
Abstract
Background
Vector populations are a key target for malaria control and elimination. In Honduras, there are at least 12 reported anopheline species, however, the definitive number of species remains uncertain. Due to the inherent limitations of morphological identification of Anopheles species, molecular approaches have been developed to provide accurate identification and robust surveillance of local malaria vectors. The aim of this study was to design and assess three PCR–RFLP assays to identify anopheline species known to presently occur in Honduras.
Methods
Mosquitoes captured between 2018 and 2022 in seven malaria-endemic and non-endemic departments in Honduras were analysed. The ITS2 ribosomal region and three restriction enzyme-based assays were evaluated in silico and experimentally.
Results
A total of 132 sequences from 12 anopheline species were analysed. The ITS2 marker showed length polymorphisms that generated products between 388 and 592 bp and no relevant intraspecies polymorphisms were found. Furthermore, the three PCR–RFLP assays were able to differentiate 11 species with sufficient precision and resolution.
Conclusion
The ITS2 region was shown to be a useful molecular marker for identifying local Anopheles species. In addition, the PCR–RFLP assays evaluated here proved to be capable of discriminating most of the anopheline species present in Honduras. These methods provide alternatives to improve entomological surveillance of Anopheles in Honduras and other Mesoamerican countries.
Similar content being viewed by others

Background
Anopheles mosquitoes are the only vectors of human malaria and are the main target of national programmes to achieve its control and elimination [1, 2]. Anopheles is a highly biodiverse genus with more than 500 recognized species and others yet to be characterized, particularly cryptic species grouped in complexes [3, 4]. Approximately 70 Anopheles species have been incriminated as vectors of Plasmodium spp. transmitting the parasite to humans [3]. Anopheles species have adapted to the ecological conditions of almost all regions of the world and their distribution is highly heterogeneous. Likewise, malaria parasites have adapted to survive immunological challenges within the vector species with which they interact in each region [5, 6].
The definitive distribution and number of Anopheles species in Honduras have not yet been determined, although several sources report the occurrence of 12 to 14 anopheline species in the country, however, none of them fully agree. According to reports from the Ministry of Health of Honduras and based on entomological surveillance activities in the last 10 years [7,8,9], there are 14 native anopheline species: four of the subgenus Nyssorhynchus (Anopheles albimanus, Anopheles darlingi, Anopheles argyritarsis, Anopheles albitarsis), nine of the subgenus Anopheles (Anopheles vestitipennis, Anopheles pseudopunctipennis, Anopheles punctimacula, Anopheles crucians, Anopheles apicimacula, Anopheles neomaculipalpus, Anopheles gabaldoni, Anopheles grabhamii, Anopheles eiseni), and one of the subgenus Kerteszia (Anopheles neivai) is the only representative of the subgenus (information taken from the reports made by the technicians in charge of entomological surveillance of the Honduran Ministry of Health). All these species have been described as malaria vectors in the Neotropics, but Honduras's dominant vector species is An. albimanus [3].
On the other hand, the database of The Walter Reed Biosystematics Unit (WRBU) [10] and the taxonomic key of female anophelines from Central America and Mexico [8] report 13 species present in Honduras, but six of them are not consistent with those described by the national authorities [11]. Anopheles albitarsis, An. gabaldoni, An. grabhamii and An. neivai are not included in the WRBU or taxonomic key, while Anopheles bradleyi, Anopheles hectoris and Anopheles strodei are included in the WRBU/taxonomic key but have not been reported by local entomologists. Added to this, three species are not included in the ‘Taxonomy’ tool of the National Centre for Biotechnology Information (NCBI) (An. gabaldoni, An. grabhamii, and An. hectoris). The NCBI taxonomy includes organism names and classifications for each sequence in the nucleotide and protein sequence databases of the International Nucleotide Sequence Database Collaboration (INSDC) and provides a framework for grouping elements within other domains for links to external web resources specific to relevant taxa and publications [12]. Also, three species have no available sequences for the internal transcribed spacer (ITS2) in the NCBI database (An. bradleyi, An. eiseni, and An. strodei).
Finally, the parasitology research group of the National University captures and identifies anophelines since 2018 in seven of the 18 departments of the country (endemic and non-endemic for malaria) and at this stage has captured and identified 10 species of Anopheles (Table 1) [7,8,9]. These discrepancies could reflect the limitations of the taxonomy of some native species, as well as the difficulty or impossibility of distinguishing anophelines based exclusively on morphological characteristics [13,14,15]. Morphological identification has demonstrated some disadvantages such as being time-consuming and requiring trained specialists; added to this, the defining anatomical structures are not always available or in good condition [16].
These limitations have led to the development and incorporation of molecular tools that facilitate and speed up the identification of vectors in areas of high diversity. Multiplex PCR, allele-specific PCR and sequencing have been successfully used to diagnose Anopheles species in several countries [17,18,19,20,21,22,23,24,25,26,27,28,29,30,31]. Other assays are based on the digestion with restriction enzymes of DNA sequences such as ITS2, the cytochrome c oxidase subunits I and II, and the D3 region of the nuclear 28S rDNA gene [21, 32,33,34,35,36,37,38,39]. In the Americas, for example, Vezenegho et al. [32] were able to discriminate between 15 anopheline species from French Guiana using a combination of two restriction enzymes; in Colombia, Zapata et al. [35] managed to diagnose seven species with a single restriction enzyme; and Cienfuegos et al. [40] found congruent morphological and molecular identification results of four Anopheles species collected in northern and western Colombia using several restriction enzymes.
The PCR–RFLP is an easy and inexpensive approach compared to Sanger sequencing, which allows a large number of individuals to be processed, even if the complete insect is not available. In addition, it allows the identification of the immature stages of the mosquito, usually lacking discriminative morphological characteristics. In this study, three PCR–RFLP assays were developed and tested to easily identify 11 of the 12 most common anopheline species reported to occur in Honduras.
Methods
Mosquito collection
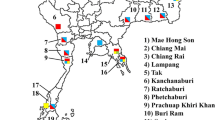
Anopheline mosquitoes were captured between 2018 and 2022 in seven departments of Honduras: Gracias a Dios, Colón, Bay Islands, Comayagua, El Paraíso, Atlántida, and Cortés (Fig. 1). The geographical coordinates and the species collected in each department are shown in Table 1. The mosquitoes were collected in the peridomicile and extradomicile during the night. Two collection methods were used: the Center for Disease Control trap (CDC-LT) [41] provided with light as an attractant, and aspiration of resting mosquitoes, sometimes complemented by human landing catch. Mosquitoes were separated and stored dry in 1.5 mL tubes and subsequently identified under a stereoscope using morphological keys for female anophelines from Central America and Mexico [11].

Map of Honduras showing the seven departments where mosquitoes were collected
Specimens of An. gabaldoni included in this study originate from Mexico (municipality of Othón P. Blanco, Quintana Roo), because none could be captured within Honduran territory over the study time frame. Additionally, it was not possible to have any specimen of An. albitarsis, thus molecular analyses were based on two sequences downloaded from the NCBI database (Acc. Numbers AF462385.1, AF462387.1) and uploaded by researchers from Brazil.
DNA extraction
Most of the mosquitoes’ DNA analysed in this study came from the DNA bank of the Genetic Research Centre of the National University of Honduras [7, 9]. For those Anopheles species for which DNA was not available, genomic DNA was extracted from freshly collected specimens. Both the DNA of the historical specimens and that of the mosquitoes captured for this study were individually extracted following the protocol of the DNeasy Blood and Tissue Kit® (QIAGEN, Hilden, Germany). The heads and thorax of each mosquito were dissected. Single maceration was carried out using a pestle and a 1.5 mL conical tube following manufacturer’s instructions. Overnight lysis at 56 °C was carried out. DNA was eluted in 150 µL of elution buffer and stored at − 20 °C until further use.
PCR
In order to assess whether it was possible to molecularly differentiate between the Anopheles species described in Honduras, the nuclear ribosomal internal transcribed spacer 2 (ITS2) was amplified [42]. PCR reactions were performed using the universal primers: 5.8S (5′-ATC ACT CGG CTC GTG GAT CG-3′) and 28S (5′-ATG CTT AAA TTT AGG GGG TAG TC-3′). The enzymatic reactions contained the following reagents: 25 μL of Taq Master Mix 2X (Promega Corp., Madison, WI, USA), 2 μL of each primer (10 μM), 2 μL of DNA (40 ng/μL), and 19 μL of nuclease-free water for a total reaction volume of 50 μL. PCR amplifications were performed under the following conditions: 94 °C for 5 min, 34 cycles of 94 °C for 30 s, 57 °C for 30 s, 72 °C for 30 s, and a final extension cycle of 72 °C for 10 min. Successful amplifications were confirmed on a 1.5% agarose gel with ethidium bromide. The size of the amplification products was assessed by comparison with a 100 bp ladder. Amplicons were sequenced in both directions using the same PCR primers. Sanger sequencing and purification services were provided by Psomagen (https://www.psomagen.com).
In-silico analyses
The sequences were trimmed and analysed with the Geneious® 9.1.7 software (Biomatters Ltd, Auckland, New Zealand). Both strands were aligned, and a consensus sequence was obtained for each specimen. Consensus sequences were edited so that they were delimited by the same primers with which they were amplified. The size of the amplicons by species was determined and subjected to three in-silico restriction analyses with the enzymes AluI (AGˆCT), DdeI (CˆTNAG), and a combination of AluI with MseI (TˆTAA), as described elsewhere [32, 33, 35] using the Geneious® 9.1.7 software. Restriction patterns were established for each Anopheles species included in this study, and the intraspecific variability was evaluated.
PCR–RFLP
To validate the prediction of the in-silico restriction profiles, experimental restriction assays were performed by digesting the ITS2 amplicons of each specimen with the above-mentioned enzymes: AluI (Promega, Corp.), DdeI (Promega, Corp.), and AluI/MseI (Thermo Scientific). The enzymatic reactions were carried out in a final volume of 20 μL, containing 2 μL of buffer (Tango buffer, Thermo Scientific for AluI and MseI, and buffer D for DdeI), 0.5 μL of each enzyme at a concentration of 10 units/μL, 10 μL of the PCR product, and 7 µL of nuclease-free water. The microtubes were incubated at 37 °C for 4 h and the digestion products were separated on a 2.5% agarose gel running at 70 V for 90 min. A molecular weight ladder of 100 bp was used. Restriction patterns were recorded on an image analyzer under ultraviolet light.
Results and discussion
As shown in Table 2, the size of the amplification products of the ITS2 region ranged between 388 and 592 bp. These size polymorphisms could be sufficient to distinguish some anopheline species, such as An. crucians, An. punctimacula, and An. neomaculipalpus with products of 388, 445, and 504 bp, respectively. However, the size differences between An. albimanus and An. pseudopunctipennis (561–565 bp), and between An. argyritarsis, An. albitarsis, An. apicimacula and An. gabaldoni (526–536 bp), as well as between An. darlingi, An. vestitipennis and An. neivai (583–592 bp), are not enough for a clear diagnosis of each of the species [35].
A heterogeneous number of individuals of each species were sequenced (from 1 to 76) as shown in Table 2. The sequences were aligned to determine the number of intraspecific polymorphic sites. Polymorphic sites were identified in three species (three for An. vestitipennis and An. pseudopunctipennis, and six for An. albitarsis). The remaining eight species (excluding An. apicimacula) had 100% identical intraspecific sequences (Table 2). None of the polymorphisms found in the three species matched any target of the three restriction enzymes used in this study. This result suggests the reproducibility of the methods regardless of the intraspecific diversity of the mosquitoes. However, considering the low number of specimens for some of the species, it is necessary to be cautious since the real diversity of the ITS2 marker could be under-represented in the present study. Although the ITS2 region generally shows low intraspecific variation, making it suitable for the identification of closely related Anopheles species [43], future studies, including more specimens collected from a larger geographic area, may confirm the robustness of the techniques proposed here.
All three assays tested in the present study showed consistent results, with unique species-specific digestion patterns allowing for the differentiation of 10 of the anopheline species (Fig. 2). Despite this, some patterns can be difficult to distinguish in practice. The assay with AluI might be insufficient to distinguish between An. albimanus (366, 162 bp) and An. argyritarsis (336, 157 bp), as well as between An. neomaculipalpus (255, 136 bp) and An. albitarsis (267, 150 bp) even in gels with a higher concentration of agarose. Determining the size of the amplification product together with analysing the AluI restriction pattern would resolve the issue in both above-mentioned cases (Table 2). The use of two of the three PCR–RFLP assays could also help to resolve those cases in which there were doubts. The same situation occurs in the profiles generated by DdeI for An. argyritarsis (417, 112 bp), An. apicimacula (408, 118 bp), and An. albitarsis (431, 118 bp), but in this case, the size of the amplicon does not resolve the conflict. Two species still exhibited a potentially confounding pattern in the two-enzyme assay (AluI and MseI). Anopheles argyritarsis (240, 157 bp), An. apicimacula (228, 120 bp) and the amplification product size do not differ between species either. In addition to this, the pattern of An. neivai/An. vestitipennis (247, 147 bp) is too similar to the pattern of An. albitarsis (242, 143 bp).

In-silico and experimental analysis of PCR–RFLP using the enzymes a AluI, b DdeI and c AluI and MseI. Lanes 1 to 12 correspond to An. albimanus, An. darlingi, An. argyritarsis, An. vestitipennis, An. pseudopunctipennis, An. punctimacula, An. crucians, An. apicimacula, An. neomaculipalpus, An. neivai, An. gabaldoni, and An. albitarsis. The restriction profile of An. albitarsis is not shown on agarose gels. A molecular weight marker of 100 bp was used
Finally, none of the tested assays allowed to differentiate between An. vestitipennis and An. neivai. When comparing the sequences of the ITS2 region of both species, no diagnostic SNPs was found, showing 579 of 583 identical sites (99.1%). This was an unexpected finding, as both species belong to different subgenera (An. (An.) vestitipennis vs An. (Kert.) neivai) and are not considered cryptic species. In fact, both species are relatively easily differentiated according to the presence, distribution and colour of the scales on the thorax and abdomen, and the patterns of spots on the femora, tibiae and scutum, among other morphological structures [11].
Fourteen sequences of the COI gene of both species’ specimens collected previously in Honduras were further compared [7, 9]. These COI sequences showed that 611 of 627 nucleotides were identical (98.7%) between the two species. Similar or identical COI sequences have been shown in other insect taxa [16], confirming the difficulties inherent in the taxonomy of arthropods, with a high fraction of species or complexes yet to be defined. This challenge is aggravated by the scarce availability of records of specimens for some species. This limitation may be due to the fact that An. neivai and An. vestitipennis are distributed in poorly studied regions of the Neotropics. Consequently, the DNA sequence reference database is poorly developed and might not reflect the intraspecific genetic diversity of the species, which restricts barcoding gap analyses. Other molecular markers should be analysed to unravel the situation.
Conclusions
Overall, the ITS2 region confirms to be an informative DNA fragment with low intraspecific variability. The three PCR–RFLP assays appear to be useful in discriminating most of the anopheline species distributed in Honduras and neighbouring Mesoamerican countries and, therefore, could be useful to medical entomologists in Southern Mexico and Central America as a tool to identify mosquitoes in the context of vector control activities. However, the AluI enzyme has the highest resolving power when the restriction profiles are analysed together with the size of the amplification product of ITS2. A combination of two or three trials might be advisable in cases where the restriction patterns of a single trial are not conclusive.
Availability of data and materials
Not applicable.
Abbreviations
- CDC:
-
Centers for Disease Control and Prevention
- ITS2:
-
Nuclear ribosomal internal transcribed spacer 2
- MASL:
-
Metres above sea level
- PCR–RFLP:
-
Polymerase chain reaction-restriction fragment length polymorphism
- SNPs:
-
Single nucleotide polymorphisms
References
Monroe A, Williams NA, Ogoma S, Karema C, Okumu F. Reflections on the 2021 World Malaria Report and the future of malaria control. Malar J. 2022;21:154.
World Health Organization. World malaria report 2021. Geneva: World Health Organization; 2021.
Sinka ME, Bangs MJ, Manguin S, Rubio-Palis Y, Chareonviriyaphap T, Coetzee M, et al. A global map of dominant malaria vectors. Parasit Vectors. 2012;5:69.
Hay SI, Sinka ME, Okara RM, Kabaria CW, Mbithi PM, Tago CC, et al. Developing global maps of the dominant Anopheles vectors of human malaria. PLoS Med. 2010;7: e1000209.
Molina-Cruz A, Barillas-Mury C. Pfs47 as a malaria transmission-blocking vaccine target. Am J Trop Med Hyg 2022;89:8.
Molina-Cruz A, Canepa GE, Alves ESTL, Williams AE, Nagyal S, Yenkoidiok-Douti L, et al. Plasmodium falciparum evades immunity of anopheline mosquitoes by interacting with a Pfs47 midgut receptor. Proc Natl Acad Sci USA. 2020;117:2597–605.
Escobar D, Archaga O, Reyes A, Palma A, Larson RT, Vasquez GM, et al. A follow-up to the geographical distribution of Anopheles species in malaria-endemic and non-endemic areas of Honduras. Insects. 2022;13:548.
Escobar D, Ascencio K, Ortiz A, Palma A, Sanchez A, Fontecha G. Blood meal sources of Anopheles spp. in malaria endemic areas of Honduras. Insects. 2020;11:450.
Escobar D, Ascencio K, Ortiz A, Palma A, Fontecha G. Distribution and phylogenetic diversity of Anopheles species in malaria endemic areas of Honduras in an elimination setting. Parasit Vectors. 2020;13:333.
WRBU. https://www.wrbu.si.edu.
Wilkerson RC, Strickman D, Litwak TR. Illustrated key to the female anopheline mosquitoes of Central America and Mexico. J Am Mosq Control Assoc. 1990;6:7–34.
NCBI Taxonomy. In: Taxonomy Help. Bethesda (MD): National Center for Biotechnology Information (US). 2011.
Kengne P, Antonio-Nkondjio C, Awono-Ambene HP, Simard F, Awolola TS, Fontenille D. Molecular differentiation of three closely related members of the mosquito species complex, Anopheles moucheti, by mitochondrial and ribosomal DNA polymorphism. Med Vet Entomol. 2007;21:177–82.
Zhang C, Luo C, Yang R, Yang Y, Guo X, Deng Y, et al. Morphological and molecular identification reveals a high diversity of Anopheles species in the forest region of the Cambodia-Laos border. Parasit Vectors. 2022;15:94.
Jeong KY, Un S, Lee J, Lee IY, Yong TS, Ree HI. Population dynamics of five Anopheles species of the Hyrcanus group in northern Gyeonggi-do. Korea Korean J Parasitol. 2010;48:351–3.
Wiemers M, Fiedler K. Does the DNA barcoding gap exist? - a case study in blue butterflies (Lepidoptera: Lycaenidae). Front Zool. 2007;4:8.
Pramasivan S, Liew JWK, Jeyaprakasam NK, Low VL, Ngui R, Vythilingam I. Multiplex PCR assay for the identification of four species of the Anopheles leucosphyrus sub-group in Malaysia. Insects. 2022;13:195.
Weitzel T, Gauch C, Becker N. Identification of Anopheles daciae in Germany through ITS2 sequencing. Parasitol Res. 2012;111:2431–8.
Bang WJ, Kim HC, Ryu J, Lee HS, Lee SY, Kim MS, et al. Multiplex PCR assay for the identification of eight Anopheles species belonging to the Hyrcanus. Barbirostris and Lindesayi groups Malar J. 2021;20:287.
Ivanescu ML, Acatrinei D, Pavel I, Sulesco T, Miron L. PCR identification of five species from the Anopheles maculipennis complex (Diptera: Culicidae) in North-Eastern Romania. Acta Parasitol. 2015;60:283–9.
Goswami G, Singh OP, Nanda N, Raghavendra K, Gakhar SK, Subbarao SK. Identification of all members of the Anopheles culicifacies complex using allele-specific polymerase chain reaction assays. Am J Trop Med Hyg. 2006;75:454–60.
Carter TE, Yared S, Hansel S, Lopez K, Janies D. Sequence-based identification of Anopheles species in eastern Ethiopia. Malar J. 2019;18:135.
Surendran SN, Sarma DK, Jude PJ, Kemppainen P, Kanthakumaran N, Gajapathy K, Peiris LB, Ramasamy R, Walton C. Molecular characterization and identification of members of the Anopheles subpictus complex in Sri Lanka. Malar J. 2013;12:304.
Ali RSM, Wahid I, Saingamsook J, Saeung A, Wannasan A, Walton C, et al. Molecular identification of mosquitoes of the Anopheles maculatus group of subgenus Cellia (Diptera: Culicidae) in the Indonesian Archipelago. Acta Trop. 2019;199: 105124.
Henry-Halldin CN, Reimer L, Thomsen E, Koimbu G, Zimmerman A, Keven JB, et al. High throughput multiplex assay for species identification of Papua New Guinea malaria vectors: members of the Anopheles punctulatus (Diptera: Culicidae) species group. Am J Trop Med Hyg. 2011;84:166–73.
Walton C, Somboon P, O’Loughlin SM, Zhang S, Harbach RE, Linton YM, et al. Genetic diversity and molecular identification of mosquito species in the Anopheles maculatus group using the ITS2 region of rDNA. Infect Genet Evol. 2007;7:93–102.
Dusfour I, Blondeau J, Harbach RE, Vythilingham I, Baimai V, Trung HD, et al. Polymerase chain reaction identification of three members of the Anopheles sundaicus (Diptera: Culicidae) complex, malaria vectors in Southeast Asia. J Med Entomol. 2007;44:723–31.
Ma Y, Li S, Xu J. Molecular identification and phylogeny of the Maculatus group of Anopheles mosquitoes (Diptera: Culicidae) based on nuclear and mitochondrial DNA sequences. Acta Trop. 2006;99:272–80.
Swain SN, Makunin A, Dora AS, Barik TK. SNP barcoding based on decision tree algorithm: a new tool for identification of mosquito species with special reference to Anopheles. Acta Trop. 2019;199: 105152.
Imanishi N, Higa Y, Teng HJ, Sunahara T, Minakawa N. Identification of three distinct groups of Anopheles lindesayi in Japan by morphological and genetic analyses. Jpn J Infect Dis. 2018;71:427–35.
Phunngam P, Boonkue U, Chareonviriyaphap T, Bangs MJ, Arunyawat U. Molecular identification of four members of the Anopheles dirus complex using the mitochondrial cytochrome C oxidase subunit I gene. J Am Mosq Control Assoc. 2017;33:263–9.
Vezenegho SB, Issaly J, Carinci R, Gaborit P, Girod R, Dusfour I, Briolant S. Discrimination of 15 Amazonian anopheline mosquito species by polymerase chain reaction-restriction fragment length polymorphism. J Med Entomol. 2022;59:1060–4.
Xu S, Zhou HY, Tang JX, Li JL, Zhu GD, Su YP, Huang GQ, Cao J, Gao Q. [Molecular identification of Anopheles hyrcanus complex by using single enzyme digestion PCR-RFLP method](in Chinese). Zhongguo Xue Xi Chong Bing Fang Zhi Za Zhi. 2012;24:435–9.
Choi KS, Coetzee M, Koekemoer LL. Simultaneous identification of the Anopheles funestus group and Anopheles longipalpis type C by PCR-RFLP. Malar J. 2010;9:316.
Zapata MA, Cienfuegos AV, Quiros OI, Quinones ML, Luckhart S, Correa MM. Discrimination of seven Anopheles species from San Pedro de Uraba, Antioquia, Colombia, by polymerase chain reaction-restriction fragment length polymorphism analysis of its sequences. Am J Trop Med Hyg. 2007;77:67–72.
Alam MT, Das MK, Dev V, Ansari MA, Sharma YD. PCR-RFLP method for the identification of four members of the Anopheles annularis group of mosquitoes (Diptera: Culicidae). Trans R Soc Trop Med Hyg. 2007;101:239–44.
Alam MT, Das MK, Dev V, Ansari MA, Sharma YD. Identification of two cryptic species in the Anopheles (Cellia) annularis complex using ribosomal DNA PCR-RFLP. Parasitol Res. 2007;100:943–8.
Goswami G, Raghavendra K, Nanda N, Gakhar SK, Subbarao SK. PCR-RFLP of mitochondrial cytochrome oxidase subunit II and ITS2 of ribosomal DNA: markers for the identification of members of the Anopheles culicifacies complex (Diptera: Culicidae). Acta Trop. 2005;95:92–9.
Fanello C, Santolamazza F, della Torre A. Simultaneous identification of species and molecular forms of the Anopheles gambiae complex by PCR-RFLP. Med Vet Entomol. 2002;16:461–4.
Cienfuegos AV, Rosero DA, Naranjo N, Luckhart S, Conn JE, Correa MM. Evaluation of a PCR-RFLP-ITS2 assay for discrimination of Anopheles species in northern and western Colombia. Acta Trop. 2011;118:128–35.
Namango IH, Marshall C, Saddler A, Ross A, Kaftan D, Tenywa F, et al. The Centres for Disease Control light trap (CDC-LT) and the human decoy trap (HDT) compared to the human landing catch (HLC) for measuring Anopheles biting in rural Tanzania. Malar J. 2022;21:181.
Yao H, Song J, Liu C, Luo K, Han J, Li Y, et al. Use of ITS2 region as the universal DNA barcode for plants and animals. PLoS ONE. 2010;5: e13102.
Collins FH, Paskewitz SM. A review of the use of ribosomal DNA (rDNA) to differentiate among cryptic Anopheles species. Insect Mol Biol. 1996;5:1–9.
Acknowledgements
The authors thank the local entomologist, Allan Reyes García, from the Honduran Ministry of Health and his collaborators. Acknowledgments to the Entomological Bioassay Unit of Quintana Roo, Mexico for the support of the entomological collections. Thanks to Dr Gabriela Matamoros for her support in the language revision.
Funding
Financial support for the development of this study was obtained from the Center for Genetic Research of the UNAH.
Author information
Authors and Affiliations
Contributions
DE conceptualized the methodology. DE collected the mosquitoes. DE and GF developed the PCR–RFLP. GF performed the in silico analyses. FP, BO, and DE performed the experiments. GF drafted the manuscript. DE helped draft the manuscript and analysed the data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Escobar, D., Pérez, F., Ortiz, B. et al. PCR–RFLP assays for the identification of Anopheles (Diptera: Culicidae) species circulating in Honduras. Malar J 22, 57 (2023). https://doi.org/10.1186/s12936-023-04494-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-023-04494-6