us insist again on the fact that the criterion used to create these shortlists is flexible and that less or more restrictive criteria could have been defined. Ultimately, the strictness of this criterion should be set by the database curator. In any case, the characteristics of these individual sequences would be the matter of further investigation, even beyond GPCRdb, in the main international protein databases.
Conclusion
Class C of GPCRs have established themselves as relevant targets in pharmacology for their role as drug targets, especially in pathologies of the central nervous system. This protein family has a heterogeneous sub-family structure, whose investigation has to be carried out from the primary structure of its members in the almost complete absence of information concerning their complete tertiary and quaternary structures.
The automatic discrimination of these sub-families is a challenging task and it has previously been shown to have clear limits. This study has investigated in some detail the hypothesis that such limits could at least be partially caused by sequence mislabeling, a problem that would fall within the remit of database curation. Mislabeling could be revealed by investigating the consistency of misclassification using ensemble techniques. In this study, we have reported experiments carried out with the RF method, which is naturally suited to the the task of measuring classification consistency with little computational effort.
Different sub-families have been shown to display very different discrimination consistency behaviors. Specific attention has been paid to the individual identification of Class C GPCR sequences that were consistently assigned by the RF base classifiers to sub-families other than their true one (i.e., misclassified). Sequences consistently misclassified across data transformations have been singled out in refined shortlists as candidates for further labeling investigation. This type of analysis is meant to provide a data engineering quality control tool that is useful not only for GPCR database curators in particular, but for curators of protein databases in general.
References
Marx V. Biology: the big challenges of big data. Nature. 2013;498(7453):255–60.
Howe D, Costanzo M, Fey P, Gojobori T, Hannick L, Hide W, Hill DP, Kania R, Schaeffer M, St Pierre S, Twigger S. Big data: the future of biocuration. Nature. 2008;455(7209):47–50.
Kniazeff J, Prézeau L, Rondard P, Pin JP, Goudet C. Dimers and beyond: the functional puzzles of class C GPCRs. Pharmacol Ther. 2011;130(1):9–25.
Katritch V, Cherezov V, Stevens RC. Structure-function of the G protein-coupled receptor superfamily. Annu Rev Pharmacol. 2013;53:531–56.
Wu H, et al. Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science. 2014;344(6179):58–64.
Doré AS, et al. Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature. 2014;551:557–62.
Gao QB, Ye XF, He J. Classifying G-protein-coupled receptors to the finest subtype level. Biochem Biophy Res Commun. 2013;439(2):303–8.
Larrañaga P, Calvo B, Santana R, Bielza C, Galdiano J, Inza I, Lozano JA, Armañanzas R, Santafé G, Pérez A, Robles V. Machine learning in bioinformatics. Brief Bioinform. 2006;7(1):86–112.
König K, Cárdenas M, Giraldo J, Alquézar R, Vellido A. Label noise in subtype discrimination of class C G-protein coupled receptors: a systematic approach to the analysis of classification errors. BMC Bioinform. 2015;16(1):314.
Cruz-Barbosa R, Vellido A, Giraldo J. The influence of alignment-free sequence representations on the semi-supervised classification of Class C G protein-coupled receptors. Med Biol Eng Comput. 2015;53(2):137–49.
Shkurin, A, Vellido A. Random forests for quality control in G-protein coupled receptor databases. In: Ortuño F, Rojas I, eds. Bioinformatics and biomedical engineering. Proceedings. of the 4th international conference (IWBBIO 2016); 2016, LNCS/LNBI 9656, p. 707-18.
Lord P, Macdonald A, Lyon L, Giaretta D. From data deluge to data curation. In: Proceedings of the UK e-science All Hands meeting; 2004. p. 371–5
Isberg V, Mordalski S, Munk C, Rataj K, Harpsøe K, Hauser AS, Vroling B, Bojarski AJ, Vriend G, Gloriam DE. GPCRdb: an information system for G protein-coupled receptors. Nucleic Acids Res. 2016;44(Database issue):D356–64.
GLISTEN COST Action CM1207. http://www.glisten-gpcr.eu. Accessed 8 Mar 2017.
IUPHAR: International Union of Basic and Clinical Pharmacology. http://www.iuphar.org. Accessed 8 Mar 2017.
Cooke RM, Brown AJ, Marshall FH, Mason JS. Structures of G protein-coupled receptors reveal new opportunities for drug discovery. Drug Discov Today. 2015;20(11):1355–64.
Sandberg M, Eriksson L, Jonsson J, Sjöström M, Wold S. New chemical descriptors relevant for the design of biologically active peptides. A multivariate characterization of 87 amino acids. J Med Chem. 1998;41:2481–91.
Cárdenas MI, Vellido A, Giraldo J. Visual interpretation of class C GPCR subtype overlapping from the nonlinear mapping of transformed primary sequences. In: Proceedings of the international conference on biomedical and health informatics (IEEE BHI 2014); 2014. p. 764–7
Davies MN, Secker A, Freitas A, Clark E, Timmis J, Flower DR. Optimizing amino acid groupings for GPCR classification. Bioinformatics. 2008;24(18):1980–6.
Can Cobanoglu M, Saygin Y, Sezerman UO. Classification of GPCRs using family specific motifs. IEEE ACM Trans Comput Biol. 2011;8(6):1495–508.
Caragea C, Silvescu A, Mitra P. Protein sequence classification using feature hashing. In: Proceedings. of the IEEE international conference on bioinformatics and biomedicine (BIBM 2011); 2011. p. 538–43
Mhamdi F, Elloumi M, Rakotomalala R. Textmining, features selection and datamining for proteins classification. In: Proceedings. of the IEEE international conference on information and communication technologies: from theory to applications, IEEE/ICTTA; 2004. p. 457–8
Breiman L. Random forests. Mach Learn. 2001;45(1):5–32.
Boulesteix A-L, Kruppa J, Konig I. Overview of random forest methodology and practical guidance with emphasis on computational biology and bioinformatics. Wiley Interdis Rev Data Mining Knowl Dis. 2012;2(6):493–507.
Strobl C, Boulesteix A-L, Kneib T, Augustin T, Zeileis A. Conditional variable importance for random forests. BMC Bioinform. 2008;9:307.
König K, Alquézar R, Vellido A, Giraldo J. Finding class C GPCR subtype-discriminating n-grams through feature selection. In: Proceedings of the 8th international conference on practical applications of computational biology and bioinformatics (PACBB 2014); 2014. p. 89–96
UniProt Database, GPCR Q5I5C3. http://www.uniprot.org/uniprot/Q5I5C3. Accessed 8 Mar 2017.
UniProt Database, GPCR B0UYJ3. http://www.uniprot.org/uniprot/B0UYJ3. Accessed 8 Mar 2017.
RefSeq: NCBI Reference Sequence Database. http://www.ncbi.nlm.nih.gov/refseq. Accessed 8 Mar 2017.
Declarations
Authors’ contributions The first author implemented the experiments that were conceptually developed by the second author. Both authors contributed to the writing and revision of the manuscript, as well as to the discussion of the experimental results. Both authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Original sequence data are publicly available from http://gpcrdb.org. For the random forest experiments, the randomForest and matrixStatsR packages of the R programming language were used.
Funding
This research, including publication costs, was funded by the Spanish TIN2016-79576-R project, under the MINECO call.
About this supplement
This article has been published as part of BioMedical Engineering OnLine Volume 16 Supplement 1, 2017: Selected articles from the 4th International Work-Conference on Bioinformatics and Biomedical Engineering-IWBBIO 2016. The full contents of the supplement are available online at https://biomedical-engineering-online.biomedcentral.com/articles/supplements/volume-16-supplement-1.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Shkurin, A., Vellido, A. Using random forests for assistance in the curation of G-protein coupled receptor databases. BioMed Eng OnLine 16 (Suppl 1), 75 (2017). https://doi.org/10.1186/s12938-017-0357-4
Published:
DOI: https://doi.org/10.1186/s12938-017-0357-4