
Abstract
Background
The duration of trial follow-up affects the ability to detect recrudescent infections following anti-malarial treatment. The aim of this study was to explore the proportions of recrudescent parasitaemia as ascribed by genotyping captured at various follow-up time-points in treatment efficacy trials for uncomplicated Plasmodium falciparum malaria.
Methods
Individual patient data from 83 anti-malarial efficacy studies collated in the WorldWide Antimalarial Resistance Network (WWARN) repository with at least 28 days follow-up were available. The temporal and cumulative distributions of recrudescence were characterized using a Cox regression model with shared frailty on study-sites. Fractional polynomials were used to capture non-linear instantaneous hazard. The area under the density curve (AUC) of the constructed distribution was used to estimate the optimal follow-up period for capturing a P. falciparum malaria recrudescence. Simulation studies were conducted based on the constructed distributions to quantify the absolute overestimation in efficacy due to sub-optimal follow-up.
Results
Overall, 3703 recurrent infections were detected in 60 studies conducted in Africa (15,512 children aged < 5 years) and 23 studies conducted in Asia and South America (5272 patients of all ages). Using molecular genotyping, 519 (14.0%) recurrences were ascribed as recrudescent infections. A 28 day artemether-lumefantrine (AL) efficacy trial would not have detected 58% [95% confidence interval (CI) 47–74%] of recrudescences in African children and 32% [95% CI 15–45%] in patients of all ages in Asia/South America. The corresponding estimate following a 42 day dihydroartemisinin-piperaquine (DP) efficacy trial in Africa was 47% [95% CI 19–90%] in children under 5 years old treated with > 48 mg/kg total piperaquine (PIP) dose and 9% [95% CI 0–22%] in those treated with ≤ 48 mg/kg PIP dose. In absolute terms, the simulation study found that trials limited to 28 days follow-up following AL underestimated the risk of recrudescence by a median of 2.8 percentage points compared to day 63 estimates and those limited to 42 days following DP underestimated the risk of recrudescence by a median of 2.0 percentage points compared to day 42 estimates. The analysis was limited by few clinical trials following patients for longer than 42 days (9 out of 83 trials) and the imprecision of PCR genotyping which overcalls recrudescence in areas of higher transmission biasing the later distribution.
Conclusions
Restricting follow-up of clinical efficacy trials to day 28 for AL and day 42 for DP will miss a proportion of late recrudescent treatment failures but will have a modest impact in derived efficacy. The results highlight that as genotyping methods improve consideration should be given for trials with longer duration of follow-up to detect early indications of emerging drug resistance.
Similar content being viewed by others


Background
Before the introduction of molecular genotyping, the World Health Organization (WHO) recommended that anti-malarial clinical trials conducted in areas of high transmission should be restricted to a short follow-up duration (2 weeks). This was in part to reduce potential confounding from new infections which then could not be distinguished from recrudescences [1]. One of the earliest indications of emerging anti-malarial drug resistance is recrudescent parasitaemia [2]. Recrudescence is delayed until the anti-malarial drug no longer suppresses parasite multiplication. As resistance increases recrudescent parasitaemias occur as parasites are able to grow in increasing concentrations of the drug [2]. As the first recrudescences occur several weeks after treatment anti-malarial therapeutic efficacy trials with two weeks of follow-up could only identify high grade parasite resistance [2, 3].
In the 1990s, the introduction of polymerase chain reaction (PCR) genotyping allowed comparison of the sizes of amplified segments of highly polymorphic Plasmodium falciparum genes, paving the way for classifying probabilistically individual recurrent infections as either homologous (identical to the initial infection, i.e. recrudescent) or heterologous (different than the initial infection, i.e. a new infection) [4]. In 2003, the WHO revised its guidelines for assessing anti-malarial efficacy. The new guideline recommended 42 days of post-treatment follow-up for artemisinin-based combination therapy (ACT), including lumefantrine, and 63 days for artemisinin-based combinations including mefloquine, in conjunction with molecular genotyping to distinguish recrudescent from new infections [5, 6]. However conducting trials with such long follow-up was associated with significant logistical difficulties, and the guideline was revised again in 2009, to recommend a minimum of 28 and 42 days follow-up for artemisinin-based combinations including lumefantrine or mefloquine, respectively [7]. It was anticipated that the revised follow-up duration would capture “most” of the treatment failures, and thereby provide a reasonable approximation of drug efficacy.
Recent studies have reported that a substantial proportion of recrudescent infections emerge in the peripheral blood beyond the currently recommended follow-up duration. A Tanzanian study reported that 28 days of follow-up missed 28% (5/18) of recrudescent infections following treatment of 206 patients with artesunate-amodiaquine (ASAQ) and 58% (7/12) following treatment of 197 patients with artemether-lumefantrine (evaluable population) [8]. In an Ethiopian study, 88% (14/16) of the recrudescent infections were detected after 28 days following treatment with AL (n = 348 patients) [9]. Other studies have reported recrudescent infections occurring after 6 weeks after treatment with artesunate-mefloquine (ASMQ) and dihydroartemisinin-piperaquine (DP) [10,11,12], and up to 6 weeks after treatment with AL [12, 13]. Together, these reports suggest that the current recommendations regarding the duration of study follow-up warrant re-examination.
The aim of this study was to use pooled data from clinical trials to assess the ability of currently recommended minimum follow-up periods to capture PCR-confirmed recrudescence following treatment of uncomplicated P. falciparum malaria with fixed dose ACT.
Methods
Study and patient data
Clinical studies with fixed dose formulations of ACT uploaded in the WorldWide Antimalarial Resistance Network (WWARN) repository were selected if they had a minimum follow-up period of 28 days with molecular genotyping carried out to differentiate recurrent parasitaemia as due to either new infection or recrudescence [14]. Early treatment failures (on or before day 7) were excluded since the focus of the analysis was to characterize the temporal distribution of late recrudescences following initial parasite clearance. Patients with missing or indeterminate genotyping outcomes were also excluded. In Africa, analysis was restricted to children less than 5 years of age, since for this population immunity is predicted to have less impact on outcomes compared to older individuals. In the studies from Asia and South America, patients of all ages were included. Further details of study inclusion and exclusion criteria are presented in Additional file 1.
The time of observed recrudescence
The time of observed recurrence (\(t_{obs} )\) was used to estimate the time to recrudescence (\(T_{R} )\) at which parasite recrudescence would exceed the threshold of detection (TOD); the latter was defined as 50 parasites per microlitre. \(T_{R}\) was estimated assuming tenfold parasite multiplication per 48 h asexual cycle using the following algorithm [15]: \(\widehat{{T_{R} }} = t_{obs} - \left( {\left( {\log_{10 } (\frac{{P_{R} }}{50}} \right)} \right) \times 2)\), where \(t_{obs}\) is the day when recrudescence was detected in the study and \(P_{R}\) is the parasite density at the observed time of recrudescence. If the estimated \(T_{R}\) was less than the time of last visit when the patient had a negative peripheral blood film examination (\(t_{neg}\)), then \(T_{R}\) was replaced as \(t_{neg} + 1\).
Estimation of temporal trend and distribution of recrudescence
The temporal trend (instantaneous risk) of recrudescence at any time during the study follow-up was quantified by its hazard function, \(h\left( t \right)\). The adjusted estimate of the hazard function was derived from a Cox proportional hazard model controlling for the body weight adjusted (mg/kg) dose of the partner drug, age of the patient, and initial parasite load (on log-scale). The baseline hazard function \(h_{0} \left( t \right) {\text{was}}\) approximated as the slope of the cumulative baseline hazard \((H_{0} \left( t \right))\). \(H_{0} \left( t \right)\) was estimated using fractional polynomial smoothing after applying log-transformation as outlined previously [16]. The fractional polynomial approach was used, as standard parametric approaches were found to provide a poor fit to the data (Additional file 1; Sections 2 and 3). The probability density \(f_{0} \left( t \right)\) of time to PCR confirmed recrudescence was then estimated as \(f_{0} \left( t \right) = h_{0} \left( t \right).S_{0} \left( t \right)\), where \(S_{0} \left( t \right)\) is the baseline survival estimate of drug efficacy (\(S_{0} \left( t \right) = e^{{ - H_{0} \left( t \right)}}\)). The constructed distribution was normalized such that the area under the curve was equal to 1. The area under the normalized probability density curve (AUC) was calculated at a given time-point and the cumulative AUC at a time-point \(t\) was reported with the associated 95% confidence interval (CI) derived using 1000 bootstrap resamples drawn from data of the same sample size. The proportion of recrudescences missed at any specific time point was calculated as 1—AUC at that time-point. For the DP regimen, the distribution was estimated for those who received a total piperaquine dose of above or below 48 mg/kg, a threshold associated with poorer therapeutic outcomes in a paediatric population [17]. For AL, the derived estimates were stratified by region as suggested previously [18].
Simulation studies
The impact of a missed proportion of recrudescences on the absolute bias (overestimation) in a derived estimate of efficacy was evaluated through two simulation studies. The first simulation study explored the impact of sub-optimal follow-up on derived efficacy in areas of low and high malaria transmission, and Kaplan–Meier estimates were generated at the end of the maximum follow-up time (day 63) and compared with the estimates derived for days 28 and 42 (Additional file 1: Section 4). The second simulation study explored how the duration of follow-up influenced the estimated efficacies of AL and DP. Hazard ratios were estimated using Cox proportional hazards regression at days 28, 42 and 63. Summarized results were reported from 1000 simulation runs. The design of the simulation studies has been described previously [19] and a step-by-step outline of the simulation protocol is presented in supplemental text (Additional file 1: Sections 4).
Sensitivity analyses
The following two sensitivity analyses were considered: (i) distributions were re-estimated by using data only from studies with 42 days of follow-up, and (ii) the 707 patients with indeterminate recurrence excluded in the primary analysis were considered as missing data, and the probability distributions were re-estimated using multiply imputed data, as previously described [20].
Software
Cumulative baseline hazard was estimated by fitting the Cox proportional hazard model using the survival library; Kaplan–Meier type hazard and kernel-smoothed hazard function were estimated using the muhaz library in R software [21].
Results
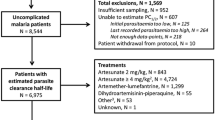
Data were available for 15,512 children aged less than 5 years from 60 studies in Africa and for 5272 patients of all ages from 23 studies in Asia and South America (Fig. 1). The duration of follow-up was 28 days in 39 (47.0%) studies, 42 days in 32 (38.6%) studies, 56 days in 3 (3.6%) studies and 63 days in 9 (10.8%) studies. The studies were published from 2001 to 2015 with 45 (54.2%) studies published before 2010. Further details on the study designs and patient characteristics are provided in Additional file 1.

Flow chart showing selection of clinical trials and participants in the study. AL = artemether-lumefantrine; DP = dihydroartemisinin-piperaquine; ASMQ = artesunate-mefloquine; ASAQ = artesunate-amodiaquine, and n = number of recrudescences; PCR = Polymerase Chain Reaction; WWARN = WorldWide Antimalarial Resistance Network; ACT = artemisinin-based combination therapy
Recurrent parasitaemia and molecular genotyping
A total of 3703 (17.8%) patients had recurrent parasitaemia between days 7 and 63 after initiation of treatment. Parasite genotyping was attempted at three or more loci (including microsatellites) in 3026 (81.7%) recurrent infections, 2 loci in 575 (15.5%) infections, one locus in 79 (2.1%) infections, and using microsatellites only in 15 (0.4%) infections. The number of markers was not documented in 8 (0.2%) infections. In total 3184 (86.0%) recurrent infections were classified as new infections and 519 (14.0%) as recrudescent infections. Among recurrent infections, the proportion of recrudescent infections was 20.3% (16/79) in studies which used a single marker, 16.5% (95/575) in studies with two markers, 13.2% (399/3026) in studies with three markers (including microsatellites), 33.3% (5/15) in studies using only microsatellites, and 50% (4/8) in studies with no description of molecular methods.
Temporal trend of P. falciparum recrudescence with AL and DP
A total of 306 recrudescent infections were confirmed after AL treatment (264 in Africa; 41 in Asia and 1 in South America) and 115 after DP treatment (94 in Africa; 19 in Asia, and 2 in South America). The observed times of recrudescence in different regions stratified by follow-up duration are presented in Fig. 2. The non-parametric estimates and the kernel smoothed estimate of the hazard function exhibited a non-monotonic trend (Additional file 1: Section 2). Covariate adjusted smoothed hazard functions obtained from Cox models exhibited a non-monotonic shape for AL and DP (Fig. 3; right panels). For AL, the instantaneous hazard function was shifted to the right for Africa compared to Asia, and the peak hazard was lower and earlier in Asia (day 21) compared to Africa (day 27). For DP, the peak hazard was earlier (day 30) in those who were under-dosed (< 48 mg/kg of piperaquine) compared to those who received a higher dose of piperaquine (day 39). There were a total of 72 recrudescent infections following treatment with fixed dose formulations of ASAQ (61 in Africa and 11 in Asia/S.America) and 26 (15 in Africa and 11 in Asia) following treatment with fixed dose ASMQ; these low numbers precluded robust modelling for these regimens. These patients were excluded in the construction of distribution of failure times.

Observed time-to-recrudescence for ACT stratified by region and duration of follow-up. The y-axis depicts the time for the recrudescent infection to reach microscopic limit of detection (50 parasites/µL). In Africa, the distribution is shown for children < 5 years age, whereas in Asia/ S. America, data are shown for patients of all ages. Each dot represents an observed recrudescence. AL = artemether-lumefantrine; DP = dihydroartemisinin-piperaquine; ASMQ = artesunate-mefloquine; ASAQ = artesunate-amodiaquine, and n = number of recrudescences. Only data from studies with at least three molecular markers are shown in the graph. For DP (Asia), the graph depicts recrudescences observed in studies from Asia and S. America combined

Fractional polynomial estimates of cumulative and instantaneous hazard for AL and DP. The cumulative baseline hazard estimated from Cox model (adjusted for age, baseline parasitaemia and mg/kg dosage of partner drug) together with fractional polynomial smoother (left panel). The temporal trend of observing recrudescence during the follow-up period, estimated by the instantaneous baseline hazard function (right panel). Data in Africa was restricted to children < 5 years whereas patients of all ages were included in Asia and S. America. Data from S. America were grouped with Asia. All studies used at least 3 locus genotypes (the usage of microsatellites was considered as a separate locus). Under-dosed was defined as total piperaquine dose ≤ 48 mg/kg (those receiving > 48 mg/kg defined as not under-dosed). AL = artemether-lumefantrine; DP = dihydroartemisinin-piperaquine. The equations for the cumulative baseline hazard functions derived using the fractional polynomial smoothing are presented in the Additional file 1
The empirical distribution of time of P. falciparum recrudescence
The probability density of the time to recrudescence in patients treated with AL and DP regimens is presented in Fig. 4. Overall the area under the curve (AUC) for AL in Africa was 0.42 [95% CI 0.26–0.53] on day 28, 0.79 [95% CI 0.53–0.97] on day 42 and 1.00 [95% CI 0.81–1.00] on day 63 (Table 1). In Asia, the distribution was shifted to the left compared to that observed in Africa, with an estimated AUC of 0.68 [95% CI 0.55–0.85] on day 28 and 0.98 [95% CI 0.93–1.00] on day 42. The distribution of recrudescence following DP could only be derived from African studies in which 85 of the 514 recurrences were categorized as recrudescence by PCR. In Asia/S. America, only 21 of 206 recurrences were categorized as recrudescences. The distribution of recrudescence following DP in Africa was shifted to the left in those who were under-dosed (receiving ≤ 48 mg/kg); the AUC by day 42 was 0.91 [95% CI 0.78–1.00] in the under-dosed group compared to 0.53 [95% CI 0.10–0.81] in those treated with a higher dose (Table 1).

Distribution of recrudescent infection for artemether-lumefantrine and dihydroartemisinin-piperaquine. Dotted line represents distribution derived from all data. Solid line represents the distribution derived by including data up to day 42 (observations beyond day 42 were censored on day 42). For Africa, the data included children < 5 years old whereas for Asia, no age restriction was applied
Simulation studies
Simulation studies were undertaken to assess correlations between duration of follow-up and estimates of drug efficacy. Compared to the “true” efficacy estimates of AL as measured at day 63, estimates derived using only the recrudescence observed until day 28 overestimated the drug efficacy by a median of 2.8 percentage points [Interquartile Range (IQR): 2.3%–3.4%; Range: 0.6%–5.5%] in areas of high transmission. With 42 days of follow-up, the overestimation in efficacy fell to 0.9 percentage points [IQR: 0.7%–1.3%; Range: 0.0%–2.9%] (Fig. 5; top-left panel). Compared to the true efficacy estimates for DP as measured at day 63, those generated at day 42 overestimated drug efficacy by 2.0 percentage points [IQR: 1.7%–2.6%; Range: 0.0%–4.6%] in high transmission settings (Fig 5; bottom left panel). The estimates for areas of low transmission intensity were similar (Fig. 5; right panels).

Absolute overestimation in efficacy due to sub-optimal follow-up duration (compared to day 63 estimates). Overestimation of efficacy relative to day 63 estimates for AL and DP regimen. In areas of high transmission, there was a median of 15% new infections in the DP arm and 30% new infections in AL arm representing. In areas of low transmission, there was a median of 8% new infections in the DP arm and 15% new infections in the AL arm. Simulation assumed 500 patients per treatment arm and was repeated for 1,000 runs. AL = artemether-lumefantrine; DP = dihydroartemisinin-piperaquine (See Additional file 1 for further details on the functions used for simulation)
Impact of follow-up duration on comparative efficacy
The effect of study duration on comparative efficacy in randomized comparative studies was investigated. In areas of low transmission in which the risk of new infections was low (8% in the DP arm and 15% in the AL arm), the median hazard ratio (HR) for AL relative to DP (for recrudescence) was 2.00 [IQR: 1.29–2.78] at day 28, 1.30 [IQR: 1.07–1.68] on day 42 and fell to parity from day 49 onwards (Fig. 6; left panel). The results were similar in areas of high transmission (Fig. 6; right panel). There was little difference in the results when the simulation was repeated with 200 and 1000 subjects per treatment arm (Additional file 1: Section 4).

Simulation study comparing hazards ratio of recrudescence for AL against DP in children < 5 years in Africa. A There were a median (across 1000 simulation runs) of 8% new infections in DP arm and 15% in AL arm representing areas of low transmission. B There were a median of 15% new infections in DP arm and 30% in AL arm representing areas of high transmission. In both simulation settings, approximately 4% recrudescence was observed by day 63 on AL and DP arm. Data for AL was simulated based on the estimated hazard function of recrudescence in children < 5 years Africa, and for DP regimen the data was simulated based on the hazard function for those who received piperaquine dose greater than 48 mg/kg (See Additional file 1 for the functions used for these simulations). The dotted horizontal line shows the line of no effect (hazard ratio = 1). The simulation assumed 500 patients per treatment arms. AL = artemether-lumefantrine; DP = dihydroartemisinin-piperaquine
Sensitivity analyses
When the analysis was restricted to outcomes at day 42 and observations beyond this time-point were censored, the distribution of the timing of recrudescence after AL in Africa shifted to the left compared to the distribution including all data up until day 63 (Fig. 4; left panel, solid lines). Similarly, the distribution of recrudescence for DP in those treated with ≥ 48 mg/kg also shifted to the left when the analysis was restricted to day 42 (Fig. 4; right panel, solid lines). These sensitivity analyses remained unchanged when indeterminate outcomes were considered as missing data (Additional file 1: Section 5).
Discussion
Using data from 83 anti-malarial clinical trials conducted in Asia, Africa and South America, this analysis characterized the temporal and cumulative distributions of recrudescence after treatment with AL or DP in patients with uncomplicated P. falciparum malaria. The temporal trend of P. falciparum recrudescent parasitaemia exhibited a non-monotonic shape, an observation consistent with anti-malarial pharmacokinetics. Soon after therapy the risk of recrudescence is extremely low, as most parasites are rapidly cleared by the highly potent artemisinin components of the ACT. Subsequently, as concentrations of the partner drug fall below the minimum inhibitory concentration (MIC), any remaining parasites can grow and expand, eventually leading to patent infection and symptomatic illness; during this period, the instantaneous risk of recrudescence rises. In a population of patients the hazard of recrudescence rises as blood concentrations of the antimalarial drug fall below MIC values and then falls again as the majority of recrudescences have occurred. New infections are also constrained by residual concentrations of the slowly eliminated antimalarial drugs and “bunch” together after blood levels fall below prevailing MIC values [22].
Characterization of the temporal trend enabled the construction of the probability distribution of the infections estimated to be recrudescences. In African children less than 5 years old treated with DP, the peak distribution of recrudescence was around day 39 after initiation of therapy in children who were adequately dosed (≥ 48 mg/kg total dose of piperaquine), but this fell to day 30 in children treated with a lower total dose of piperaquine (< 48 mg/kg). This is consistent with patients treated with a lower dose having blood concentrations which fall below the MIC sooner, allowing the parasite biomass to become patent earlier.
In patients treated with AL there were significant regional differences in the probability distributions of recrudescent infections with a shift to the left in Asia and South America (in patients of all ages) compared to Africa (in children < 5 years). This may reflect suppression of the parasite growth by host immunity, which is acquired earlier in life in Africa, where children often have multiple infections per year, whilst immunity is acquired more slowly in low endemic settings in Asia and South America [23]. However this explanation does not explain adequately why that suppression should then decrease to allow recrudescence. The derived distribution of recrudescences in high transmission settings, such as Africa, needs to be interpreted with caution, as late recrudescence beyond day 42 may also have arisen through misclassification of new infections as recrudescences. In higher transmission settings, the proportion of recurrent infections that are due to reinfection increases in studies with longer follow up, until eventually all recurrent infections are newly acquired. The uncertainty is particularly pertinent for individuals treated with AL; only a small proportion of patients (11.4%) were followed for more than 42 days and the infections ascribed as recrudescences detected between days 42 and 63 in 9 patients influenced the estimation (See Fig. 4 on the impact of follow-up duration on the estimated distribution) [12, 24]. In areas of high transmission polyclonal infections are common and these confound the interpretation of genotyping data [25, 26]. In multilocus genotyping using separate PCRs the amplified sequences are not phased so haplotypes cannot be inferred if there are multiple genotypes. The proportion of recurrent infections can be overestimated or underestimated depending on the background prevalence and distribution of the polymorphic markers, the technique used, the criteria set, and the number of reinfections (transmission intensity). In the one African study of AL, in which patients were followed until day 84, recrudescence was defined as persistence of at least one baseline clone, and this is likely to have led to a substantial proportion of late new infections being misclassified as recrudescent infections [24]. When the distributions of recrudescence following AL in Africa were derived using only data from studies with 42 days follow up the distribution shifted to the left and this was far more apparent in Africa than Asia (Fig. 4).
To estimate the degree to which absolute efficacy might be overestimated, simulation studies for AL and DP regimens were undertaken. Compared to the efficacy estimates at day 63, studies of AL restricted to 28 days follow-up overestimated drug efficacy by a median of 2.8% (range: 0.6%–5.5%), whereas studies of DP restricted to 42 day follow-up overestimated efficacy in adequately dosed DP by a median of 2.0% (range: 0%–4.6%). These results suggest that follow-up of 28 days for AL and 42 days for DP may result in failure to detect early signs of recrudescent parasitaemia occurring after the end of follow-up. Overall, the impact of follow-up on estimated treatment efficacy was modest, and any additional gain in accuracy in deriving these estimates must be weighed against the increased logistical challenge of studies with longer follow-up and a higher risk of misclassification when defining recrudescences and reinfections.
Recrudescent parasitaemia is the primary determinant for the selection and onward transmission of de novo resistant parasites [15]. Such selection and propagation of resistance is predicted to occur within a “window” determined by a drug’s pharmacokinetic profile [27]. For the standard AL regimen, this hypothetical window lies between days 24 to 27 for emergence of de novo resistance and between days 20 to 39 for acquired resistance [27]. The corresponding window for mefloquine (a drug with a longer elimination half-life) is estimated to be between 73 to 87 days and 65 to 113 days for the emergence of de novo and acquired resistance, respectively [27].
The analysis presented has a number of important limitations. First, the estimation of the hazard function is vulnerable to the method used for estimation, especially on the distal part of the distribution (See Additional file 1: Section 2). Second, only 9 studies included in the analysis had a follow-up greater than 42 days and there were only 255 recurrent events (25 recrudescences and 230 new infections) detected beyond this period, affecting the tail-end of the derived distributions. When the analysis was restricted to studies with only 42 days of follow-up, the estimated distribution of recrudescence shifted to the left compared to that derived using all available data (Fig. 4; solid lines). Despite the shifts in the distributions, the results indicated that the currently recommended follow-up times do not capture all late recrudescence. Third, the definition of recrudescence is dependent upon the molecular methods applied and their interpretation. Current genotyping methods are imprecise and, in areas of high transmission, overcall late recrudescent infections [25]. Whilst data on the number of polymorphic loci used to genotype were documented in all but 8 of the 3026 recurrent infections, no data were available on the population allele frequencies of these markers or the multiplicity of infection, and hence it was not possible to discern the degree to which late recrudescing parasites may have been misclassified. This will have affected the tail of the temporal distribution, particularly in high transmission settings, where reinfection is common. Accounting for this error, when constructing the empirical distribution and in the simulation studies, was beyond the scope of this work, and hence the tail of the distribution of recrudescence following AL in Africa should be interpreted with caution. Finally, the estimated time of recrudescence assumed tenfold parasite multiplication per 48-h asexual cycle for estimating the time when the parasite density would have first reached 50 parasites/µl, and yet this is likely affected by host characteristics, such as acquired immunity, and by circulating drug concentrations, which could not be quantified in this analysis [15, 28].
Conclusions
The derived empirical distribution of recrudescence indicates that the currently recommended minimum follow-up for anti-malarial efficacy trials do not capture a significant proportion of PCR confirmed recrudescences occurring after AL and DP treatment. Whilst the overall impact of this limitation on the estimated efficacy of these anti-malarial regimens was modest in absolute terms, extension of the duration of follow-up to 42 days for AL and 63 days for DP, particularly with more precise methods of genotyping, would facilitate detection of early signs of emerging drug resistance, which can manifest through delayed parasite recrudescence.
Availability of data and materials
The data included during in the current study is available from the WorldWide Antimalarial Resistance Network (WWARN), a controlled open access repository of anti-malarial studies, on reasonable request through the Data Access Committee: http://www.wwarn.org/about-us/governance-people/data-access-committee.
Abbreviations
- ACT:
-
Artemisinin-based combination therapy
- AL:
-
Artemether-lumefantrine
- ASAQ:
-
Artesunate-amodiaquine
- ASMQ:
-
Artesunate-mefloquine
- AUC:
-
Area under the density curve
- CI:
-
Confidence interval
- DP:
-
Dihydroartemisinin-piperaquine
- HR:
-
Hazards ratio
- PCR:
-
Polymerase chain reaction
- PIP:
-
Piperaquine
- WWARN:
-
WorldWide Antimalarial Resistance Network
References
WHO. Assesment of therapeutic efficacy of antimalarial drugs for uncomplicated falciparum malaria in areas with intense transmission. Geneva, World Health Organization; 1996.
White NJ. The assessment of antimalarial drug efficacy. Trends Parasitol. 2002;18:458–64.
Stepniewska K, Taylor WRJ, Mayxay M, Price R, Smithuis F, Guthmann J-P, et al. In vivo assessment of drug efficacy against Plasmodium falciparum malaria: duration of follow-up. Antimicrob Agents Chemother. 2004;48:4271–80.
Snounou G, Beck HP. The use of PCR genotyping in the assessment of recrudescence or reinfection after antimalarial drug treatment. Parasitol Today. 1998;14:462–7.
WHO. Monitoring Antimalarial Drug Resistance. Geneva: World Health Organization; 2001.
WHO. Assessment and monitoring of antimalarial drug efficacy for the treatment of uncomplicated falciparum malaria. Geneva: World Health Organization; 2003.
WHO. Methods for surveillance of antimalarial drug efficacy. Geneva: World Health Organization; 2009.
Mårtensson A, Strömberg J, Sisowath C, Msellem MI, Gil JP, Montgomery SM, et al. Efficacy of artesunate plus amodiaquine versus that of artemether-lumefantrine for the treatment of uncomplicated childhood Plasmodium falciparum malaria in Zanzibar. Tanzania Clin Infect Dis. 2005;41:1079–86.
Eshetu T, Abdo N, Bedru KH, Fekadu S, Wieser A, Pritsch M, et al. Open-label trial with artemether-lumefantrine against uncomplicated Plasmodium falciparum malaria three years after its broad introduction in Jimma Zone, Ethiopia. Malar J. 2012;11:240.
Janssens B, van Herp M, Goubert L, Chan S, Uong S, Nong S, et al. A randomized open study to assess the efficacy and tolerability of dihydroartemisinin-piperaquine for the treatment of uncomplicated falciparum malaria in Cambodia. Trop Med Int Health. 2007;12:251–9.
Valecha N, Phyo AP, Mayxay M, Newton PN, Krudsood S, Keomany S, et al. An open-label, randomised study of dihydroartemisinin-piperaquine versus artesunate-mefloquine for falciparum malaria in Asia. PLoS One. 2010;5:e11880.
Sirima SB, Ogutu B, Lusingu JPA, Mtoro A, Mrango Z, Ouedraogo A, et al. Comparison of artesunate-mefloquine and artemether-lumefantrine fixed-dose combinations for treatment of uncomplicated Plasmodium falciparum malaria in children younger than 5 years in sub-Saharan Africa: a randomised, multicentre, phase 4 trial. Lancet Infect Dis. 2016;16:1123–33.
Ursing J, Kofoed PE, Rodrigues A, Blessborn D, Thoft-Nielsen R, Björkman A, et al. Similar efficacy and tolerability of double-dose chloroquine and artemether-lumefantrine for treatment of Plasmodium falciparum infection in Guinea-Bissau: a randomized trial. J Infect Dis. 2011;203:109–16.
WWARN Data Inventory. 2020 (cited 2020 Feb 26). www.wwarn.org; https://app-live.wwarn.org/DataInventoryExplorer/#1.
White NJ, Pongtavornpinyo W, Maude RJ, Saralamba S, Aguas R, Stepniewska K, et al. Hyperparasitaemia and low dosing are an important source of anti-malarial drug resistance. Malar J. 2009;8:253.
Royston P. Estimating a smooth baseline hazard function for the Cox model. London; 2011. https://www.ucl.ac.uk/statistics/research/pdfs/rr314.pdf. Accessed 27 Jan 2016.
The WorldWide Antimalarial Resistance Network (WWARN) DP Study Group. The effect of dosing regimens on the antimalarial efficacy of dihydroartemisinin-piperaquine: a pooled analysis of individual patient data. PLoS Med. 2013;10:e1001564.
Worldwide Antimalarial Resistance Network (WWARN) AL Dose Impact Study Group. The effect of dose on the antimalarial efficacy of artemether–lumefantrine: a systematic review and pooled analysis of individual patient data. Lancet Infect Dis. 2015;15:692–702.
Dahal P, Guerin PJ, Price RN, Simpson JA, Stepniewska K. Evaluating antimalarial efficacy in single-armed and comparative drug trials using competing risk survival analysis : a simulation study. BMC Med Res Methodol. 2019;19:107.
Dahal P, Stepniewska K, Guerin PJ, D’Alessandro U, Price RN, Simpson JA. Dealing with indeterminate outcomes in antimalarial drug efficacy trials: a comparison between complete case analysis, multiple imputation and inverse probability weighting. BMC Med Res Methodol. 2019;19:215.
R Development Core Team R. R: A Language and Environment for Statistical Computing. R Found. Stat Comput; 2017.
Watson J, Chu CS, Tarning J, White NJ. Characterizing blood-stage antimalarial drug MIC values in vivo using reinfection patterns. Antimicrob Agents Chemother. 2018;62:e02476-e2517.
Covell G. Relationship between malarial parasitaemia and symptoms of the disease: a review of the literature. Bull World Health Organ. 1960;22:605–19.
Borrmann S, Sasi P, Mwai L, Bashraheil M, Abdallah A, Muriithi S, et al. Declining responsiveness of Plasmodium falciparum infections to artemisinin-based combination treatments on the Kenyan coast. PLoS One. 2011;6:e26005.
Greenhouse B, Dokomajilar C, Hubbard A, Rosenthal PJ, Dorsey G. Impact of transmission intensity on the accuracy of genotyping to distinguish recrudescence from new infection in antimalarial clinical trials. Antimicrob Agents Chemother. 2007;51:3096–103.
WHO. Methods and techniques for clinical trials on antimalarial drug efficacy: genotyping to identify parasite populations. Geneva: World Health Organization; 2007.
Stepniewska K, White NJ. Pharmacokinetic determinants of the window of selection for antimalarial drug resistance. Antimicrob Agents Chemother. 2008;52:1589–96.
Simpson JA, Aarons L, Collins WE, Jeffery GM, White NJ. Population dynamics of untreated Plasmodium falciparum malaria within the adult human host during the expansion phase of the infection. Parasitology. 2002;124:247–63.
OxTREC. Oxford Tropical Research Ethics Committee (OxTREC). 2016 (cited 2019 Mar 23).https://researchsupport.admin.ox.ac.uk/governance/ethics/committees/oxtrec.
Acknowledgements
We would like to thank the patients and all the staff who participated in the clinical trials included in this IPD meta-analysis. We would like to thank Dr. Birgit Schramm, Dr. Mary Oguike, Dr. Maman Laminou, Dr. Patrick Sawa, Dr. Ingrid van den Broek and Prof. Christopher Whitty for their help with data access. We thank the Novartis team (Dr. Caroline Boulton and Dr Guoqin Su) for thoroughly reviewing the manuscript. We thank the Sanofi team (Dr.Andrew Hockey, Sanofi-Global Health) for providing support through making patient level data available.
Study group members: The complete list of author’s contribution is listed in Additional file 2. The members of the WorldWide Antimalarial Resistance Network (WWARN) Methodology Study Group are the authors of this paper: Prabin Dahal, World Wide Antimalarial Resistance Network (WWARN), Oxford, UK; Infectious Diseases Data Observatory (IDDO), Oxford, UK; Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom; Julie Anne Simpson, Centre for Epidemiology and Biostatistics, Melbourne School of Population and Global Health, University of Melbourne, Melbourne, VIC, Australia; Salim Abdulla, Ifakara Health Institute, Dar es Salaam, Tanzania; Jane Achan, Malaria Consortium, London, UK; Ishag Adam, Faculty of Medicine, University of Khartoum, Khartoum, Sudan; Aarti Agarwal, New York City Dept of Health and Mental Hygiene, New York, USA; Richard Allan, The Mentor Initiative; Anupkumar R. Anvikar, National Institute of Malaria Research, Sector 8, Dwarka, New Delhi 110077, India, Emmanuel Arinaitwe, Infectious Diseases Research Collaboration, Kampala, Uganda; Elizabeth A. Ashley, Myanmar Oxford Clinical Research Unit, Myanmar; Centre for Tropical Medicine, Nuffield Department of Clinical Medicine, University of Oxford, Oxford, UK; Ghulam Rahim Awab, Medical Faculty of Nangarhar University, Afghanistan; MORU tropical Health Network; Quique Bassat, Centro de Investigação em Saúde de Manhiça (CISM), Maputo, Mozambique; ISGlobal, Hospital Clínic - Universitat de Barcelona, Barcelona, Spain; ICREA, Pg. Lluís Companys 23, 08010 Barcelona, Spain; Consorcio de Investigación Biomédica en Red de Epidemiología y Salud Pública (CIBERESP), Madrid, Spain; Anders Björkman, Depatment Microbiology,Tumour and Cell Biology, Karolinska institutet, Stockholm Sweden; Steffen Borrmann, Institute for Tropical Medicine, University of Tübingen, Germany & Kenya Medical Research Institute/Wellcome Trust Research Programme, Kilifi, Kenya; Heidelberg University School of Medicine, Dept. of Infectious Diseases, Heidelberg, Germany; Teun Bousema, Radboud Institute for Health Sciences, Radboudumc Nijmegen, The Netherlands; Department of Medical Microbiology, Radboud University Nijmegen Medical Centre, Njimegen, The Netherlands; Hasifa Bukirwa, African Field Epidemiology Network, Kampala, Uganda; Verena I. Carrara, Shoklo Malaria Research Unit, Mae Sot, Thailand; Mahidol Oxford University Research Unit, Bangkok, Thailand; Marco Corsi, Private consultancy for Drug Development in Tropical Diseases, formerly Sigma Tau SpA Industrie Farmaceutiche Riunite, Pomezia (Rome) Italy; Michel Cot, MERIT, IRD, Université de Paris, Paris, 75006, France; Umberto D’Alessandro, Medical Research Council Unit The Gambia at the London School of Hygiene and Tropical Medicine, Fajara, The Gambia; London School of Hygiene and Tropical Medicine, London, UK; Timothy M. E. Davis, Medical School, University of Western Australia,Crawley, WA, Australia; Philippe Deloron, MERIT, IRD 216, Université de Paris, Paris, 75006, France; Meghna Desai, Malaria Branch, Division of Parasitic Diseases and Malaria, Centers for Disease Control and Prevention, Atlanta, GA, USA; Pedro Rafael Dimbu, National Malaria Control Program, Luanda, Angola; Djibrine Djalle, Institut Pasteur de Bangui, BP 923 Bangui, Central African Republic; Abdoulaye Djimde, Malaria Research and Training Center, Department of Epidemiology of Parasitic Diseases, Faculty of Pharmacy, University of Science, Techniques and Technologies of Bamako, Bamako, Mali; Grant Dorsey, Department of Medicine, University of California San Francisco, San Francisco, USA; Chris J. Drakeley, Department of Infection Biology, London School of Hygiene and Tropical Medicine, London, UK; Stephan Duparc, Medicines for Malaria Venture, Geneva, Switzerland; Michael D. Edstein, Australian Defence Force Malaria and Infectious Disease Institute, Brisbane, QLD, Australia; Emmanuelle Espie, GSK Vaccines, R&D center, Clinical and Epidemiology department (avenue Fleming 20, 1300 Wavre); Formerly: Epicentre, 8, rue Saint Sabin, 75011, Paris, France; Abul Faiz, Malaria Research Group, Chittagong, Bangladesh; Dev Care Foundation, Dhaka, Bangladesh; Catherine Falade, Department of Pharmacology & Therapeutics, College of Medicine, University of Ibadan,Ibadan, Nigeria; Caterina Fanello, Centre for Global Health, Nuffield Department of Medicine, University of Oxford, UK; Jean-Francois Faucher, INSERM, U1094, Tropical Neuroepidemiology, Limoges, France; Department of Infectious Diseases and Tropical Medicine, 87042 Limoges, France; Babacar Faye, Department of Medical Parasitology, Medical Faculty, Université Cheikh Anta Diop, Dakar, Senegal; Filomeno de Jesus Fortes, Institute of Hygiene and Tropical Medicine, Universidade Nova de Lisboa-Portugal; Nahla B. Gadalla, Sudanese American Medical Association, Fairfax, VA, US; Oumar Gaye, Department of Medical Parasitology, Medical Faculty, Université Cheikh Anta Diop, Dakar, Senegal; J. Pedro Gil, Department of Microbiology, Tumor and Cell Biology (MTC), Karolinska Institutet (Biomedicum), Solnavägen 9, C9, 17165 Solna, Sweden; Julius Gilayeneh, National Malaria Control Program, Ministry of Health, Republic of Liberia; Brian Greenwood, Faculty of Infectious and Tropical Diseases, London School of Hygiene and Tropical Medicine, London, UK; Anastasia Grivoyannis, The Johns Hopkins Hospital, Baltimore, Maryland, USA; Tran Tinh Hien, Oxford University Clinical Research Unit (OUCRU), Centre for Tropical Medicine, Wellcome Trust Major Overseas Program (MOP); Jimee Hwang, US President’s Malaria Initiative, Malaria Branch, Division of Parasitic Diseases and Malaria, US Centers for Disease Control and Prevention, Atlanta, GA, USA; Global Health Group, University of California San Francisco, San Francisco, USA; Bart Janssens, Medecins Sans Frontieres, Brussels, Belgium; Elizabeth Juma, Kenya Medical Research Institute, Nairobi, Kenya; Erasmus Kamugisha, Catholic University of Health and Allied Sciences, Mwanza, Tanzania; Corine Karema, Swiss Tropical and Public Health Institute, Basel, Switzerland; University of Basel, Basel, Switzerland; Harin A. Karunajeewa, Melbourne Medical School – Western Health, The University of Melbourne; Western Health Chronic Disease Alliance, Sunshine Hospital, Furlong Road, St Albans, VIC 3021, Australia; Jean R. Kiechel, Drugs for Neglected Diseases initiative, Geneva, Switzerland; Fred Kironde, Department of Microbiology, Faculty of Health Sciences, Islamic University in Uganda; Poul-Erik Kofoed, Bandim Health Project, Indepth Network, Apartado 861, Bissau, Guinea-Bissau; Department of Paediatrics, Kolding Hospital, Denmark/IRS University of Southern Denmark; Peter G. Kremsner, Institute of Tropical Medicine, University of Tübingen, Germany; Centre de Recherches Medicales de Lambarene, Gabon; Sue J. Lee, Faculty of Tropical Medicine, Mahidol University, Bangkok, Thailand; Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom; Kevin Marsh, Centre for Tropical Medicine, Nuffield Department of Clinical Medicine, University of Oxford, Oxford, United Kingdom; Kenya Medical Research Institute/Wellcome Trust Research Programme, Kilifi, Kenya; Andreas Mårtensson, Department of Women’s and Children’s Health, International Maternal and Child Health (IMCH), Uppsala University, Uppsala, Sweden; Mayfong Mayxay, Lao-Oxford-Mahosot Hospital-Wellcome Trust Research Unit, Microbiology Laboratory, Mahosot Hospital, Vientiane, Laos; Centre for Tropical Medicine, Nuffield Department of Clinical Medicine, Churchill Hospital, Oxford, UK; Hervé Menan, Department of Parasitology, Faculty of Pharmacy, University of Cocody, Abidjan, Côte d'Ivoire; Petra Mens, Medical Microbiology, Parasitology, Amsterdam University Medical Centers, Amsterdam, The Netherlands; Theonest K. Mutabingwa, Hubert Kairuki Memorial University, Dar-es-Salaam, Tanzania; Department of Infectious and Tropical Diseases, London School of Hygiene and Tropical Medicine, London, UK; Jean-Louis Ndiaye, Department of Parasitology and Mycology, Health Sciences Research and Training Unit, University of Thies, Senegal; Billy E. Ngasala, Department of Parasitology, Muhimbili University of Health and Allied Sciences, Dar es Salaam, Tanzania; Department of Women’s and Children’s Health, International Maternal and Child Health (IMCH), Uppsala University, Uppsala, Sweden; Harald Noedl, Malaria Research Initiative, Bandarban; Francois Nosten, Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine Research building, University of Oxford Old Road campus, Oxford, UK; Shoklo Malaria Research Unit, Mahidol-Oxford Tropical Medicine Research Unit, Faculty of Tropical Medicine, Mahidol University, Mae Sot, Thailand; Andre Toure Offianan, Malariology department, Institut Pasteur de Côte d'Ivoire, Abidjan, Côte d'Ivoire; Bernhards R. Ogutu, Kenya Medical Research Institute/United States Army Medical Research Unit, Kisumu, Kenya; Piero L. Olliaro, Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom; Jean Bosco Ouedraogo, Direction Regionale de l’Ouest, Institut de Recherche en Sciences de la Santé (IRSS) , Bobo-Dioulasso, Burkina Faso; Non Transmissible disease department, Centre Muraz Bobo-Dioulasso, Bobo-Dioulasso, Burkina Faso; Non Transmissible disease department, Centre Muraz Bobo-Dioulasso, Bobo-Dioulasso, Burkina Faso; Patrice Piola, Institut Pasteur du Cambodge, Phnom Penh, Cambodia; Christopher V. Plowe, Duke Global Health Institute, Duke University, Durham, NC, USA; Mateusz M. Plucinski, US President’s Malaria Initiative, Malaria Branch, Division of Parasitic Diseases and Malaria, US Centers for Disease Control and Prevention, Atlanta, GA, USA; Epidemic Intelligence Service, Centers for Disease Control and Prevention, Atlanta, Georgia, USA; Oliver James Pratt, National Malaria Control Program, Ministry of Health and Social Welfare, Liberia; Zulfikarali Premji, Muhimbili University College of Health Sciences, Dar es Salaam, Tanzania; Michael Ramharter, Department of Tropical Medicine, Bernhard Nocht Institute for Tropical Medicine & I Dep. of Medicine University Medical Center Hamburg-Eppendorf, Hamburg, Germany; Institut für Tropenmedizin, Universität Tübingen, Germany; Christophe Rogier, French Armed Forces Biomedical Research Institute (IRBA), Marseille, France; Institut Pasteur de Bangui, Bangui, Central African Republic; Primum Vitare !, Paris, France; Lars Rombo, Clinical research centre, sormland county council, Uppsala University, Sweden; Philip J. Rosenthal, Department of Medicine, University of California San Francisco, San Francisco, USA; Carol Sibley, World Wide Antimalarial Resistance Network (WWARN), Oxford, UK; Department of Genome Sciences, University of Washington, Seattle, USA; Sodiomon Sirima, Groupe de Recherche Action en Santé (GRAS) 06 BP 10248, Ouagadougou 06, Burkina Faso; Frank Smithuis, Myanmar Oxford Clinical Research Unit; Medical Action Myanmar; Sarah G. Staedke, Department of Clinical Research, London School of Hygiene & Tropical Medicine, London, UK; Infectious Diseases Research Collaboration, Kampala, Uganda; Inge Sutanto, Department of Parasitology, Faculty of Medicine, University of Indonesia, 6 Salemba Raya, Jakarta 10430, Indonesia; Ambrose Otau Talisuna, Regional Office for Africa, World Health Organization, Brazzaville, Congo; Joel Tarning, WorldWide Antimalarial Resistance Network, Oxford, United Kingdom; Mahidol Oxford Tropical Medicine Research Unit, Faculty of Tropical Medicine, Mahidol University, Bangkok, Thailand; Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom; Walter R. J. Taylor, Faculty of Tropical Medicine, Mahidol University, Bangkok, Thailand; Emmanuel Temu, UNOPS/RBM Partnership to End Malaria, Geneva Switzerland; Kamala Thriemer, Global and Tropical Health Division, Menzies School of Health Research, Charles Darwin University, Darwin, Northern Territory, Australia; Nguyen Thuy-Nhien, Oxford University Clinical Research Unit (OUCRU), Centre for Tropical Medicine, Wellcome Trust Major Overseas Program (MOP); Venkatachalam Udhayakumar, Malaria Branch and President’s Malaria Initiative, Division of Parasitic Diseases and Malaria, Center for Global Health, Centers for Disease Control and Prevention, Atlanta, Georgia, USA; Johan Ursing, Department of Clinical Sciences, Karolinska Institutet, Danderyd Hospital, Stockholm, Sweden; Department of Infectious Diseases, Danderyd Hospital, Stockholm, Sweden; Bandim Health Project, Indepth Network, Apartado 861, Bissau, Guinea-Bissau; Michel van Herp, Medecins Sans Frontieres, Operational Centre Brussels, Brussels, Belgium; Marit van Lenthe, Médecins Sans Frontières-Operational Centre Amsterdam; Michele van Vugt, Division of Infectious Diseases, Center for Tropical Medicine & Travel Medicine, Academic medical Center, University of Amsterdam, The Netherlands; Yavo William, Department of Parasitology, Faculty of Pharmacy, University of Cocody,Abidjan, Côte d’Ivoire; Cornelis Winnips, Novartis International AG, Basel, Switzerland; Sophie Zaloumis, Centre for Epidemiology and Biostatistics, Melbourne School of Population and Global Health, University of Melbourne, Melbourne, VIC, Australia; Issaka Zongo, Direction Regionale de l’Ouest, Institut de Recherche en Sciences de la Santé (IRSS) , Bobo-Dioulasso, Burkina Faso; Nick J. White, Mahidol-Oxford Research Unit, Bangkok, Thailand; Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom; Philippe J. Guerin, World Wide Antimalarial Resistance Network (WWARN), Oxford, UK; Infectious Diseases Data Observatory (IDDO), Oxford, UK; Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom; Kasia Stepniewska, World Wide Antimalarial Resistance Network (WWARN), Oxford, UK; Infectious Diseases Data Observatory (IDDO), Oxford, UK; Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom; Ric N. Price, World Wide Antimalarial Resistance Network (WWARN), Oxford, UK; Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom; Menzies School of Health Research and Charles Darwin University, Darwin, Australia.
Funding
This work was funded by Tropical Network Fund, Nuffield Department of Clinical Medicine, University of Oxford to PD. The WorldWide Antimalarial Resistance Network (PD, KS, RNP and PJG) is funded by a Bill and Melinda Gates Foundation grant. JAS is an Australian National Health and Medical Research Council Senior Research Fellow (1104975). RNP is a Wellcome Trust Senior Fellow in Clinical Science (200909). The funders did not participate in the study development, the writing of the paper, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Consortia
Contributions
Study conception: PD, PJG, RNP, JAS and KS. Statistical methodology: PD, JAS, KS. Identification and collation of individual participant data: PD. Wrote the first draft of the manuscript: PD, PJG, JAS, KS, RNP. All authors critically edited the first draft, read and approved the final version. The complete list of author’s contribution is listed in Additional file 2. All authors read and approved the final manuscript.
Ethics declarations
Ethics approval and consent to participate
All studies included in this analysis were obtained in accordance with the laws and ethical approvals applicable to the countries in which the studies were conducted, and were obtained with the knowledge and consent of the individual to which they relate. Individual participant data meta-analysis met the criteria for waiver of ethical review as defined by Oxford Tropical Research Ethics Committee (OxTREC) since the research consisted of secondary analysis of existing, anonymous data [29].
Consent for publication
Not applicable.
Competing interests
The findings and conclusions in this report are those of the author(s) (Jimee Hwang, Mattaeusz Plucinski, Meghna Desai, Udhayakumar Venkatachalam) and do not necessarily represent the official position of the US Centers for Disease Control and Prevention. The opinions expressed are those of the authors and do not necessarily reflect those of the Australian Defence Force, Joint Health Command or any extant Australian Defence Force policy (Michael Edstein). The opinions expressed are those of the authors and do not necessarily reflect those of the Novartis Pharmaceuticals (Cornelius Winnips). The rest of the authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Further methodological details and results.
Additional file 2.
List of studies used.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
The WorldWide Antimalarial Resistance Network Methodology Study Group. Temporal distribution of Plasmodium falciparum recrudescence following artemisinin-based combination therapy: an individual participant data meta-analysis. Malar J 21, 106 (2022). https://doi.org/10.1186/s12936-021-03980-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-021-03980-z