
Abstract
Background
The routine surveillance of asymptomatic malaria using nucleic acid-based amplification tests is essential in obtaining reliable data that would inform malaria policy formulation and the implementation of appropriate control measures.
Methods
In this study, the prevalence rate and the dynamics of Plasmodium species among asymptomatic children (n = 1697) under 5 years from 30 communities within the Hohoe municipality in Ghana were determined.
Results and discussion
The observed prevalence of Plasmodium parasite infection by polymerase chain reaction (PCR) was 33.6% (571/1697), which was significantly higher compared to that obtained by microscopy [26.6% (451/1697)] (P < 0.0001). Based on species-specific analysis by nested PCR, Plasmodium falciparum infection [33.6% (570/1697)] was dominant, with Plasmodium malariae, Plasmodium ovale and Plasmodium vivax infections accounting for 0.1% (1/1697), 0.0% (0/1697), and 0.0% (0/1697), respectively. The prevalence of P. falciparum infection among the 30 communities ranged from 0.0 to 82.5%. Following artesunate-amodiaquine (AS + AQ, 25 mg/kg) treatment of a sub-population of the participants (n = 184), there was a substantial reduction in Plasmodium parasite prevalence by 100% and 79.2% on day 7 based on microscopy and nested PCR analysis, respectively. However, there was an increase in parasite prevalence from day 14 to day 42, with a subsequent decline on day 70 by both microscopy and nested PCR. For parasite clearance rate analysis, we found a significant proportion of the participants harbouring residual Plasmodium parasites or parasite genomic DNA on day 1 [65.0% (13/20)], day 2 [65.0% (13/20)] and day 3 [60.0% (12/20)] after initiating treatment. Of note, gametocyte carriage among participants was low before and after treatment.
Conclusion
Taken together, the results indicate that a significant number of individuals could harbour residual Plasmodium parasites or parasite genomic DNA after treatment. The study demonstrates the importance of routine surveillance of asymptomatic malaria using sensitive nucleic acid-based amplification techniques.
Similar content being viewed by others

Background
Human malaria remains a global health concern despite the introduction of several control measures over the past decades [1]. In 2020, a global estimate of 241 million malaria cases was recorded, which is comparable to malaria cases reported in 2000 [1]. Among the malaria-vulnerable groups, children under 5 years are the most affected accounting for about 80% of all malaria deaths in 2020 [1]. In sub-Saharan Africa, the region accounting for approximately 95% of total malaria cases [1], four Plasmodium species (Plasmodium falciparum, Plasmodium malariae, Plasmodium ovale and Plasmodium vivax) have been implicated in clinical malaria with P. falciparum being the dominant species [2]. The other non-falciparum species have persisted in the background with limited geographical distribution and are usually detected as co-infection with P. falciparum [3].
Towards the attainment of malaria elimination and subsequent global eradication, one major setback is the sustained transmission of Plasmodium parasites among asymptomatic carriers [3, 4]. These subclinical individuals harbour Plasmodium parasites and serve as a reservoir for continuous transmission of the parasites [5]. Most of these subclinical malaria cases are detected as low-density parasitaemia that is often missed or undetected by the commonly used malaria diagnostic tools such as microscopy and antigen-based rapid diagnostic tests (RDTs) [6, 7] due to their limited sensitivity and poor specificity in distinguishing the different Plasmodium species [8]. As such, the true extent of asymptomatic malaria infections is usually underestimated in epidemiological studies that involve the use of only microscopy and/or RDTs [9]. Nucleic acid-based amplification tests such as polymerase chain reaction (PCR), on the other hand, provide relatively high analytical performance for species-specific identification of Plasmodium species even at low parasite densities [9].
In sub-Saharan Africa, cross-sectional studies in different countries, including Ghana, Uganda, Angola, Nigeria, Malawi and Equatorial Guinea, using PCR-based techniques have reported varying prevalence rates of falciparum and non-falciparum species [10,11,12,13,14,15,16]. These reports suggest that the transmission dynamics of both falciparum and non-falciparum species vary across different study populations and age groups [3, 17, 18]. In some malaria-endemic settings, changing prevalence rates of the different Plasmodium species have been observed following malaria interventions. Notably, a study in Tanzania observed an increasing prevalence of P. malariae and P. ovale with decreasing prevalence of P. falciparum [19]. This observation was also reported in a more recent study in Kenya, where P. ovale prevalence increased following a decreased prevalence of P. falciparum [20]. In addition, a recent study in Nigeria observed a surprisingly high prevalence of non-falciparum Plasmodium species [21]. Other studies have also reported the emergence of non-falciparum Plasmodium species after the administration of artemisinin-based combination therapy (ACT) in Ghana [15] and Gabon [22]. These reports highlight the need for routine surveillance of both falciparum and non-falciparum species to improve malaria management.
In Ghana, few PCR-based epidemiological studies have assessed the distribution of falciparum and non-falciparum species among asymptomatic individuals [14, 15, 23,24,25]. The microscopic prevalence of the genus Plasmodium in a cross-sectional study of asymptomatic children living in 30 communities within the Hohoe Municipality of Ghana have been previously reported [26]. In this study, the prevalence rates of P. falciparum, P. malariae, P. ovale and P. vivax among the same study population were determined using nested PCR, which provides relatively high sensitivity and specificity. In addition to this, the temporal dynamics of Plasmodium species and the rate of parasite clearance after ACT were assessed.
Methods
Ethical approval
The study was approved by the ethical review committee of the Ghana Health Service (GHS-ERC:14/05/15). In addition, permission was obtained from the Municipal Health directorate, chiefs and elders of the participating communities. Written informed consent was given by the parent or guardian of each participant before enrolment.
Study area, study design and sample collection
The study was conducted in 30 communities within the Hohoe municipality in the Volta Region of Ghana (Fig. 1). Hohoe is a hyper-endemic region with all-year malaria transmission which peaks in two rainy seasons: one peak in the major rainy season (April to July) and another peak during the minor rainy season (September to November). The entomological inoculation rate (EIR) for the study area is approximately 65 infectious bites per person per year [27]. The current work involved both cross-sectional and longitudinal cohort studies. In the cross-sectional study, a total of 1697 children aged 6–59 months were recruited from the 30 communities in November 2015. Finger-pricked blood samples were collected from the participants for microscopy examination of malaria parasites and dried blood spot (DBS) preparation using Whatman filter paper. A volume of 50 µL of whole blood was used per blood spot. DBS were kept in a desiccant-containing zip-lock bags and stored at room temperature until ready for DNA extraction.

A Google Map of the study sites with insert showing the extended view of the location of the 30 study communities within the Hohoe municipality, Volta Region, Ghana. The 30 study sites have been depicted with blue marks and numbered from 1 to 30, in no particular order
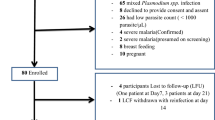
Among the study participants who tested positive (541/1697) for Plasmodium species by microscopy, 184 eligible participants were further recruited in the longitudinal cohort study (Cohort 1). The Cohort 1 study participants received a supervised treatment of a 3-day regimen of Artesunate-Amodiaquine (AS + AQ, 25 mg/kg) (IPCA Laboratories, India). The first dose of AS + AQ was given on day 0 by the study team, while subsequent doses were given on day 1 and day 2 by the parent or guardian of each participant. The participants were followed for a period of 70 days, with follow-up on days 7, 14, 28, 42, 56 and 70 after initiating treatment. Finger-pricked blood samples were collected during the follow-up to prepare blood smears for microscopy examination and DBS for PCR analysis.
To assess the rate of malaria parasite clearance, 20 other participants (Cohort 2) from the cross-sectional study who harboured asexual Plasmodium parasites, based on microscopy examination, were recruited and treated with a 3-day regimen of AS + AQ (25 mg/kg) under supervision. Baseline samples were taken on day 0 (before treatment). The study participants were then treated on day 0, day 1 and day 2. Additional finger-prick blood samples were collected on days 1, 2 and 3 after initiating treatment to prepare DBS for quantitative PCR (qPCR) assay. These 20 participants (Cohort 2) are independent of the 184 participants (Cohort 1) who were followed over the 70-day period. All DBS were stored at room temperature with desiccant until ready for DNA extraction.
Microscopy examination of asexual parasites and gametocytes
The details of microscopy preparation and examination have been described previously [27]. Briefly, thick and thin blood smears were prepared at the time of blood sample collection and stained with 10% Giemsa. Asexual parasite count was determined per 200 white blood cells (WBCs), while gametocyte count was estimated per 500 WBCs. Both asexual parasite and gametocyte count per microlitre of blood was determined using the standard count of 8000 leucocytes per microlitre of blood as previously described [28].
Molecular analysis
DNA extraction
Chelex DNA extraction method was used for genomic DNA extraction following previously described protocol [29] with few modifications. Briefly, the DBS were cut and placed in Eppendorf tubes, soaked in 0.5% saponin and incubated overnight at room temperature. The samples were centrifuged and the saponin solution was discarded. The resulting DBS discs were washed three times in excess of 1X PBS. A volume of 200 µL of 2% Chelex-100 solution was added to the DBS discs and incubated at 95ºC for 30 min. The samples were then centrifuged and approximately 150 µL of the resulting supernatant containing the genomic DNA was collected for each sample and stored at − 20 ℃ until ready for PCR assays.
Nested PCR (PCRn) assays for the detection of Plasmodium species
The identification of Plasmodium species was determined retrospectively by nested PCR (PCRn) using previously reported primer sets targeting the small sub-unit ribosomal genes [30] with few modifications. Briefly, the detection of genus Plasmodium (outer PCR) was performed in a total volume of 20 µL containing 1X Standard Taq reaction buffer, 200 µM dNTPs, 0.2 µM of each of the forward and the reverse primers, 0.1 µL of OneTaq DNA polymerase (BioLabs, New England) and 2 µL of the template genomic DNA (which is approximately 0.7 µL of blood sample). For species-specific analysis, one microlitre of the resulting outer PCR amplicons was used as template in the inner PCR. The cycling conditions for both outer and inner PCR assays included an initial denaturation at 95 ℃ for 5 min followed by 35 cycles of 15 s at 95 ℃, 60 s at 58 ℃ and 90 s at 68 ℃ and a final extension at 68 ℃ for 5 min. A volume of 5 µL of the inner PCR product was separated on 1.5% agarose gel for each sample using the Quick-Load 100 bp DNA Ladder (New England, Biolabs) as the molecular marker. The resulting gels were processed using the Amersham Imager 600 (General Electric Healthcare Life Sciences, Chicago, IL, USA). PCRn assay was performed for the samples obtained from participants in the cross-sectional study (n = 1697) and Cohort 1 (n = 184).
Rate of parasite clearance using quantitative PCR (qPCR) analysis
The rate of parasite clearance after ACT for samples from Cohort 2 (n = 20) was determined using SYBR Green-based qPCR assay. This qPCR assay was performed using pan-Plasmodium primer set targeting the Plasmodium methionine transfer RNA gene which were previously used in a Taqman probe-based qPCR assay developed by Beshir et al. [31]. The detection limit of the SYBR Green-based qPCR assay was not determined in this study. The assays were performed on the Quantstudio5 system (Applied Biosystems). Each of the 20 samples had a day 0 (before treatment) sample and the corresponding day 1, day 2 and day 3 samples after initiating AS + AQ treatment. All reactions were performed in a total volume of 15 µL consisting of 1X Luna qPCR master mix (Bio Labs, New England), 0.2 µM of each primer, and 2 µL of the purified genomic DNA as template. The cycling conditions included an initial denaturation of 5 min at 95 ℃ followed by 40 cycles of 15 s at 95 ℃, and 60 s at 60 ℃. The specificity of the resulting qPCR products was analysed using the melting curves. The mean Ct-values from technical replicates were used to estimate the relative quantity of parasite genomic DNA. The relative quantity of parasite genomic DNA was estimated from the formula 2−∆∆ct using the day 0 samples (before treatment) as the reference and the human β-tubulin gene for normalization.
MSP genotyping
Plasmodium falciparum clonal diversity analysis was performed for Cohort 2 samples (day 0, day 1, day 2 and day 3) to investigate possible treatment failure using merozoite-surface protein (MSP) genotyping. MSP1 (K1, MAD20 and RO33) and MSP2 (FC27 and 3D7) allelic families were analysed by nested PCR (PCRn) using previously described protocol and primer sets [32]. Briefly, the outer PCR was performed in a 20 µL reaction volume consisting of 1X Standard Taq reaction buffer, 150 µM dNTPs, 0.15 µM of each primer, 0.1 µL of OneTaq DNA polymerase (New England, BioLabs) and 2 µL of the template DNA. The cycling conditions for the outer PCR included an initial denaturation at 95 ℃ for 5 min followed by 30 cycles of 15 s at 95 ℃, 60 s at 55 ℃ and 90 s at 68 ℃ and a final extension at 68 ℃ for 5 min. The inner PCR consisted of 0.5 µL of the outer PCR product as template DNA. Similar cycling conditions for the outer PCR were used for the inner PCR with varying annealing temperature: K1 and MAD20 at 62 ℃, RO33 at 58 ℃ and MSP2 (FC27 and 3D7) at 57 ℃.
Statistical analyses
The resulting data were analysed using the IBM SPSS Statistics v26, GraphPad Prism 8.0.2 and Microsoft Excel 2016 Software. Statistical significance for the proportion of positive samples was determined using the Chi-Square test or Fisher’s exact test. Parasite densities were log10 transformed and compared across three or more groups using one-way ANOVA test, and where differences were observed, pairwise comparison was conducted using the unpaired t-test. A multivariate analysis was performed to determine the association between age and Plasmodium infection, controlling for gender and study site. One-way ANOVA test was used to compare the mean number of clonal infections across the four-time points (days 0, 1, 2, and 3), and differences in the groups were compared by pair-wise analysis using Student’s t-test. All statistical analyses were considered significant for P < 0.05.
Results
The prevalence of Plasmodium species
The demographic, and clinical characteristics of the study participants (asymptomatic children, n = 1697) have been described in detail in the previous study [26]. The prevalence rate of Plasmodium parasite infection among the study participants in the cross-sectional study as reported in the previous study [26] by microscopy was 26.6% (451/1697) (Fig. 2A). In this study, the total prevalence rate of Plasmodium parasite infection by nested PCR (PCRn) was 33.6% (571/1697) (Fig. 2A), which was significantly higher compared to that of microscopy (Chi-square, χ2 = 20.16, P < 0.0001). Among the microscopy-positive samples, 16.2% (73/451) were negative by PCRn. Also, 33.6% (192/571) of the PCRn-positive samples were missed by microscopy. For species-specific analysis, the prevalence rates of P. falciparum, P. malariae, P. ovale and P. vivax infections by PCRn were 33.6% (570/1697), 0.1% (1/1697), 0.0% (0/1697), and 0.0% (0/1697), respectively (Fig. 2B). Of note, the distribution of P. falciparum varied from 0.0% to 82.5% among the 30 study communities (Fig. 2C). Interestingly, no Plasmodium species infection was detected among participants from three (3) of the 30 study communities by PCRn (Fig. 2C).

The prevalence rates of Plasmodium species among the study participants (n = 1697). A The prevalence of Plasmodium parasite infection determined by microscopy and nested PCR (PCRn). B Species-specific distribution of Plasmodium parasites by PCRn. C The distribution of P. falciparum across the 30 study sites by nested PCR. The 30 study sites have been numbered 1 to 30, in no particular order, with the corresponding geographical location indicated in Fig. 1
The temporal dynamics of Plasmodium species infection after treatment
To assess the dynamics of Plasmodium species infection after ACT, a total of 184 participants (Cohort 1) who harboured Plasmodium parasites, determined by microscopy, were treated with AS + AQ and followed at weekly or biweekly intervals over a period of 70 days. The prevalence rate of asexual Plasmodium parasites by microscopy on days 0, 7, 14, 28, 42, 56 and 70 were 100%, 0.0%, 1.8%, 11.7%, 12.7%, 9.9%, and 10.4%, respectively (Fig. 3A). Using PCRn, the prevalence rate of Plasmodium parasites on days 0, 7, 14, 28, 42, 56 and 70 were 83.8%, 17.4%, 51.3%, 47.3%, 52.8%, 38.8% and 21.7%, respectively (Fig. 3A). As expected, the observed prevalence rates on all the follow-up days were relatively higher by PCRn compared to the corresponding prevalence by microscopy. There was a substantial reduction in parasite prevalence on day 7 by 100% and 79.2% after treatment by microscopy and PCRn, respectively. However, an increase in parasite infection was observed from day 14 to day 42 with a subsequent decline by day 70 by both microscopy and PCRn (Fig. 3A). Based on parasite quantification analysis by microscopy, it was observed that the mean parasitaemia for individuals who harboured parasites was significantly higher on day 28 (P = 0.0003), day 42 (P = 0.0004), day 56 (P < 0.0001) and day 70 (P < 0.0001) compared to the mean parasitaemia before treatment (day 0) (Fig. 3B).

The dynamics of Plasmodium parasites after initiating artesunate-amodiaquine (AS-AQ) treatment (n = 184). A The overall prevalence of Plasmodium parasites over the 70-day period after ACT treatment by microscopy and nested PCR (PCRn). B Comparison of parasitaemia as determined by microscopy before and after treatment. Data have been presented as box plot and the geometric mean of the parasite load is denoted by the plus symbol ( +). Parasite densities were log10 transformed and compared using one-way ANOVA test for three or more groups and unpaired t-test for two groups (***P < 0.001, ****P < 0.0001 and ns represents not significant). C The prevalence of P. falciparum by age (months) over 70 days after ACT treatment. D The prevalence rate of gametocyte before and after ACT treatment
The trends in the distribution of P. falciparum by PCRn following ACT over the 70-day period were similar when participants were grouped by age (Fig. 3C). However, participants between the ages of 6–12 and 49–59 months had sporadic trends of P. falciparum distribution (Fig. 3C).
Also, the odds of P. falciparum infection within 28 days after treatment was neither associated with age nor gender (P > 0.05 for all comparisons, Table 1). None of the 184 study cohorts was found to harbour non-falciparum species before and after ACT. The prevalence rate of gametocytes as determined by microscopy on days 0, 7, 14, 28, 42, 56 and 70 were 3.8%, 1.6%, 0.0%, 0.0%, 1.1%, 0.0% and 0.5%, respectively (Fig. 3D).
The rate of parasite clearance after ACT
To determine the rate of parasite clearance, 20 participants (Cohort 2) who harboured Plasmodium parasite as determined by microscopy in the cross-sectional study were treated with AS + AQ on days 0, 1 and 2. Samples were obtained from participants on day 0 (before treatment) and days 1, 2 and 3 after initiating treatment. Using qPCR analysis, all the 20 participants were confirmed to harbour Plasmodium parasite on day 0 (before treatment). After initiating treatment, the proportion of participants that harboured detectable Plasmodium parasite genomic DNA on day 1, day 2 and day 3 were 65.0% (13/20), 65.0% (13/20) and 60.0% (12/20), respectively (Fig. 4A). Compared to day 0 (before treatment), the relative mean quantity of parasite genomic DNA on day 1, day 2, and day 3 were 0.26, 0.14, and 0.11, respectively (Fig. 4B). These indicate percentage reduction in parasite genomic DNA by 74.2%, 85.8% and 88.4% on day 1, day 2 and day 3, respectively. Although there was a decreasing trend in the mean residual parasite genomic DNA levels from day 1 to day 3 (Fig. 4B), the differences were not statistically significant (P = 0.283).

Parasite prevalence before treatment (day 0) and days 1, 2 and 3 after initiating artesunate-amodiaquine (AS-AQ) treatment using SYBR Green-based quantitative real-time PCR (qPCR) (n = 20). A Prevalence of Plasmodium parasites before treatment (day 0) and after initiating treatment. B The relative abundance of parasite genomic DNA before treatment (day 0) and days 1, 2 and 3 after initiating treatment
Plasmodium falciparum clonal diversity analysis
Following the detection of residual parasite genomic DNA on day 3 after initiating treatment, P. falciparum parasite clonal diversity (multiplicity of infection) analysis was performed to determine the likelihood of parasite recrudescence or new infection as previously reported [33,34,35,36,37,38]. Recrudescence was defined as the presence of an allele on day 3 if the same allele was present on either day 0, day 1 or day 2, while new infection was defined as the presence of an allele on day 3 if the same allele was not present on either day 0, day 1 or day 2 [33, 39]. The mean multiplicity of infection on day 0 (before treatment), day 1, day 2 and day 3 were 1.9 [Range: 1–4], 1.4 [Range: 0–3], 1.1 [Range: 0–3], and 0.8 [Range: 0–2], respectively (Fig. 5A). This result indicates a gradual reduction in parasite clonal diversity from day 0 to day 3. As expected, the number of parasite clones for all the five MSP allelic families (K1, MAD20, RO33, FC27 and 3D7) was lowest on day 3 after initiating treatment (Fig. 5B).

P. falciparum clonal infection determined by merozoite-surface protein (MSP) genotyping after initiating artesunate-amodiaquine (AS-AQ) treatment. A The average number of P. falciparum clones detected per sample before treatment (day 0) and days 1, 2 and 3 after initiating treatment. Error bars represent the standard deviation. B The relative proportion of MSP genotypes before treatment (day 0) and days 1, 2 and 3 after initiating treatment
On day 0, a total of 32 P. falciparum genotypes (K1 = 10, MAD20 = 4, RO33 = 6, FC27 = 4 and 3D7 = 8) were detected among the 20 participants. Of these, 81.2% (26/32) had adequate treatment response, while the remaining 18.8% (6/32) were classified as recrudescence infection. Five of the recrudescence infections [83.3% (5/6)] were of the K1 allelic family. In addition, eleven [11] new infections were detected on day 3, most of which were of the 3D7 allelic family [54.5% (6/11)]. Participants with residual parasite genomic DNA on day 3 resulting from either recrudescence and/or new infection generally had higher multiplicity of infection on day 0 than those with adequate treatment response, however, the difference was not statistically significant (P = 0.87).
Discussion
In this study, a cross-sectional analysis was first performed to determine the prevalence of Plasmodium species by nested PCR. As expected, the prevalence rate of Plasmodium parasites by nested PCR was significantly higher than that of microscopy. Compared to nested PCR, 33.6% of the PCR positives were misdiagnosed by microscopy. Plasmodium falciparum was the most prevalent (33.6%) Plasmodium species among the study participants. Following ACT, a significant proportion of the participants was found to harbour Plasmodium parasites, with a fluctuating prevalence rate that increased from day 14 to day 42 and a subsequent decline by day 70. Interestingly, the mean parasitaemia determined by microscopy from day 28 to day 70 was significantly higher compared to pre-treatment. Further analysis of the rate of parasite clearance indicates the presence of residual parasites or parasite genomic DNA, with a decline in clonal diversity from day 0 (before treatment) to day 3 after initiating ACT. These observations highlight the importance of routine surveillance of Plasmodium species using nucleic acid-based amplification assays to obtain reliable data that would inform the implementation of effective control measures and interventions [40].
This present study observed a considerable high number of PCRn-positive samples [33.6% (192/571)] that were undetected by microscopy and microscopy-positive samples [16.2% (73/451)] that were also missed by PCRn. These observed discrepancies may be explained by factors including the lower detection limit of PCR compared to microscopy [9], and the technical expertise and high quality control required for microscopy [41,42,43,44]. This means that even a slight fall in standards can lead to false positive or false negative results [43, 44]. For species-specific analysis, the observed prevalence rate of P. falciparum (33.6%) in this study is comparable to reports from previous studies involving asymptomatic children under 5 years in Ghana (36.8%) (25), Burkina Faso (38.2%) [45], and Nigeria (29.0%) [46]. However, other studies in Ghana reported higher prevalence among older children in the Ashanti region (66%) [15], and the Eastern region (63.8%) [47]. The differences in the observed prevalence may be due to age-dependant immunity since the pattern in the distribution of Plasmodium species infection has been associated with age [48,49,50]. In addition, variation in malaria transmission intensity across the study sites could also account for the observed differences in the prevalence of P. falciparum [10, 51, 52].
The current study also observed a very low carriage of non-falciparum species (< 0.1%) among the study participants. These frequencies are within the range of the estimated national prevalence of < 10% for P. malariae, < 2% for P. ovale and 0.0% for P. vivax in Ghana [53]. This observation corroborates findings in the Volta region where this study was performed [54] and in other studies in Ghana [16], Burkina Faso [55], Zambia [56] and Uganda [57]. On the contrary, other studies conducted elsewhere in Ghana have reported higher frequencies of non-falciparum species among asymptomatic children [14, 15]. Also, a more recent study in Nigeria among adolescents aged 10–19 years observed an unexpectedly high prevalence of 66.4% and 30.5% for P. malariae and P. ovale, respectively [22]. The differences in the dynamics of non-falciparum species across different sites could be due to the variations in population characteristics, geographic and seasonal transmission [3, 18]. In addition, the use of other PCR-based methods including cooperative primer-based assays [17] and primers targeting mitochondria DNA [58] may be suitable or optimal for the detection of non-falciparum malaria parasites since they are usually detected as low-density infection.
Using PCR analysis, we observed a significant proportion of the study participants harbouring detectable parasite genomic DNA after ACT for both the 70-day (Cohort 1) and the 3-day (Cohort 2) follow-up participants. Previous studies have also reported sub-patent parasite loads after ACT in Ghana [15], Kenya [33], Uganda [35], Angola [36] and Tanzania [38]. Another study in Zanzibar associated sub-patent parasite that was observed post-treatment with factors including parasite density at enrolment, age, baseline temperature and haemoglobin levels [59]. Due to the limited sample size (n = 20, Cohort 2) of these treated participants in the current study, these previously reported factors were not investigated. It is important to highlight that a 3-day regimen of ACT is generally known to rapidly clear asexual malaria parasites and is also expected to clear both new or developing Plasmodium infections [60,61,62]. Based on the microscopy analysis in this study, one possible explanation for the observed parasites or parasite genomic DNA during the follow-up could be due to the presence of gametocytes that were not cleared after treatment as reported in previous studies [15, 33]. In addition, there is the possibility of parasite re-infection or recrudescence after treatment [33,34,35,36,37,38]. Also, circulating parasite genomic DNA released from dead parasites could account for the observed parasite genomic DNA detected during and after treatment [63, 64].
The study further performed parasite clonal analysis using MSP genotyping to investigate the possibility of parasite re-infection or recrudescence. It is important to highlight that MSP genotyping was done for only the 3-day follow-up participants (Cohort 2) and not the 70-day follow-up participants (Cohort 1) due to financial constraints. Based on the parasite clonal diversity analysis, it was observed that the residual parasites or parasite genomic DNA detected on day 3 after initiating treatment was due to both recrudescence and new infections. This observation is in line with previous studies in Kenya [33] and Tanzania [38], where recurrent infections were associated with recrudescence and new infections. Of note, there is the possibility of underestimating recrudescent infections due to undetected parasite clones in the pre-treatment sample which become detectable after drug exposure reduced parasite density, as previously noted [33]. Over-estimating the number of recrudescent infections is theoretically possible, especially within a specific geographic location where some genetic relatedness is possible, but this is far less likely in high transmission, and high complexity settings such as Hohoe [65,66,67,68]. In addition, circulating genomic DNA from dead parasites could potentially lead to over-estimation of parasite recrudescence or re-infections [62, 63]. Other possible factors that may explain the observed “new” infections have been described in details elsewhere [33]. A further study is recommended to accurately determine the source of the residual parasites or genomic DNA after treatment.
Taken together, the results presented here suggest a high transmission intensity at the study site amidst malaria control measures and interventions [27]. These observations have been associated with resistance to anti-malarial drugs such as chloroquine and sulfadoxine-pyrimethamine [68, 69]. This calls for the routine application of sensitive molecular-based tools for monitoring anti-malarial drug efficacy.
Conclusion
The study demonstrates the importance of routine community-based surveillance of Plasmodium species using sensitive nucleic acid-based amplification assays. As expected, P. falciparum was the dominant Plasmodium species among the study participants. The residual parasites or parasite genomic DNA observed after treatment necessitates further studies among a larger study population to properly establish the efficacy of ACT in Ghana. In addition, the use of RNA-based detection methods, parasite culturing and in vitro drug assays will be necessary to accurately assess the efficacy of ACT against both asexual and sexual stage parasites in human blood.
Availability of data and materials
All data presented in this manuscript are available on reasonable request from the corresponding author.
References
WHO. World malaria report. Geneva: World Health Organization; 2021.
White NJ. Plasmodium knowlesi: the fifth human malaria parasite. Clin Infect Dis. 2008;46:172–3.
Hawadak J, Dongang Nana RR, Singh V. Global trend of Plasmodium malariae and Plasmodium ovale spp. malaria infections in the last two decades (2000–2020): a systematic review and meta-analysis. Parasit Vectors. 2021;14:297.
Andolina C, Rek JC, Briggs J, Okoth J, Musiime A, Ramjith J, et al. Sources of persistent malaria transmission in a setting with effective malaria control in eastern Uganda: a longitudinal, observational cohort study. Lancet Infect Dis. 2021;21:1568–78.
Vantaux A, Samreth R, Piv E, Khim N, Kim S, Berne L, et al. Contribution to malaria transmission of symptomatic and asymptomatic parasite carriers in Cambodia. J Infect Dis. 2018;217:1561–8.
Chiodini PL. Malaria diagnostics: now and the future. Parasitology. 2014;141:1873–9.
Tedla M. A focus on improving molecular diagnostic approaches to malaria control and elimination in low transmission settings. Parasite Epidemiol Control. 2019;6:e00107.
Gimenez AM, Marques RF, Regiart M, Bargieri DY. Diagnostic methods for non-falciparum malaria. Front Cell Infect Microbiol. 2021;11:681063.
Hofmann N, Mwingira F, Shekalaghe S, Robinson LJ, Mueller I, Felger I. Ultra-sensitive detection of Plasmodium falciparum by amplification of multi-copy subtelomeric targets. PLoS Med. 2015;12:e1001788.
Bruce MC, Macheso A, Kelly-Hope LA, Nkhoma S, McConnachie A, Molyneux ME. Effect of transmission setting and mixed species infections on clinical measures of malaria in Malawi. PLoS ONE. 2008;3:e2775.
Oboh MA, Badiane AS, Ntadom G, Ndiaye YD, Diongue K, Ndiaye D. Molecular identification of Plasmodium species responsible for malaria reveals Plasmodium vivax isolates in Duffy negative individuals from southwestern Nigeria. Malar J. 2018;17:1–2.
Fançony C, Gamboa D, Sebastião Y, Hallett R, Sutherland C, Sousa-Figueiredo JC, et al. Various pfcrt and pfmdr1 genotypes of Plasmodium falciparum cocirculate with P. malariae, P. ovale spp, and P. vivax in northern Angola. Antimicrob Agents Chemother. 2012;56:5271–7.
Oguike MC, Betson M, Burke M, Nolder D, Stothard JR, Kleinschmidt I, et al. Plasmodium ovale curtisi and Plasmodium ovale wallikeri circulate simultaneously in African communities. Int J Parasitol. 2011;41:677–83.
Amoah LE, Donu D, Abuaku B, Ahorlu C, Arhinful D, Afari E, Malm K, Koram KA. Probing the composition of Plasmodium species contained in malaria infections in the Eastern region of Ghana. BMC Public Health. 2019;19:1–1.
Dinko B, Oguike MC, Larbi JA, Bousema T, Sutherland CJ. Persistent detection of Plasmodium falciparum, P. malariae, P. ovale curtisi and P. ovale wallikeri after ACT treatment of asymptomatic Ghanaian school-children. Int J Parasitol Drugs Drug Resist. 2013;3:45–50.
Owusu ED, Brown CA, Grobusch MP, Mens P. Prevalence of Plasmodium falciparum and non-P. falciparum infections in a highland district in Ghana, and the influence of HIV and sickle cell disease. Malar J. 2017;16:167.
Ansah F, Suurbaar J, Darko D, Anabire NG, Blankson SO, Domson BK, et al. Development of cooperative primer-based real-time PCR assays for the detection of Plasmodium malariae and Plasmodium ovale. J Mol Diagnostics. 2021;23:1393–403.
Mueller I, Zimmerman PA, Reeder JC. Plasmodium malariae and Plasmodium ovale—the ‘bashful’malaria parasites. Trends Parasitol. 2007;23:278–83.
Yman V, Wandell G, Mutemi DD, Miglar A, Asghar M, Hammar U, et al. Persistent transmission of Plasmodium malariae and Plasmodium ovale species in an area of declining Plasmodium falciparum transmission in eastern Tanzania. PLoS Negl Trop Dis. 2019;13:e0007414.
Akala HM, Watson OJ, Mitei KK, Juma DW, Verity R, Ingasia LA, et al. Plasmodium interspecies interactions during a period of increasing prevalence of Plasmodium ovale in symptomatic individuals seeking treatment: an observational study. Lancet Microbe. 2021;2:e141–50.
Abdulraheem MA, Ernest M, Ugwuanyi I, Abkallo HM, Nishikawa S, Adeleke M, et al. High prevalence of Plasmodium malariae and Plasmodium ovale in co-infections with Plasmodium falciparum in asymptomatic malaria parasite carriers in southwestern Nigeria. Int J Parasitol. 2022;52:23–33.
Groger M, Veletzky L, Lalremruata A, Cattaneo C, Mischlinger J, Manego Zoleko R, et al. Prospective clinical and molecular evaluation of potential Plasmodium ovale curtisi and wallikeri relapses in a high-transmission setting. Clin Infect Dis. 2019;69:2119–26.
Ayanful-Torgby R, Quashie NB, Boampong JN, Williamson KC, Amoah LE. Seasonal variations in Plasmodium falciparum parasite prevalence assessed by varying diagnostic tests in asymptomatic children in southern Ghana. PLoS ONE. 2018;13:e0199172.
Verner O, Kokou AA, Jones OA, James OY, Ibrahim J, Innocent A, et al. Asymptomatic Plasmodium falciparum infection and poor school performance in primary school children in the Volta Region of Ghana. Ethiop J Health Sci. 2018;28:749–58.
Okyere B, Owusu-Ofori A, Ansong D, Buxton R, Benson S, Osei-Akoto A, et al. Point prevalence of asymptomatic Plasmodium infection and the comparison of microscopy, rapid diagnostic test and nested PCR for the diagnosis of asymptomatic malaria among children under 5 years in Ghana. PLoS ONE. 2020;15:e0232874.
Tarkang E, Kweku M, Takramah W, Axame WK, Owusu R, Mohammed TM, et al. Prevalence and factors associated with malaria infection among children under five in urban and rural communities in the Hohoe municipality of Ghana. Int J Case Stud Clin Res. 2017;1:78–88.
Kweku M, Appiah EK, Takramah W, Enuameh Y, Norman I, Binka F. Effect of a malaria control program on the prevalence of malaria, fever and anaemia in children under five years in the Hohoe municipality of Ghana: a comparative analysis of cross-sectional surveys. Adv Infect Dis. 2015;5:180.
Greenwood BM, Armstrong JR. Comparison of two simple methods for determining malaria parasite density. Trans R Soc Trop Med Hyg. 1991;85:186–8.
Plowe CV, Djimde A, Bouare M, Doumbo O, Wellems TE. Pyrimethamine and proguanil resistance-conferring mutations in Plasmodium falciparum dihydrofolate reductase: polymerase chain reaction methods for surveillance in Africa. Am J Trop Med Hyg. 1995;52:565–8.
Snounou G, Viriyakosol S, Zhu XP, Jarra W, Pinheiro L, do Rosario VE, et al. High sensitivity of detection of human malaria parasites by the use of nested polymerase chain reaction. Mol Biochem Parasitol. 1993;61:315–20.
Beshir KB, Hallett RL, Eziefula AC, Bailey R, Watson J, Wright SG, et al. Measuring the efficacy of anti-malarial drugs in vivo: quantitative PCR measurement of parasite clearance. Malar J. 2010;9:312.
Soulama I, Nébié I, Ouédraogo A, Gansane A, Diarra A, Tiono AB, et al. Plasmodium falciparum genotypes diversity in symptomatic malaria of children living in an urban and a rural setting in Burkina Faso. Malar J. 2009;8:135.
Beshir KB, Sutherland CJ, Sawa P, Drakeley CJ, Okell L, Mweresa CK, et al. Residual Plasmodium falciparum parasitemia in Kenyan children after artemisinin-combination therapy is associated with increased transmission to mosquitoes and parasite recurrence. J Infect Dis. 2013;208:2017–24.
Carlsson AM, Ngasala BE, Dahlström S, Membi C, Veiga IM, Rombo L, et al. Plasmodium falciparum population dynamics during the early phase of anti-malarial drug treatment in Tanzanian children with acute uncomplicated malaria. Malar J. 2011;10:380.
Chang HH, Meibalan E, Zelin J, Daniels R, Eziefula AC, Meyer EC, et al. Persistence of Plasmodium falciparum parasitemia after artemisinin combination therapy: evidence from a randomized trial in Uganda. Sci Rep. 2016;6:26330.
Kiaco K, Teixeira J, Machado M, Do Rosário V, Lopes D. Evaluation of artemether-lumefantrine efficacy in the treatment of uncomplicated malaria and its association with pfmdr1, pfatpase6 and K13-propeller polymorphisms in Luanda, Angola. Malar J. 2015;14:504.
Mwaiswelo R, Ngasala B. Evaluation of residual submicroscopic Plasmodium falciparum parasites 3 days after initiation of treatment with artemisinin-based combination therapy. Malar J. 2020;19:162.
Mwaiswelo R, Ngasala B, Jovel I, Xu W, Larsson E, Malmberg M, et al. Prevalence of and risk factors associated with Polymerase Chain Reaction-determined Plasmodium falciparum positivity on Day 3 after initiation of artemether–lumefantrine treatment for uncomplicated malaria in Bagamoyo District, Tanzania. Am J Trop Med Hyg. 2019;100:1179–86.
WHO. Methods and techniques for clinical trials on antimalarial drug efficacy: genotyping to identify parasite populations: informal consultation organized by the Medicines for Malaria Venture and cosponsored by the World Health Organization, 29–31 May 2007, Amsterdam, The Netherlands. Geneva, World Health Organization; 2008.
WHO. Disease surveillance for malaria control. Malaria treatment guidelinestment guidelines. Geneva: World Health Organization. 2013.
Maguire JD, Lederman ER, Barcus MJ, O’Meara WA, Jordon RG, Duong S, et al. Production and validation of durable, high quality standardized malaria microscopy slides for teaching, testing and quality assurance during an era of declining diagnostic proficiency. Malar J. 2006;5:92.
Durrhelm DN, Becker PJ, Billinghurst K, Brink A. Diagnostic disagreement–the lessons learnt from malaria diagnosis in Mpumalanga. South African Med J. 1997;87:609–11.
Prescott WR, Jordan RG, Grobusch MP, Chinchilli VM, Kleinschmidt I, Borovsky J, et al. Performance of a malaria microscopy image analysis slide reading device. Malar J. 2012;11:155.
WHO. Malaria microscopy quality assurance manual-version 2. Geneva: World Health Organization; 2016.
Ouédraogo M, Samadoulougou S, Rouamba T, Hien H, Sawadogo JE, Tinto H, et al. Spatial distribution and determinants of asymptomatic malaria risk among children under 5 years in 24 districts in Burkina Faso. Malar J. 2018;17:460.
Akinbo FO, Emekaili DO, Mbarie AM, Ogbogu MI. Asymptomatic malaria in children under 5 years old in Benin City, Nigeria Savannah. J Med Res Pract. 2015;4:66–71.
Owusu ED, Buabeng V, Dadzie S, Brown CA, Grobusch MP, Mens P. Characteristics of asymptomatic Plasmodium spp. parasitaemia in Kwahu-Mpraeso, a malaria endemic mountainous district in Ghana, West Africa. Malar J. 2016;15:38.
Stanisic DI, Fowkes FJ, Koinari M, Javati S, Lin E, Kiniboro B, et al. Acquisition of antibodies against Plasmodium falciparum merozoites and malaria immunity in young children and the influence of age, force of infection, and magnitude of response. Infect Immun. 2015;83:646–60.
Nhabomba AJ, Guinovart C, Jiménez A, Manaca MN, Quintó L, Cisteró P, et al. Impact of age of first exposure to Plasmodium falciparum on antibody responses to malaria in children: a randomized, controlled trial in Mozambique. Malar J. 2014;13:121.
Aponte JJ, Menendez C, Schellenberg D, Kahigwa E, Mshinda H, Vountasou P, et al. Age interactions in the development of naturally acquired immunity to Plasmodium falciparum and its clinical presentation. PLoS Med. 2007;4:e242.
Betson M, Clifford S, Stanton M, Kabatereine NB, Stothard JR. Emergence of non-falciparum Plasmodium infection despite regular artemisinin combination therapy in an 18-month longitudinal study of Ugandan children and their mothers. J Infect Dis. 2018;217:1099–109.
Bruce MC, Donnelly CA, Packer M, Lagog M, Gibson N, Narara A, et al. Age-and species-specific duration of infection in asymptomatic malaria infections in Papua New Guinea. Parasitology. 2000;121:247–56.
WHO. Malaria country profiles 2011. Geneva: World Health Organization; 2011.
Dinko B, Ayivor-Djanie R, Abugri J, Agboli E, Kye-Duodu G, Tagboto S, et al. Comparison of malaria diagnostic methods in four hospitals in the Volta region of Ghana. Malar World J. 2016;7:5.
Geiger C, Agustar HK, Compaoré G, Coulibaly B, Sié A, Becher H, Lanzer M, Jänisch T. Declining malaria parasite prevalence and trends of asymptomatic parasitaemia in a seasonal transmission setting in north-western Burkina Faso between 2000 and 2009–2012. Malar J. 2013;12:27.
Sitali L, Miller JM, Mwenda MC, Bridges DJ, Hawela MB, Hamainza B, et al. Distribution of Plasmodium species and assessment of performance of diagnostic tools used during a malaria survey in Southern and Western Provinces of Zambia. Malar J. 2019;18:130.
Pullan RL, Bukirwa H, Staedke SG, Snow RW, Brooker S. Plasmodium infection and its risk factors in eastern Uganda. Malar J. 2010;9:2.
Msellem M, Morris U, Soe A, Abbas FB, Ali AW, Barnes R, et al. Increased sensitivity of Plasmodium falciparum to artesunate/amodiaquine despite 14 years as first-line malaria treatment, Zanzibar. Emerg Infect Dis. 2020;26:1767.
Djimdé A, Lefèvre G. Understanding the pharmacokinetics of Coartem®. Malar J. 2009;8(Suppl 1):S1.
Ezzet F, Mull R, Karbwang J. Population pharmacokinetics and therapeutic response of CGP 56697 (artemether+ benflumetol) in malaria patients. Br J Clin Pharmacol. 1998;46:553–61.
Ezzet F, Van Vugt M, Nosten F, Looareesuwan S, White NJ. Pharmacokinetics and pharmacodynamics of lumefantrine (benflumetol) in acute falciparum malaria. Antimicrob Agents Chemother. 2000;44:697–704.
Haanshuus CG, Mørch K. Detection of remaining Plasmodium DNA and gametocytes during follow up after curative malaria treatment among returned travellers in Norway. Malar J. 2020;19:296.
Homann MV, Emami SN, Yman V, Stenström C, Sondén K, Ramström H, et al. Detection of malaria parasites after treatment in travelers: a 12-months longitudinal study and statistical modelling analysis. EBioMedicine. 2017;25:66–72.
Juliano JJ, Gadalla N, Sutherland CJ, Meshnick SR. The perils of PCR: can we accurately ‘correct’ antimalarial trials? Trends Parasitol. 2010;26:119–24.
Juliano JJ, Ariey F, Sem R, Tangpukdee N, Krudsood S, Olson C, et al. Misclassification of drug failure in Plasmodium falciparum clinical trials in southeast Asia. J Infect Dis. 2009;200:624–8.
Greenhouse B, Dokomajilar C, Hubbard A, Rosenthal PJ, Dorsey G. Impact of transmission intensity on the accuracy of genotyping to distinguish recrudescence from new infection in antimalarial clinical trials. Antimicrob Agents Chemother. 2007;51:3096–103.
Kwiek JJ, Alker AP, Wenink EC, Chaponda M, Kalilani LV, Meshnick SR. Estimating true antimalarial efficacy by heteroduplex tracking assay in patients with complex Plasmodium falciparum infections. Antimicrob Agents Chemother. 2007;51:521–7.
Sutherland CJ, Alloueche A, Curtis J, Drakeley CJ, Ord R, Duraisingh M, et al. Gambian children successfully treated with chloroquine can harbor and transmit Plasmodium falciparum gametocytes carrying resistance genes. Am J Trop Med Hyg. 2002;67:578–85.
Bousema JT, Gouagna LC, Drakeley CJ, Meutstege AM, Okech BA, Akim IN, et al. Plasmodium falciparum gametocyte carriage in asymptomatic children in western Kenya. Malar J. 2004;3:18.
Acknowledgements
The authors are grateful to the Hohoe Municipal Health Management Team for assisting with the survey. Authors are also grateful to the study communities, field workers, the study participants and their parents or guardians for the samples.
Funding
This study was supported by a DELTAS Africa grant (DEL-15-007: Awandare). The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS)’s Alliance for Accelerating Excellence in Science in Africa (AESA) and supported by the New Partnership for Africa's Development Planning and Coordinating Agency (NEPAD Agency) with funding from the Wellcome Trust (107755/Z/15/Z: Awandare) and the UK government. BD and CJS are funded by Wellcome Trust (110090/Z/15/Z). The views expressed in this publication are those of the authors and not necessarily those of AAS, NEPAD Agency, Wellcome Trust or the UK government.
Author information
Authors and Affiliations
Contributions
BD, MK, GAA, FNB, and CS designed the study; F.A, KN, RL, RO, DA, JB, DA, NR, VEK, SA, COA, J.D.C, JA, YA and NO performed molecular analysis and data acquisition; FA, NO and BD participated in data analysis; FA and BD participated in the initial drafting and revision of the manuscript. All authors read and approved the final draft.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval to conduct this study was obtained from the ethical review committee of the Ghana Health Service, Accra (GHS-ERC:14/05/15) and a written consent to participate in the study was obtained from the parents and guardians of all participants before enrolment into the study.
Consent for publication
All authors have consented to publishing these data.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ansah, F., Nyame, K., Laryea, R. et al. The temporal dynamics of Plasmodium species infection after artemisinin-based combination therapy (ACT) among asymptomatic children in the Hohoe municipality, Ghana. Malar J 22, 271 (2023). https://doi.org/10.1186/s12936-023-04712-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-023-04712-1