
Abstract
Background
Cape Verde is an archipelago located off the West African coast and is in a pre-elimination phase of malaria control. Since 2010, fewer than 20 Plasmodium falciparum malaria cases have been reported annually, except in 2017, when an outbreak in Praia before the rainy season led to 423 autochthonous cases. It is important to understand the genetic diversity of circulating P. falciparum to inform on drug resistance, potential transmission networks and sources of infection, including parasite importation.
Methods
Enrolled subjects involved malaria patients admitted to Dr Agostinho Neto Hospital at Praia city, Santiago island, Cape Verde, between July and October 2017. Neighbours and family members of enrolled cases were assessed for the presence of anti-P. falciparum antibodies. Sanger sequencing and real-time PCR was used to identify SNPs in genes associated with drug resistance (e.g., pfdhfr, pfdhps, pfmdr1, pfk13, pfcrt), and whole genome sequencing data were generated to investigate the population structure of P. falciparum parasites.
Results
The study analysed 190 parasite samples, 187 indigenous and 3 from imported infections. Malaria cases were distributed throughout Praia city. There were no cases of severe malaria and all patients had an adequate clinical and parasitological response after treatment. Anti-P. falciparum antibodies were not detected in the 137 neighbours and family members tested. No mutations were detected in pfdhps. The triple mutation S108N/N51I/C59R in pfdhfr and the chloroquine-resistant CVIET haplotype in the pfcrt gene were detected in almost all samples. Variations in pfk13 were identified in only one sample (R645T, E668K). The haplotype NFD for pfmdr1 was detected in the majority of samples (89.7%).
Conclusions
Polymorphisms in pfk13 associated with artemisinin-based combination therapy (ACT) tolerance in Southeast Asia were not detected, but the majority of the tested samples carried the pfmdr1 haplotype NFD and anti-malarial-associated mutations in the the pfcrt and pfdhfr genes. The first whole genome sequencing (WGS) was performed for Cape Verdean parasites that showed that the samples cluster together, have a very high level of similarity and are close to other parasites populations from West Africa.
Similar content being viewed by others

Background
Malaria, caused by Plasmodium parasites, is a global public health problem. Almost half of world population is at risk of malaria, and in 2018 there were 228 million cases and 405,000 deaths, globally [1]. In Africa, where Plasmodium falciparum infections dominate, 6 countries (Nigeria, Democratic Republic of the Congo, Uganda, Côte d’Ivoire, Mozambique, Niger) accounted for more than half of all malaria cases worldwide. Further, 94% of all malaria deaths occurred in the African continent. Due to malaria control activities, such as improved case management and roll-out of insecticide-treated nets and indoor residual spraying, the number of countries moving towards disease elimination has increased. In particular, the number of countries with < 100 indigenous cases increased from 17 in 2010 to 27 in 2018 [1].
Cape Verde (population size: ~ 500,000) is one of the African countries in a pre-elimination phase of malaria control, with < 1 case per 1000 population per year [2]. There were 583 indigenous cases and 5 deaths between 2010 and 2018. However, in 2017, 423 (72.6%) cases occurred in an outbreak. This year was an outlier as it corresponds to an increase of 89.9% of cases compared to previous years, and excluding it, the number of cases reported yearly since 2010 has varied between 1 and 48 (average < 20 cases) [1].
The research and studies conducted within the scope of this epidemic revealed that the probable factors associated with the increase in the number of cases were: (1) the reduction of indoor residual spraying; (2) the reduction of regular vector control interventions [3]; and, (3) ecological and environmental factors such as unusual high rainfall during 2016 [4]. In response to the 2017 outbreack, authorities have implemented measures to improve vector control interventions, which has contributed to reducing autochthonous malaria cases and mortality since 2018 [1, 4].
Despite the control efforts implemented by health authorities, autochthonous cases persist and could delay elimination targets. Malaria prevalence is unstable and autochthonous cases are restricted to the islands of Santiago (96%) and Boavista (4%), while imported cases from countries with disease transmission are recorded in all 9 islands. In recent years, local transmission has been restricted to the island of Santiago, especially in Praia city, capital of the country, where 158 cases were recorded, more than 90% of autochthonous cases from 2010 to 2016 [2]. There is typically low malaria endemicity, but there are fluctuations in morbidity depending on rainfall, with transmission normally occurring between the months of September and November [5]. However, in 2017, the malaria outbreak occurred before the rainy season, where all 423 autochthonous P. falciparum cases were reported in Praia city [3, 4]. Eighteen patients had at least two relapse episodes in that year, 23 further imported cases were registered [4].
All non-complicated malaria cases in Cape Verde are hospitalized and treated with artemisin-based combination therapy (ACT): artemether and lumefantrine, which targets the parasite erythrocytic asexual stage. All cases also receive the gametocytocidal primaquine drug at the start of treatment to prevent transmission and interrupt the spread of the disease. Severe cases are treated with intravenous artesunate. Levels of parasitaemia are monitored at health facilities during the period of the disease and followed up on multiple occasions, up to 42 days [6]. Success in the control and treatment of malaria depends on the clinical efficiency of ACT and avoiding drug resistance [7]. Therefore, understanding the epidemiology of drug resistance is vital for an effective drug policy, especially within the Cape Verde elimination settings [8].
The non-temporary movement of people between Cape Verde and malaria-endemic countries, particularly to and from West Africa, increases the potential for case importation, and poses a challenge to malaria elimination on the archipelago [9]. The circulation of parasites between regions also increases the risk of importing drug resistance, which is underpinned by mutations in the P. falciparum genome. For example, polymorphisms associated with resistance to several anti-malarial drugs have been identified in P. falciparum genes pfdhfr (target for anti-malaria drug pryrimethamine), pfdhps (target for sulfadoxine), pfcrt (target for chloroquine), pfk13 (target for artemisin) and pfmdr1 (target for mefloquine, chloroquine). Similarly, P. falciparum genetics (e.g., pfama1 gene) also gives the parasite the ability to evade the immune response of the host. Therefore, there is a need to understand the genetic diversity of circulating parasites to inform on drug resistance and transmission networks identified by finding near-identical parasite genomic sequences. To support this, epidemiological data and blood samples were collected from close contacts of patients, including neighbours and family members, thereby informing on potential risk factors for malaria susceptibility, transmission and the emergence and spread of outbreaks. Further, this study on the molecular characterization of P. falciparum isolates collected from patients during the 2017 outbreak will provide a baseline assessment of malaria parasite drug resistance profile and genetics in Cape Verde, from which to design population-specific diagnostics, and contribute to strengthening the country’s measures for prevention and control, in order to achieve its elimination targets.
Methods
Malaria patients
Malaria patients admitted to Dr Agostinho Neto Hospital at Praia city, Santiago island, Cape Verde, between July and October 2017 were enrolled. A total of 190 (from 446; 187 indigenous, 3 imported) gave informed consent to participate in the present study. The laboratory diagnosis of malaria was performed by rapid diagnostic tests and microscopy.
All 190 patients that agreed to participate remained hospitalized for at least 3 days and were discharged from hospital only after laboratory confirmation of Plasmodium spp. negative blood smear and clinical evaluation; follow-up assessments were performed on days 7, 14, 21, 28, 35, and 42 post-start of treatment. The management of participating patients strictly followed the guidelines from the Ministry of Health [6].
Data and sample collection
Data were obtained from laboratory analysis and from a questionnaire. Of the 190 malaria cases enrolled in this study, 131 (68.9%) cases and their neighbours and family members (n = 137; without malaria symptoms), answered a questionnaire in a community setting. This questionnaire was designed in both Portuguese and Crioulo, which captured sociodemographic data, present and past about malaria and fever, travel information and signs and symptoms consistent with malaria. Species identification and parasite quantification was performed by microscopic observations of thick and thin blood smears. Blood drops of malaria cases and their neighbours/family obtained by finger prick were collected on filter paper and stored at − 20 °C for later molecular characterization of parasites and antibody (AMA1 and MSP1) detection. Parasite DNA was extracted using Chelex [10] and species confirmation by PCR [11].
AMA1 and MSP1-19 indirect ELISA
Dried blood spot elution was performed as previously described [12]. In brief, 137 blood spots from samples collected in 2017 were equilibrated to ambient room temperature before opening, and 2.5-mm diameter discs were cut from Whatman 3 MM paper using a leather punch. Each disc was then eluted in a 96-deep-well plate containing 150 μl PBS/0.05% (v/v) Tween 20/0.05% (w/v) sodium azide and incubated at ambient room temperature for > 18 h with gentle rotary agitation ~ 100 rpm. Serum was diluted to a 1:100 dilution. IgG antibodies were detected by indirect ELISA using MSP1-19 (Wellcome Genotype) and AMA1 (3D7) antigens expressed in Escherichia coli strain BL21, as described by Okech et al. [13], and by Hoder [14]. AMA1 and MSP1-19 antigens were coated onto 96-well ELISA plates (Immulon 4HBX, Thermo) with coating buffer (carbonate-bicarbonate) in a 50-µl volume at a concentration of 0.5 µg/m and incubated a 40 °C overnight. Plates were washed several times using PBS plus 0.05% Tween 20 (PBS/T) and blocked with 1% (w/v) skimmed milk powder in PBS/T (block solution). The previously eluted serum was used at 1:400 working dilution into block solution. European confirmed negative and pooled hyperimmune positive serum were used as negative/positive control samples. Serum was incubated at 40˚C overnight and washed in PBS/T. Rabbit anti-human HRP conjugated secondary antibody (Dako) was used at a concentration of 1:15,000 in PSB/T. TMB substrate was added followed by a 15-min room temperature (RT) incubation. H2SO4 stop solution was added, followed by plate reading at 450 nm.
Candidate gene sequencing and analysis
A total of 38 samples with the highest parasitaemia (2000–274,000 parasites/μl) were selected for sequencing of anti-malarial drug resistance loci, including pfmdr1 (PF3D7_0523000; including codons 86, 184 and 1246), pfk13 (PF3D7_1343700; including codons 417 to codon 714), pfdhfr (PF3D7_0417200.1; including codons 50, 51, 59, 108 and 164) and pfdhps (PF3D7_0810800.1; including codons 431, 436, 437, 540). Genes were PCR amplified and products cleaned using SureClean (Bioline, USA) following manufacturer instructions. The PCR products were analysed by electrophoresis on a 1.5% agarose gel stained with GreenSafe Premium (Nzytech) to confirm amplification. The primer pairs and thermocycling conditions are summarized in Additional file 1: Table S1. The PCR products were sequenced using Sanger capillary platform, and the resulting sequences were analysed using Geneious software (v4.8.5) and the laboratory-adapted strain 3D7 was used as reference. A fragment of the pfcrt gene (PF3D7_0709000; including codons 72, 74, 75, 76) was also examined. Three dual-labelled probes with sequences complementary to each of the pfcrt 72–76 haplotypes (denoted as CVIET, CVMNK, SVMNT), were included in the real-time PCR reaction, each containing a different fluorescent molecule [15, 16]. Two amplicons (479 and a 516 bp) of the gene pfama1 (PF3D7_1133400) were amplified in 20 samples from 2017 and 4 samples from the 2016, using protocols published elsewhere [17]. Amplicons were amplified with primer pairs and thermocycling conditions summarized in Additional file 1: Table S1. PCR products were analysed by electrophoresis on a 1.5% agarose gel stained with ethidium bromide and sequencing using the same methodology described above.
Selective whole genome amplification and whole genome sequencing
Seven samples from 2017 with the highest parasitaemias (3141–126,690 parasites/μl) and meeting whole genome sequencing (WGS)-required DNA quality were selected for WGS, together with 4 samples from 2016. Genomic DNA was extracted from dried blood spots using the QIAamp DNA Investigator Kit (QIAGEN, Germany) as per manufacturer’s directions. DNA concentrations were measured using the Invitrogen Qubit Fluorometer. A selective whole genome amplification (SWGA) strategy was applied prior to WGS, following previously published protocol [18]. Oligonucleotides that preferentially bind with high frequency to the P. falciparum DNA, and rarely bind to the human DNA, were used for SWGA. All SWGA reactions were carried out in a UV Cabinet for PCR Operations (UV-B-AR, Grant-Bio) to eliminate potential contamination. Briefly, a maximum of 60 ng of gDNA (minimum of 5 ng) was added to a total 50 µl reaction alongside with 1X phi-29 buffer (New England Biolabs), 1X bovine serum albumin, 1 mM dNTPs, 2.5 µM of primer mix, 30 U phi-29 DNA polymerase (New England Biolabs) and water. The reaction was carried out on a thermocycler with the following step-down programme: 5 min at 35 °C, 10 min at 34 °C, 15 min at 33 °C, 20 min at 32 °C, 25 min at 31 °C, 16 h at 30 °C, and 10 min at 65 °C. The 5 samples, selected for SWGA, were then whole genome sequenced on a MiSeq (Illumina). The QIAseq FX DNA Library Kit (QIAGEN) was used for library preparation according to the manufacturer’s protocol, with a 20-min fragmentation step. Library DNA concentration was analysed using a Qubit 2.0 fluorometer. All sequencing reactions were performed using 2 × 150 bp reads.
Whole genome sequence data analysis
Raw fastq files obtained after the MiSeq run were trimmed using trimmomatic set to default parameters [19], and aligned to the P. falciparum 3D7 reference genome (PlasmoDB) using bwa-mem software [20]. SNPs were identified using samtools software [21] and filtered for quality based on previously described methods [22, 23]. The coverage of each nucleotide was analysed using sambamba [24], which was set to include SNPs with only a coverage of fivefold or above. To investigate the population structure of P. falciparum parasites, a distance matrix was created which was based on a matrix of pair-wise identity calculated from the SNPs present in each sample. Using the distance matrix, a neighbourhood-joining tree was produced and visualized in iTOL [25]. WGS data from an extra 400 publicly available samples from Africa, South America, South Asia and Southeast Asia were also used for analysis [22, 23, 26]. A neighbourhood-joining tree was used to investigate the clustering of samples from West Africa.
For genetic distance comparisons between samples, a clustering approach using a Manhattan distance matrix of pair-wise identity by state values was calculated from the sub-set of SNPs available for each pair-wise comparison using the R software.
Statistical analysis
Regarding data collected by questionnaire (among other sociodemographic data, present and past about malaria and fever, travel information and signs and symptoms consistent with malaria), two groups were compared: malaria cases (n = 131) and neighbours/family members (n = 137).
Differences in categorical variables were tested by Chi-Square or Fisher exact tests. Differences in continuous variables were assessed using t-tests (after checking for normality and the homogeneity of variances), or alternatively, Mann–Whitney-Wilcoxon non-parametric test. Malaria cases and neighbours and family were analysed following an unmatched design, this decision was also supported by recent discussion about the efficiency of matched and unmatched studies [27, 28], balancing sample size and efficiency. Thus, unconditional multiple logistic regression models were used to to explore simultaneously the association of different independent variables with the binary dependent variable (malaria case vs close contacts), controlling for potential confounding. Multiple models included all variables with p < 0.20 in the simple logistic regression models. Adjusted odds ratios (OR) were presented with 95% confidence intervals (95% CI).
Results
Characterization of malaria cases
From the 446 cases in the 2017 outbreak, 190 blood samples collected from malaria patients before treatment administration, including 187 indigenous and 3 imported infections, were analysed. All samples were P. falciparum positive (PCR confirmed) and one patient presented with a mixed P. falciparum and Plasmodium malariae infection, undetected by microscopy. Considering indigenous cases group, only 27 (14.4%) were children (≤ 16 years), and the majority of participants were adults (n = 160, 86.6%; range: 17–91 years old). All imported cases were young adults, 2 were 33 years and one 31 years of age.
Malaria cases were distributed throughout Praia city (Fig. 1) and consistent with official statistics. Varzea, Paiol and Achadinha were the neighbourhoods with highest number of cases (n = 13) (Fig. 1). There were no cases of severe malaria, nonetheless 18 patients presented with hyperparasitaemia (> 100,000 parasites/µl blood) ranging from 105,840 to 400,000 parasites/µl blood. One fatality was registered, but malaria was not the cause of death. The 3 most common self-reported symptoms in malaria cases were fever, headache and chills. At day 3 of hospital treatment, Giemsa-stained smears were negative for Plasmodium spp. for all patients. The length of hospital stay of malaria cases varied between 3 days to 1 month, with a median of 4 days (Table 1). None of the patients evidenced signs of severe malaria. Extended hospitalizations were clinically justified by other medical conditions.

Number of malaria cases in Praia city per neighbourhood (N). N.1—São Filipe; N.2—Fonton; N.3—Tira Chapéu; N.4—Monte Vermelho; N.5—Achadinha Pires; N.6—Chã de Areia; N.7—São Pedro Latada; N.8—Bela Vista; N.27—Várzea; N.9—Achada Grande Frente; N.10—Tahiti Chã de Areia; N.11—Achada Eugénio Lima; N.12—Jamaica/Agua Funda; N.13—Palmarejo; N.14—Lém Cachorro; N.15—Achada Mato; N.16—Achada Santo António; N.17—Lém Ferreira; N.18—Ponta de Agua; N.19—Safende; N.20—Fazenda Sucupira; N.21—Vila Nova; N.22—Coqueiro Castelão; N.23—Calabaceira; N.24—Pensamento. N.25—Paiol; N.26—Achadinha
Sociodemographic and epidemiological characterization
Table 1 (and Additional file 1: Table S1) shows sociodemographic and epidemiological information of a set of 131 patients and 137 close contacts (family members or neighbours, without malaria symptoms). Almost all participants (265/268; 98.9%) were Cape Verde nationals. Malaria cases were predominantly single (73.3%), male (65.6%) and younger than non-malaria cases (all p < 0.001). Malaria cases tended to travel less in the previous 6 months (5.6%) than their close contacts (11.8%), but this difference was marginally non-significant (p = 0.060). Students and tertiary-sector activities accounted for 80.2% malaria cases and 73.0% non-malaria group. Geographical locations of the cases suggested (Fig. 1) some clusters in Varzea, Paiol, Achadinha, Calabaceira, and Pensamento, corresponding to 43.5% of all cases.
Multiple logistic regression models suggested increased risk of malaria from being male (OR = 4.99; 95% CI 2.90–8.58) and single (OR = 1.92, 95% CI 1.02–3.58), with potentially lower risk from higher age (OR = 0.98; 95% CI 0.97–1.00). All malaria cases were PCR positive for P. falciparum, while all close contacts (n = 137) were negative. Serologic detection of P. falciparum anti-AMA1 and MSP-1-19 IgG/IgM was also negative for all 137 neighbours and family members.
Drug resistance profile of malaria cases
A sub-set of the malaria cases (n = 38/190) was investigated for the presence of alleles associated with drug resistance. The chloroquine-resistant CVIET haplotype (pfcrt gene) was detected in all samples analysed except one that carried CVMNK (wild type haplotype). No mutations in the pfk13 gene, associated with artemisinin resistance, were identified. All pfk13 sequences analysed were identical to that of 3D7 reference strain except one that carried two mutations R645T and E668K, which have not been observed in other isolates [22]. Sulfadoxine-pyrimethamine (SP) drug-resistant associated alleles were also surveyed. For the pfdhps gene the wild-type haplotype (S436/A437/K540) was present in all samples analysed. On the other hand, for pfdhfr the triple mutation S108N/N51I/C59R was detected in all analysed samples (n = 12). The codons 86, 184 and 1246 from the pfmdr1 gene were also analysed. The majority of samples (89.7%) carried the haplotype NFD (meaning N86/F184/D1246) that is associated with artemether + lumefantrine tolerance and the others were wild-type for the 3 codons (N86/Y184/D1246). No other polymorphisms were detected for the samples and regions analysed in the 5 genes.
Population genetic analysis
WGS was performed in 7 autochthonous samples with the highest parasitaemia and 4 samples from 2016. The resulting data were compared to publicly available P. falciparum global genomic sequences [23, 26], in order to explore their genetic diversity. Prior to WGS, SWGA was used to increase the amount of parasite DNA. The success of the SWGA is very dependent on parasitaemia and the quality of the DNA. After WGS, the samples from 2016 were excluded for analysis due to having less than 1% of genome sequenced. The WGS data confirmed the drug resistance profile results, with all samples having the pfcrt CVIET haplotype, the Y184F mutation in pfmdr1 gene, the triple mutation S108N/N51I/C59R in pfdhfr and wild type alleles for pfdhps.
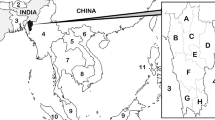
A neighbour-joining tree was constructed using SNP data and demonstrates that the Cape Verdean samples group together, and cluster with samples from West African (Fig. 2). To investigate the genetic similarity between samples, an analysis of SNP sharing was performed, including samples from other locations in Africa and Asia. The samples from Cape Verde shared > 99% of SNPs, while when compared with other samples on average 97% (maximum 97.6%) of SNPs were shared, a value normally observed between samples from different regions (e.g., African and Asian samples share on average 96.6% SNPs). To further explore the genetic diversity, two fragments of the PfAMA1 gene were sequenced for 20 samples, including the samples from 2016. The results show that all amplified samples that were successfully sequenced (n = 18 for 2017; n = 3 from 2006) share the same haplotype in both amplicons.

Neighbour-joining trees constructed using SNP data from Cape Verde samples using global samples. Cape Verde (pink) clusters together with other samples from Africa (a) particularly West Africa (b)
Discussion
Insights into the epidemiology of Cape Verde malaria
There is a lack of studies into the genetics and epidemiology of malaria cases in Cape Verde [2, 4]. The data contradict the typical profile based on women and children under-five years of age. Official data reports [2, 4] have indicated that more males are affected by malaria, and identified high-risk occupations, including construction or agricultural workers, security guards and the homeless. Only one agricultural worker and 4 security guards appeared in this data, which was dominated by tertiary-sector activities, students and unemployed. The geographical pattern of the samples overlaps with those previous malaria hotspots, including Várzea, Chã de Areia, Fazenda, Achadinha Paiol, and Lém Ferreira neighbourhoods [2, 4]. Further, the results showed that all neighbours and family members tested were negative for malaria (serology and PCR data), which reinforces that infection might be occurring outside of households.
The first WGS for Cape Verdean parasites was performed, and it clearly showed that the analysed samples cluster together and have a very high level of similarity, which suggests that possible the outbreak resulted from clonal expansion of local parasites. The Cape Verdean samples also cluster with isolates from West Africa, as expected. It would be possible to evaluate the robustness of these findings by increasing the number of Cape Verdean parasite samples undergoing WGS, including by considering samples from other years.
Drug resistance associated molecular markers
Artemether + lumefantrine (AL), is the recommended first treatment line for uncomplicated P. falciparum malaria in Cape Verde [6]. Apart from therapeutic efficacy studies (TES), the WHO recommends the use of molecular markers to monitor the emergence of mutations associated with resistance to anti-malarial drugs to detect emerging resistance and prevent potential future treatment failure [29]. Anti-malarial resistance genes polymorphism were assessed by Sanger and Next Generation Sequencing (NGS), the methods were used to screen for mutations in a set of malaria-positive blood samples targeting the pfcrt, pfmdr1, pfk13, pfdhps, and pfdhfr genes, which have previously been associated with anti-malarial resistance. The prevalence of the different resistance alleles in Cape Verde was not known, either after or before the introduction of ACT in the country. The prevalence of pfcrt (chloroquine resistance marker)-resistant haplotype CVIET remains high, only one of the analysed isolates carried the wild-type haplotype CVMNK. Despite the implementation of ACT in Cape Verde (since 2006) and the withdrawal of chloroquine, the frequency of the resistant allele 76T remains high. Although chloroquine resistant pfcrt genotype prevalence follows the trend of continental Africa [30,31,32], the origin is probably different. In Cape Verde it possibly reflects the limited recombination opportunity, due to the very low malaria transmission over the recent decades. Nevertheless, the identification of the wild-type allele K76 is in line with findings in other malaria regions, where AL is the predominant ACT [33,34,35,36]. Probably, due to lumefantrine, regimens tend to select wild-type pfcrt alleles [37,38,39]. It is generally reported that the mutant haplotype 51I/59R/108N in pfdhfr confers resistance to pyrimethamine while 437G/540E in pfdhps confers resistance to sulfadoxine. Combined, known as the quintuple mutant, they confer resistance to SP [40,41,42,43]. For the pfdhps gene the wild-type (S436/A437/K540) haplotype was identified in all analysed samples and for pfdhfr the triple mutation S108N/N51I/C59R was detected also in all samples successfully analysed, consistent with other African populations, such as Malawi [22] or Senegal [44]. SP has never been used as first-line treatment for P. falciparum uncomplicated malaria in Cape Verde, nor has it ever been used as intermittent preventive treatment (IPT) in infants and pregnant women.
The pfmdr1 amino acid mutations that have been implicated in multidrug-resistant phenotypes include N86Y, Y184F and D1246Y [45,46,47,48], where the NFD haplotype is suspected to be involved in parasite tolerance to AL [49,50,51]. In this study there was high prevalence of the 184F (89.7%) allele as well as the NFD haplotype (detected in 80% of the clinical isolates analysed). The NFD haplotype has also been identified as the most commonly reported pfmdr1 haplotype in continental Africa, where AL is widely used as first-line ACT to treat falciparum malaria [11, 47, 52,53,54]. Regarding the pfk13, all samples were identical to 3D7 strain, only one sample carried 2 mutations R645T and E668K, which are not thought to be resistance related. This outcome was expected as the overall prevalence of SNPs in the pfk13 is reported to have low prevalence outside Southeast Asia and high diversity among African parasites [1, 55]. The 2 SNPs identified in pfk13 gene in this study do not coincide either with the most reported or with the non-validated SNPs associated with delayed artemisinin parasite clearance in Africa [55, 56]. Although in Africa, artemisinin partial resistance has not yet been confirmed [1], there have been unconvincing reports of treatment failure in travellers returning from Africa [57, 58]. Mutations in pfk13, occurring in more than one African region affected by malaria, are increasing [35, 59,60,61,62,63,64,65], evidencing the need to close monitor this gene.
The Cape Verdean population has low immunity to malaria, 57% live on Santiago and Boavista islands where local malaria transmission occurs, the rest of the population lives in islands where no transmission occurs or abroad [6, 9]. Prevention measures are essential to reduce the risk of malaria transmission and the potential for malaria epidemics. Cape Verde is a country in the elimination phase of malaria, with indoor residual spraying with insecticides a major control strategy. This is supported by good case management with diagnostic tests and in-patient treatment of all cases [66]. Other actions recommended by the WHO are chemoprophylaxis and the use of mosquito nets. However, insecticide-impregnated mosquito nets are less used in Cape Verde, where a recent study shows that only 19% of the population use them, but if offered them, 91% would use it [4]. There is the potential to reduce the rate of infection, morbidity and mortality, through the very cost-effective mosquito net malaria prevention strategy [67].
Conclusion
The majority of the parasite samples analysed shared the same polymorphisms in the drug resistance-associated genes. Polymorphisms in the pfk13 gene associated with ACT tolerance in Southeast Asia were not detected, but the majority of the tested samples carried the pfmdr1 haplotype NFD. The first WGS for Cape Verdean parasites was performed and showed that all samples cluster together, have a high level of similarity and are close to West African parasite populations.
Availability of data and materials
The P. falciparum genome sequences used in this study are available in Genbank with the following accession numbers: MW712599-MW712662,MW579763-MW579766.
References
WHO. World malaria report 2019. Geneva, World Health Organization, 2019. https://www.who.int/news-room/fact-sheets/detail/malaria.
Depina AJ, Niang EHA, Barbosa Andrade AJ, Dia AK, Moreira A, Faye O, et al. Achievement of malaria pre-elimination in Cape Verde according to the data collected from 2010 to 2016. Malar J. 2018;17:236.
WHO. Update on the E-2020 initiative of 21 malaria-eliminating countries. Geneva, World Health Organization, 2018. http://apps.who.int/iris/bitstream/handle/10665/272724/WHO-CDS-GMP-2018.10-eng.pdf?ua=1%0Ahttp://www.who.int/malaria/publications/atoz/e-2020-progress-report/en/.
Depina AJ, Dia AK, De Ascenção Soares Martins A, Ferreira MC, Moreira AL, Leal SV, et al. Knowledge, attitudes and practices about malaria in Cabo Verde: A country in the pre-elimination context. BMC Public Health. 2019;19:850.
Arez AP, Snounou G, Pinto J, Sousa CA, Modiano D, Ribeiro H, et al. A clonal Plasmodium falciparum population in an isolated outbreak of malaria in the Republic of Cabo Verde. Parasitology. 1999;118:347–55.
Ministério de Saúde de Cabo Verde. Programa integrado de luta contra as doenças transmitidas por vectores e problemas da saúde associados ao meio ambiente: Protocolo de Tratamento Do Paludismo. 2015; 1–40.
Lubell Y, Dondorp A, Guerin PJ, Drake T, Meek S, Ashley E, et al. Artemisinin resistance-modelling the potential human and economic costs. Malar J. 2014;13:452.
Khatoon L, Baliraine FN, Bonizzoni M, Malik SA, Yan G. Prevalence of antimalarial drug resistance mutations in Plasmodium vivax and P. falciparum from a malaria-endemic area of Pakistan. Am J Trop Med Hyg. 2009;81:525–8.
WHO. Eliminating Malaria: Moving towards sustainable elimination in Cape Verde. Geneva: World Health Organization; 2012.
Bereczky N, Mårtensson A, Gil JP, Färnert A. Rapid DNA extraction from archive blood spots on filter paper for genotyping of Plasmodiumfalciparum. Am J Trop Med Hyg. 2005;72:249–51.
Lobo E, De Sousa B, Rosa S, Figueiredo P, Lobo L, Pateira S, et al. Prevalence of pfmdr1 alleles associated with artemether-lumefantrine tolerance/resistance in Maputo before and after the implementation of artemisinin-based combination therapy. Malar J. 2014;13:300.
Corran PH, Cook J, Lynch C, Leendertse H, Manjurano A, Griffin J, et al. Dried blood spots as a source of anti-malarial antibodies for epidemiological studies. Malar J. 2008;7:195.
Okech BA, Corran PH, Todd J, Joynson-Hicks A, Uthaipibull C, Egwang TG, et al. Fine specificity of serum antibodies to Plasmodium falciparum merozoite surface protein, PfMSP-1(19), predicts protection from malaria infection and high-density parasitemia. Infect Immun. 2004;72:1557–67.
Hodder AN, Crewther PE, Anders RF. Specificity of the protective antibody response to apical membrane antigen 1. Infect Immun. 2001;69:3286–94.
Sutherland CJ, Haustein T, Gadalla N, Armstrong M, Doherty JF, Chiodini PL. Chloroquine-resistant Plasmodium falciparum infections among UK travellers returning with malaria after chloroquine prophylaxis. J Antimicrob Chemother. 2007;59:1197–9.
He Y, Campino S, Benavente ED, Warhurst DC, Beshir KB, Lubis I, et al. Artemisinin resistance-associated markers in Plasmodium falciparum parasites from the China-Myanmar border: predicted structural stability of K13 propeller variants detected in a low-prevalence area. PLoS ONE. 2019;14:e0213686.
Lerch A, Koepfli C, Hofmann NE, Messerli C, Wilcox S, Kattenberg JH, et al. Development of amplicon deep sequencing markers and data analysis pipeline for genotyping multi-clonal malaria infections. BMC Genom. 2017;18:864.
Oyola SO, Ariani CV, Hamilton WL, Kekre M, Amenga-Etego LN, Ghansah A, et al. Whole genome sequencing of Plasmodium falciparum from dried blood spots using selective whole genome amplification. Malar J. 2016;15:597.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Ravenhall M, Benavente ED, Mipando M, Jensen ATR, Sutherland CJ, Roper C, et al. Characterizing the impact of sustained sulfadoxine/pyrimethamine use upon the Plasmodium falciparum population in Malawi. Malar J. 2016;15:575.
Ravenhall M, Benavente ED, Sutherland CJ, Baker DA, Campino S, Clark TG. An analysis of large structural variation in global Plasmodium falciparum isolates identifies a novel duplication of the chloroquine resistance associated gene. Sci Rep. 2019;9:8287.
Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics. 2015;31:2032–4.
Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44:W242–5.
Manske M, Miotto O, Campino S, Auburn S, Almagro-Garcia J, Maslen G, et al. Analysis of Plasmodium falciparum diversity in natural infections by deep sequencing. Nature. 2012;487:375–9.
Pearce N. Analysis of matched case-control studies. BMJ. 2016;352:i969.
Kuo C-L, Duan Y, Grady J. Unconditional or conditional logistic regression model for age-matched case–control data? Front Public Health. 2018;6:57.
WHO. Minutes of the Expert Review Committee on K13 molecular marker of artemisinin resistance. Geneva: World Health Organization; 2014.
Voumbo-Matoumona DF, Kouna LC, Madamet M, Maghendji-Nzondo S, Pradines B, Lekana-Douki JB. Prevalence of Plasmodium falciparum antimalarial drug resistance genes in southeastern Gabon from 2011 to 2014. Infect Drug Resist. 2018;11:1329–38.
Ocan M, Akena D, Nsobya S, Kamya MR, Senono R, Kinengyere AA, et al. Persistence of chloroquine resistance alleles in malaria endemic countries: a systematic review of burden and risk factors. Malar J. 2019;18:76.
Dhingra SK, Gabryszewski SJ, Small-Saunders JL, Yeo T, Henrich PP, Mok S, et al. Global spread of mutant PfCRT and its pleiotropic impact on Plasmodium falciparum multidrug resustance and fitness. mBio. 2019;10:e02731-18.
Sondo P, Derra K, Tarnagda Z, Nakanabo SD, Zampa O, Kazienga A, et al. Dynamic of Plasmodiumfalciparum chloroquine resistance transporter gene Pfcrt K76T mutation five years after withdrawal of chloroquine in Burkina Faso. Pan Afr Med J. 2015;21:101.
Mbogo GW, Nankoberanyi S, Tukwasibwe S, Baliraine FN, Nsobya SL, Conrad MD, et al. Temporal changes in prevalence of molecular markers mediating antimalarial drug resistance in a high malaria transmission setting in Uganda. Am J Trop Med Hyg. 2014;91:54–61.
Conrad MD, Bigira V, Kapisi J, Muhindo M, Kamya MR, Havlir DV, et al. Polymorphisms in K13 and falcipain-2 associated with artemisinin resistance are not prevalent in Plasmodium falciparum isolated from Ugandan children. PLoS ONE. 2014;9:e0105690.
Tumwebaze P, Conrad MD, Walakira A, LeClair N, Byaruhanga O, Nakazibwe C, et al. Impact of antimalarial treatment and chemoprevention on the drug sensitivity of malaria parasites isolated from Ugandan children. Antimicrob Agents Chemother. 2015;59:3018–30.
Somé AF, Séré YY, Dokomajilar C, Zongo I, Rouamba N, Greenhouse B, et al. Selection of known Plasmodium falciparum resistance-mediating polymorphisms by artemether-lumefantrine and amodiaquine-sulfadoxine-pyrimethamine but not dihydroartemisinin-piperaquine in Burkina Faso. Antimicrob Agents Chemother. 2010;54:1949–54.
Venkatesan M, Gadalla NB, Stepniewska K, Dahal P, Nsanzabana C, Moriera C, et al. Polymorphisms in Plasmodiumfalciparum chloroquine resistance transporter and multidrug resistance 1 genes: Parasite risk factors that affect treatment outcomes for P.falciparum malaria after artemether-lumefantrine and artesunate-amodiaquine. Am J Trop Med Hyg. 2014;91:833–43.
Sisowath C, Petersen I, Veiga MI, Mårtensson A, Premji Z, Björkman A, et al. In vivo selection of Plasmodium falciparum parasites carrying the chloroquine-susceptible pfcrt K76 allele after treatment with artemether-lumefantrine in Africa. J Infect Dis. 2009;199:750–7.
Omar SA, Adagu IS, Warhurst DC. Can pretreatment screening for dhps and dhfr point mutations in Plasmodium falciparum infections be used to predict sulfadoxine-pyrimethamine treatment failure? Trans R Soc Trop Med Hyg. 2001;95:315–9.
Staedke SG, Sendagire H, Lamola S, Kamya MR, Dorsey G, Rosenthal PJ. Relationship between age, molecular markers, and response to sulphadoxine-pyrimethamine treatment in Kampala, Uganda. Trop Med Int Health. 2004;9:624–9.
Kublin JG, Dzinjalamala FK, Kamwendo DD, Malkin EM, Cortese JF, Martino LM, et al. Molecular markers for failure of sulfadoxine-pyrimethamine and chlorproguanil-dapsone treatment of Plasmodium falciparum malaria. J Infect Dis. 2002;185:380–8.
Desai M, Gutman J, Taylor SM, Wiegand RE, Khairallah C, Kayentao K, et al. Impact of sulfadoxine-pyrimethamine resistance on effectiveness of intermittent preventive therapy for Malaria in pregnancy at clearing infections and preventing low birth weight. Clin Infect Dis. 2016;62:323–33.
Okell LC, Griffin JT, Roper C. Mapping sulphadoxine-pyrimethamine-resistant Plasmodium falciparum malaria in infected humans and in parasite populations in Africa. Sci Rep. 2017;7:7389.
Ferreira PE, Holmgren G, Veiga MI, Uhlén P, Kaneko A, Gil JP. PfMDR1: mechanisms of transport modulation by functional polymorphisms. PLoS ONE. 2011;6:e0023875.
Idowu AO, Oyibo WA, Bhattacharyya S, Khubbar M, Mendie UE, Bumah VV, et al. Rare mutations in Pfmdr1 gene of Plasmodium falciparum detected in clinical isolates from patients treated with anti-malarial drug in Nigeria. Malar J. 2019;18:319.
Muiruri P, Juma DW, Ingasia LA, Chebon LJ, Opot B, Ngalah BS, et al. Selective sweeps and genetic lineages of Plasmodium falciparum multi-drug resistance (pfmdr1) gene in Kenya. Malar J. 2018;17:398.
Gil JP, Krishna S. pfmdr1 (Plasmodium falciparum multidrug drug resistance gene 1): a pivotal factor in malaria resistance to artemisinin combination therapies. Expert Rev Anti Infect Ther. 2017;15:527–43.
Nsobya SL, Kiggundu M, Nanyunja S, Joloba M, Greenhouse B, Rosenthal PJ. In vitro sensitivities of Plasmodium falciparum to different antimalarial drugs in Uganda. Antimicrob Agents Chemother. 2010;54:1200–6.
Nkhoma S, Nair S, Mukaka M, Molyneux ME, Ward SA, Anderson TJC. Parasites bearing a single copy of the multi-drug resistance gene (pfmdr-1) with wild-type SNPs predominate amongst Plasmodium falciparum isolates from Malawi. Acta Trop. 2009;111:78–81.
Uhlemann AC, McGready R, Ashley EA, Brockman A, Singhasivanon P, Krishna S, et al. Intrahost selection of Plasmodium falciparum pfmdr1 alleles after antimalarial treatment on the northwestern border of Thailand. J Infect Dis. 2007;195:134–41.
Duah NO, Matrevi SA, De Souza DK, Binnah DD, Tamakloe MM, Opoku VS, et al. Increased pfmdr1 gene copy number and the decline in pfcrt and pfmdr1 resistance alleles in Ghanaian Plasmodium falciparum isolates after the change of anti-malarial drug treatment policy. Malar J. 2013;12:377.
Veiga MI, Dhingra SK, Henrich PP, Straimer J, Gnädig N, Uhlemann AC, et al. Globally prevalent PfMDR1 mutations modulate Plasmodium falciparum susceptibility to artemisinin-based combination therapies. Nat Commun. 2016;7:11553.
Asua V, Vinden J, Conrad MD, Legac J, Kigozi SP, Kamya MR, et al. Changing molecular markers of antimalarial drug sensitivity across Uganda. Antimicrob Agents Chemother. 2019;63:e01818-e1918.
Ocan M, Akena D, Nsobya S, Kamya MR, Senono R, Kinengyere AA, et al. K13-propeller gene polymorphisms in Plasmodium falciparum parasite population in malaria affected countries: a systematic review of prevalence and risk factors. Malar J. 2019;18:60.
Ménard D, Khim N, Beghain J, Adegnika AA, Shafiul-Alam M, Amodu O, et al. A worldwide map of Plasmodium falciparum K13-propeller polymorphisms. N Engl J Med. 2016;374:2453–64.
Van Hong N, Amambua-Ngwa A, Tuan NQ, Cuong DD, Giang NTH, Van Dung N, et al. Severe malaria not responsive to artemisinin derivatives in man returning from Angola to Vietnam. Emerg Infect Dis. 2014;20:1199–202.
Lu F, Culleton R, Zhang M, Ramaprasad A, von Seidlein L, Zhou H, et al. Emergence of indigenous artemisinin-resistant Plasmodium falciparum in Africa. N Engl J Med. 2017;376:991–3.
Kamau E, Campino S, Amenga-Etego L, Drury E, Ishengoma D, Johnson K, et al. K13-propeller polymorphisms in Plasmodium falciparum parasites from sub-Saharan Africa. J Infect Dis. 2015;211:1352–5.
Taylor SM, Parobek CM, De Conti DK, Kayentao K, Coulibaly SO, Greenwood BM, et al. Absence of putative artemisinin resistance mutations among Plasmodium falciparum in sub-Saharan Africa: a molecular epidemiologic study. J Infect Dis. 2015;211:680–8.
Isozumi R, Uemura H, Kimata I, Ichinose Y, Logedi J, Omar AH, et al. Novel mutations in k13 propeller gene of artemisinin-resistant Plasmodium falciparum. Emerg Infect Dis. 2015;21:490–2.
Escobar C, Pateira S, Lobo E, Lobo L, Teodosio R, Dias F, et al. Polymorphisms in Plasmodium falciparum K13-propeller in angola and mozambique after the introduction of the ACTs. PLoS ONE. 2015;10:e0119215.
Tacoli C, Gai PP, Bayingana C, Sifft K, Geus D, Ndoli J, et al. Artemisinin resistance-associated K13 polymorphisms of Plasmodium falciparum in Southern Rwanda, 2010–2015. Am J Trop Med Hyg. 2016;95:1090–3.
Boussaroque A, Fall B, Madamet M, Camara C, Benoit N, Fall M, et al. Emergence of mutations in the K13 propeller gene of Plasmodium falciparum isolates from Dakar, Senegal, in 2013–2014. Antimicrob Agents Chemother. 2016;60:624–7.
Yang C, Zhang H, Zhou R, Qian D, Liu Y, Zhao Y, et al. Polymorphisms of Plasmodium falciparum k13-propeller gene among migrant workers returning to Henan Province, China from Africa. BMC Infect Dis. 2017;17:560.
WHO. Global technical strategy for malaria 2016–2030. Geneva, World Health Organization, 2015. http://apps.who.int/iris/bitstream/10665/176712/1/9789241564991_eng.pdf?ua=1.
Kabir MM, Naher S, Islam A, Karim A, Rasid MH-O, Laskar SI. Vector control using LLIN/ITN: reduction of malaria morbidity in Bangladesh. Malar J. 2014;13:47.
Acknowledgements
We are grateful to the population of Praia city, Cape Verde who accepted to collaborate in this study. We thank the Malaria Pre elimination Program (António Lima Moreira), Health Delegate (Ullardina Furtado) and laboratory technicians Health delegation of Praia (especially Admilson Ramos and Maria Rosa Mendonça) and laboratory technicians of Hospital Dr. Agostinho Neto (Josefa Varela and Elisa Veiga) for sample collection. Especially acknowledgment to Celivianne Marisia Ramos de Sousa for laboratory support, Manuel Tavares, driver in the National Institute of Public Health and Jailson Jose Tavares Varela, technician of National Institute of Territory Management for the maps development. This publication uses some data generated and published by the Pf3k project (www.malariagen.net/pf3k) [26].
Funding
This research was partially supported by National Institute of Public Health through Government of Cape Verde funds to 65.06.01.04.37-Promoçao Da Capacidade Institucional e Tecnica Do INSP. It was also partially supported by Fundação para a Ciência e a Tecnologia for funds to GHTM-UID/04413/2020, Portugal. TGC received funding from the MRC UK (Grant no. MC_PC_15103, MR/K000551/1, MR/M01360X/1, MR/N010469/1, MR/R020973/1) and BBSRC UK (BB/R013063/1). SC received funding from the Medical Research Council UK grants (MC_PC_15103, MR/R020973/1) and the BBSRC UK (BB/R013063/1).
Author information
Authors and Affiliations
Contributions
SL, HS, TV, FN and LG were responsible for the study design. SL, DM and IV collected the blood samples. SL, SC, FN, and LG interpreted the data. IV, TC, SL, FN, SC performed the polymerase chain reaction. DW, FN and SC Performed the ELISA test. SC, AI, EDB and TGC performed and analysed the whole genome sequencing. SL, FN, and HS wrote the first version of the manuscript. EBD, AI, SC, TGC, MLLM, and FN contributed to the writing, review and editing of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was reviewed and approved by the Ethical Committee of the Ministry of Health of Cape Verde (Reference no. 39/2017) and by the National Data Protection Commission (reference no. 30/2019). Written informed consent of all participants was then obtained after giving respondents adequate information on the aim of the study, potential risks and benefits of being participant, and the rights of the respondents. The privacy and confidentiality issues had been secured throughout the processes.
Consent for publication
Received from all co-authors.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Number of malaria cases in Praia city per neighbourhood (N).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Da Veiga Leal, S., Ward, D., Campino, S. et al. Drug resistance profile and clonality of Plasmodium falciparum parasites in Cape Verde: the 2017 malaria outbreak. Malar J 20, 172 (2021). https://doi.org/10.1186/s12936-021-03708-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-021-03708-z