
Abstract
Background
The production of properly folded, recombinant sub-unit Plasmodium falciparum malaria vaccine candidates in sufficient quantities is often a challenge. Success in vaccine immunogenicity studies in small animal models does not always predict immunogenicity in non-human primates and/or human subjects. The aim of this study was to assess the immunogenicity of a chimeric blood-stage malaria vaccine in Aotus monkeys. This vaccine candidate includes the neutralizing B cell epitopes of P. falciparum merozoite surface protein 1 (rPfMSP119) genetically linked to a highly immunogenic, well-conserved P. falciparum merozoite surface protein 8 (rPfMSP8 (ΔAsn/Asp)) partner.
Methods
Aotus nancymaae monkeys were immunized with purified rPfMSP1/8 or rPfMSP8 (ΔAsn/Asp) formulated with Montanide ISA 720 as adjuvant, or with adjuvant alone. Antibody responses to MSP119 and MSP8 domains were measured by ELISA following primary, secondary and tertiary immunizations. The functionality of vaccine-induced antibodies was assessed in a standard P. falciparum blood-stage in vitro growth inhibition assay. Non-parametric tests with corrections for multiple comparisons when appropriate were used to determine the significance of differences in antigen-specific IgG titres and in parasite growth inhibition.
Results
The chimeric rPfMSP1/8 vaccine was shown to be well tolerated and highly immunogenic with boost-able antibody responses elicited to both PfMSP8 and PfMSP119 domains. Elicited antibodies were highly cross-reactive between FVO and 3D7 alleles of PfMSP119 and potently inhibited the in vitro growth of P. falciparum blood-stage parasites.
Conclusions
Similar to previous results with inbred and outbred mice and with rabbits, the PfMSP1/8 vaccine was shown to be highly effective in eliciting P. falciparum growth inhibitory antibodies upon immunization of non-human primates. The data support the further assessment of PfMSP1/8 as a component of a multivalent vaccine for use in human subjects. As important, the data indicate that rPfMSP8 (ΔAsn/Asp) can be used as a malaria specific carrier protein to: (1) drive production of antibody responses to neutralizing B cell epitopes of heterologous vaccine candidates and (2) facilitate production of properly folded, recombinant P. falciparum subunit vaccines in high yield.
Similar content being viewed by others


Background
There have been measurable gains in reducing the global burden of malaria through the integration of control programmes involving insecticide-treated bed nets, indoor residual spraying of pesticides, and intermittent preventive therapy [1]. Nevertheless, the malaria burden remains high, with the World Health Organization reporting 198 million cases and an estimated 367,000–755,000 deaths worldwide in 2013. It is expected that the addition of an effective malaria vaccine to the battery of malaria control strategies would accelerate the decline in disease and promote long-term sustainable control. RTS,S, the first malaria vaccine to reach phase III clinical trials, is a pre-erythrocytic-stage vaccine based on the circumsporozoite protein of Plasmodium falciparum [2]. Initial reports suggest that vaccine efficacy may only be around 30 % in the most vulnerable target population of infants [3], with a higher efficacy of approximately 50 % in young children [4], but with concerns regarding the durability of protection. The efficacy of other malaria vaccine candidates that have been evaluated in phase II trials has not been impressive [5–10].
There is mounting agreement in the field that an effective malaria vaccine will require induction of immune responses to multiple, distinct target antigens. This concept is central in the development of whole parasite-based vaccines, including radiation (PfSPZ) [11, 12] and genetically (PfGAS) [12–14] attenuated sporozoite vaccines, infection-treatment pre-erythrocytic-stage vaccines [15, 16], and chemically inactivated whole blood-stage vaccines [17, 18]. However, these whole parasite approaches face significant challenges related to production, formulation, standardization, delivery, and safety that are less problematic for sub-unit-based vaccines. Other significant challenges associated with sub-unit malaria vaccine development have been encountered. These include difficulties in producing properly folded candidate antigens, polymorphism in T and B cell epitopes, and poor immunogenicity. Several years ago while working in the Plasmodium yoelii model, the additional problem of antigenic competition was encountered when combining just two blood-stage vaccine components, merozoite surface protein 1 (MSP142) and MSP8 [19]. This problem has also impeded the development of other multi-antigen malaria vaccine formulations [20–24].
For sub-unit malaria vaccines, a well-established strategy to enhance the immunogenicity of neutralizing B cell epitopes was adopted, namely the use of a highly immunogenic carrier protein. Taking advantage of the immunogenicity of MSP8, a chimeric protein with the conformational, protective B cell epitopes of MSP119, fused to MSP8 was generated. Immunization with the chimeric rPyMSP1/8 vaccine induced high and comparable antibody responses against both PyMSP119 and PyMSP8, resulting in nearly complete protection against lethal P. yoelii 17XL malaria [19]. The enhanced efficacy of the rPyMSP1/8 vaccine, in comparison to a combined formulation of rPyMSP142 and rPyMSP8, was not due to an improved conformation of protective B cell epitopes or the generation of novel epitopes in the chimeric molecule [19, 25]. The key finding was that immunization with rPyMSP1/8 elicited an MSP8-restricted T cell response that provided effective help for both PyMSP119 and PyMSP8 specific B cells to produce high and sustained levels of protective antibodies [25].
Based on the proof-of-concept studies in the P. yoelii model, P. falciparum MSP8 was pursued as a parasite-specific carrier protein to overcome challenges associated with the production of recombinant antigen vaccines (quality, yield) and with the sub-optimal immunogenicity of relevant neutralizing B cell epitopes [26, 27]. Among different P. falciparum isolates, MSP8 is highly conserved, exhibiting 95 % amino acid identity with slight variations in an N-terminal Asn/Asp-rich domain [28]. The remaining C-terminal sequence is invariant. Following codon harmonization [29] and genetic fusion of PfMSP119 and PfMSP8 (ΔAsn/Asp) coding sequences, a properly folded chimeric antigen was produced in high yield, utilizing an Escherichia coli expression system [27]. Immunogenicity studies in both inbred and outbred mice demonstrated a strong T cell response restricted to epitopes within PfMSP8 sequence and the production of high titres of antibodies to both MSP119 and MSP8 domains. Comparable PfMSP1/8 immunogenicity studies in rabbits demonstrated the induction of high titres of PfMSP119 specific antibodies that very effectively inhibited the in vitro growth of P. falciparum parasites of both the 3D7 and FVO strains. While these data are encouraging, results of vaccine studies in mice and rabbits do not always predict outcomes upon immunization of human subjects. In this study, the immunogenicity of PfMSP1/8 was evaluated in non-human primates, assessing the magnitude, specificity and functional activity of elicited antibodies.
Methods
Recombinant antigens
The production and purification of the chimeric rPfMSP1/8 and rPfMSP8 (ΔAsn/Asp) (P. falciparum FVO strain) followed the same protocol, using codon-harmonized, synthetic gene sequences cloned into pET-28 (EMD Biosciences, San Diego, CA, USA) and SHuffle™ T7 Express lysY E. coli cells (New England Biolabs, Ipswich, MA, USA) as host. Expression of the recombinant proteins was accomplished using a BioFLo115 bench-top bioreactor (New Brunswick Scientific, Edison, NJ, USA). Protocols for the expression and purification of recombinant antigens have been reported previously [26, 27]. For this study, rPfMSP1/8 and rPfMSP8 (ΔAsn/Asp) were further purified by gel filtration (Superdex 75, GE Healthcare Bio-Sciences Corp, Piscataway, NJ, USA) followed by a second round of Ni–NTA affinity chromatography (Qiagen, Valencia, CA, USA). The eluted material was dialyzed into 20 mM Tris–HCl, pH 7.2, 0.5 M NaCl. The final preparations of rPfMSP1/8 and rPfMSP8 (ΔAsn/Asp) were filter sterilized (0.22 µm), aliquoted and stored at −80 °C. The final protein concentration was determined by bicinchoninic acid protein assay (BCA; Thermo Scientific, Rockford, IL, USA). Endotoxin levels were quantitated using the ToxinSensor Chromogenic LAL Endotoxin Assay (GenScript, Piscataway, NJ, USA). Protein purity and conformation were assessed by Coomassie blue staining following SDS-PAGE on 10 % gels, run under both reduced and non-reduced conditions. Purity (reduced lanes) and % monomer (doublet, non-reduced lanes) were estimated by densitometry using ImageJ processing and analysis software [30].
Animals and immunizations
Adult Aotus nancymaae monkeys were housed at a Centers for Disease Control (CDC) primate facility, fully-accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC). Animal studies were reviewed, approved and conducted in compliance with the Institutional Animal Care and Use Committee (IACUC) of CDC. Eighteen Aotus monkeys were stratified according to weight and sex into three groups of six animals, which were then randomly assigned to vaccine and control groups. On day 0, groups of animals were immunized by intramuscular injection (0.5 ml) of 50 µg of rPfMSP1/8 or rPfMSP8 (ΔAsn/Asp) emulsified in Montanide ISA 720 (Seppic Inc, Paris, France) at a ratio of 70:30 (vol/vol). Control animals were immunized with saline/Montanide alone. Monkeys were boosted on day 28 and on day 84 with the same antigen/adjuvant formulation that was used for the priming immunization.
Safety assessment
All monkeys were in good health, free of tuberculosis and weighed between 0.8 and 1.2 kg at the start of the study. Animals were weighed at weekly intervals. Blood and serum were collected at biweekly intervals beginning 2 weeks prior to the first immunization and until the completion of the study. Immunization sites were monitored continuously for adverse local reactions. Systemic reactions were evaluated by monitoring a panel of haematologic parameters and serum chemistry values that included measurements of white blood cells, red blood cells, haemoglobin, haematocrit, platelets, blood urea nitrogen, glucose, alkaline phosphatase, total protein, alanine aminotransferase, and creatinine.
Measurement of antigen-specific antibody responses by ELISA
Antigen-specific antibody responses induced by immunization with rPfMSP1/8 and rPfMSP8 (ΔAsn/Asp) were measured by ELISA as previously described [27]. Plates were coated with 0.25 µg per well of purified rPfMSP1/8, rPfMSP8 (ΔAsn/Asp), rGST-PfMSP19 (FVO) or rGST-PfMSP19 (3D7). Pre-immune sera and sera collected 2 weeks following primary, secondary and tertiary immunizations were serially diluted twofold starting at 1:1250. Bound antibodies were detected using horseradish peroxidase conjugated goat anti-Aotus IgG [31] diluted 1:7500 and with ABTS [2,2′-azinobis(3-ethylbenzthiazolinesulfonic acid)] as substrate. A 405 values between 1.0 and 0.1 were plotted and titre calculated as the reciprocal of the dilution that yielded an A 405 of 0.5. A high titre pool of serum obtained from rPfMSP1/8 immunized monkeys (n = 5) was included on each assay plate as an internal reference to normalize the data between assays. Pre-immune sera were used to establish background reactivity of each animal to each antigen and subtracted as background.
Plasmodium falciparum growth inhibition assays
The growth inhibitory activity (GIA) of protein G purified IgG from rPfMSP1/8 to rPfMSP8 (ΔAsn/Asp) immunized and adjuvant control monkeys was assessed in vitro by the measurement of parasite lactate dehydrogenase activity [32] using standard protocols. Pre-bleed and adjuvant control IgG served as negative controls. Immune IgG was tested at a concentration of 10 mg/ml. GIA was calculated relative to P. falciparum FVO blood-stage parasites growing in complete media in the absence of any added Aotus IgG.
Statistical analysis
When comparing data from two groups, the statistical significance of the differences in antigen-specific IgG titres or parasite growth inhibition was determined by the Mann–Whitney test. The statistical significance of increases in antigen-specific titres between paired primary, secondary and tertiary immunization sera was determined using the Friedman test followed by Dunn’s multiple comparison correction. Probability (p) values ≤ 0.05 were considered significant.
Results
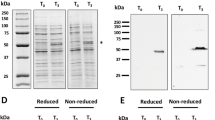
The production and purification of rPfMSP1/8 and rPfMSP8 (ΔAsn/Asp) was reported previously [26, 27]. Figure 1 shows a Coomassie-blue stained polyacrylamide gel of reduced (lanes 1 and 2) and non-reduced (lanes 3 and 4) purified recombinant antigens used in this Aotus immunogenicity study. As previously observed, the two recombinant vaccine antigens migrate as a predominant doublet in the absence of reducing agent [26, 27]. Purity of the final product was estimated to be 93–94 % with ≥85 % monomer. Endotoxin levels in the final vaccine preparation were less than 1 EU/mg of protein. Prior studies showed that immunization of Aotus monkeys with rPfMSP142 elicited antibodies that inhibited the in vitro growth of P. falciparum blood-stage parasites [31]. rPfMSP142 vaccines formulated with Complete Freund’s adjuvant elicited higher titres of functional antibodies in comparison to formulations with Montanide ISA 720. For this study, Montanide ISA 720 was selected as adjuvant in an effort to balance potency with safety. Aotus monkeys (six per group) were immunized and boosted twice (days 0, 28 and 84) with 50 µg/dose of rPfMSP1/8 or rPfMSP8 (ΔAsn/Asp) formulated with Montanide ISA 720 or with adjuvant alone.

Purified recombinant chimeric rPfMSP1/8 and rPfMSP8 (ΔAsn/Asp) vaccine antigens. A Coomassie blue-stained 10 % SDS–polyacrylamide gel containing purified rPfMSP1/8 (3 μg, lanes 1 and 3) or rPfMSP8 (ΔAsn/Asp) (3 μg, lanes 2 and 4) was run under reducing (lanes q and 2) and non-reducing (lanes 3 and 4) conditions. Molecular weight markers in kilodaltons (kDa) are shown. Endotoxin levels and % purity for each antigen preparation are also indicated
Immunization with rPfMSP1/8 or rPfMSP8 (ΔAsn/Asp) was reasonably well tolerated. Local injection site reactions were noted following the second and third immunizations (Table 1). These consisted primarily of small nodules at the injection site and/or local swelling, which typically resolved. Prior to the first immunization, baseline haematology and blood chemistry profiles were determined for each animal. Subsequently, blood and serum were collected biweekly through the course of the study for monitoring. Fluctuations from baseline values were noted in individual animals across groups. Alterations attributable to a systemic response to a specific vaccine formulation were not apparent.
Sera collected 2 weeks after each immunization were evaluated by ELISA to determine the titres of IgG antibodies specific for the rPfMSP8 (ΔAsn/Asp) carrier and for PfMSP119 epitopes of both the FVO and 3D7 alleles (Fig. 2). Immunization with rPfMSP8 (ΔAsn/Asp) or rPfMSP1/8 elicited comparable IgG responses to rPfMSP8 specific epitopes following the primary immunization (p > 0.5). Two additional immunizations markedly boosted the rPfMSP8-specific IgG response in both groups by >100-fold (p < 0.05, primary vs tertiary mean titre). Significant titres of antibodies to PfMSP119 epitopes were not detected in rPfMSP8 (ΔAsn/Asp) immunized animals following primary and secondary immunization. Following a third immunization with the rPfMSP8 (ΔAsn/Asp) carrier, a low titre of antibodies cross-reactive with PfMSP119 was detected in two animals, presumably due to some similarity in the C-terminal EGF-like domains of PfMSP1 and PfMSP8 [33]. In contrast, immunization with rPfMSP1/8 induced a strong and boost-able antibody response to PfMSP119 that was highly cross-reactive between the FVO and 3D7 alleles of PfMSP119.

Specificity of antibody response induced by immunization with rPfMSP8 (ΔAsn/Asp) versus rPfMSP1/8 vaccines. Antigen-specific IgG titres (mean ± standard deviation) in sera collected from Aotus monkeys (6 animals/group) immunized with a rPfMSP8 (ΔAsn/Asp) or b rPfMSP1/8 vaccines formulated with Montanide ISA 720 were determined by ELISA. Sera collected 2 weeks following primary, secondary and tertiary immunization were evaluated. ELISA plates were coated with either rPfMSP1/8, rPfMSP8 (ΔAsn/Asp), rGST-PfMSP119 (FVO), or rGST-PfMSP119 (3D7) as indicated. For each animal, reactivity of pre-immunization serum was subtracted as background
To assess functionality, antibodies elicited by immunization with rPfMSP8 (ΔAsn/Asp) or rPfMSP1/8 were evaluated for the ability to inhibit the in vitro growth of P. falciparum blood-stage parasites. As shown in Fig. 3, purified IgGs obtained following the third immunization of Aotus monkeys with rPfMSP1/8 effectively inhibited parasite growth. In contrast, no growth inhibitory activity was observed with IgG from rPfMSP8 (ΔAsn/Asp) immunized or adjuvant control animals (p < 0.01). PfMSP8 is not believed to be a significant target of growth inhibitory antibodies due to its low and transient expression on the merozoite surface [26, 34]. These data from Aotus monkeys are also consistent with the previous findings in antigen reversal assays, demonstrating that the inhibitory activity of rabbit anti-rPfMSP1/8 IgG was due to antibodies specific for PfMSP119 epitopes [27]. However, there was no correlation between GIA and the magnitude of the antibody response to rPfMSP1/8, rPfMSP8 (ΔAsn/Asp) or rPfMSP119 in rPfMSP1/8 immunized animals. As such, the fine specificity of the PfMSP119 response and/or a potential interaction between PfMSP119 and PfMSP8 specific antibodies may be a factor.

In vitro inhibition of Plasmodium falciparum growth by Aotus IgG induced by immunization with rPfMSP8 (ΔAsn/Asp) versus rPfMSP1/8 vaccines. In vitro growth inhibitory activity of IgG from immunized Aotus monkeys for P. falciparum FVO blood-stage parasites was based on measurement of parasite lactate dehydrogenase levels. Purified IgG (10 mg/ml) from animals immunized three times with Montanide alone (n = 6), rPfMSP8 (ΔAsn/Asp) + Montanide (n = 5) or rPfMSP1/8 + Montanide (n = 5) was evaluated. Two immunized animals (1 rPfMSP8 (ΔAsn/Asp), 1 rPfMSP1/8) tested at less than 10 mg/ml of IgG were not included in the Figure. Per cent growth inhibition in the presence of Aotus IgG relative to controls in the absence of IgG is shown
Following immunization, rPfMSP8 (ΔAsn/Asp), rPfMSP1/8 and adjuvant control groups were challenged intravenously with 50,000 P. falciparum FVO parasitized red blood cells (pRBCs). Three rPfMSP8 (ΔAsn/Asp) immunized animals developed moderate parasitaemia (<200,000 pRBC/µl) and self-cured. The remaining three animals in this group developed high-density parasitaemia (>200,000 pRBC/µl) requiring drug treatment to clear the infection. Similarly, three rPfMSP1/8 immunized animals controlled parasitaemia (<200,000 pRBC/µl), with two of these clearing their infection and one requiring drug treatment due to anaemia. In the Montanide control group, three animals developed high-density parasitaemia (>200,000 pRBC/µl) and were drug treated to clear the infection. However, the remaining two adjuvant control animals exhibited greatly extended prepatent periods, developed unexpectedly low peak parasitaemia (<5000 pRBC/µl) and rapidly self-cured. The reason for this large and unusual variability in parasitaemia in the control group with this highly virulent strain was not apparent. This line of P. falciparum FVO strain in almost every previous instance has produced rapidly rising parasitaemia in control animals that required drug treatment to prevent death. Nevertheless, because of this unexpected variability, the ability to draw meaningful conclusions regarding vaccine efficacy was compromised.
In a previous study, Lyon et al. [31] completed an immunogenicity study in Aotus monkeys with rPfMSP142 formulated with CFA/IFA, AS02A or Montanide 720. With the exception of adjuvant choice, immunization protocols were similar to the present study with respect to immunizing dose, route, number of immunizations, and interval between immunizations. However, antibodies elicited by immunization with PfMSP142 formulated with AS02A or Montanide 720 only modestly inhibited P. falciparum growth. The induction of more effective inhibitory antibodies required immunization with PfMSP142 formulated with Freund’s adjuvant. In contrast, IgG elicited following immunization with the PfMSP1/8 chimeric vaccine formulated with Montanide ISA 720, an adjuvant which has been used in human clinical trials, potently inhibited parasite growth (52.0 ± 11.3 %).
These data, in conjunction with prior studies [26, 27], demonstrate that rPfMSP8 (ΔAsn/Asp) is an effective malaria-specific carrier protein that elicits strong CD4+ T cell help for the production of merozoite neutralizing antibodies to linked PfMSP119 epitopes. The potency of rPfMSP8 (ΔAsn/Asp) has been demonstrated in mice and rabbits and now in non-human primates and does not require Freund’s adjuvant. Upon immunization of human subjects, similarly strong CD4+ T cell responses to provide B cell help are expected as rPfMSP8 (ΔAsn/Asp) is predicted to contain epitopes that will bind with high affinity to multiple HLA class II alleles [35]. It is also notable that rPfMSP8 (ΔAsn/Asp) has been engineered to facilitate production and purification of chimeric antigens at high levels and in native conformation utilizing readily scalable protocols and procedures. By fusing relevant neutralizing B cell epitopes of other vaccine candidates to PfMSP8 (ΔAsn/Asp), a conserved, highly immunogenic P. falciparum-specific carrier protein, it may be possible to similarly to overcome challenges associated with the production of recombinant antigen vaccines (quality, yield) and sub-optimal immunogenicity.
Conclusions
Extending previous work in small animal models, these studies now show that the chimeric PfMSP1/8 vaccine is highly immunogenic in non-human primates and induces potent parasite neutralizing antibodies. These data justify additional effort to incorporate PfMSP1/8 as a component of a multivalent P. falciparum malaria vaccine to augment control efforts. rPfMSP8 (ΔAsn/Asp) may have broader use as a fusion partner for poorly immunogenic and/or difficult to produce sub-unit vaccine candidates.
References
WHO. World Malaria Report 2014. Geneva: World Health Organization; 2014.
Cohen J, Nussenzweig V, Nussenzweig R, Vekemans J, Leach A. From the circumsporozoite protein to the RTS, S/AS candidate vaccine. Hum Vaccin. 2010;6:90–6.
Agnandji ST, Lell B, Fernandes JF, Abossolo BP, Methogo BG, Kabwende AL, et al. A phase 3 trial of RTS, S/AS01 malaria vaccine in African infants. N Engl J Med. 2012;367:2284–95.
Agnandji ST, Lell B, Soulanoudjingar SS, Fernandes JF, Abossolo BP, Conzelmann C, et al. First results of phase 3 trial of RTS, S/AS01 malaria vaccine in African children. N Engl J Med. 2011;365:1863–75.
Ellis RD, Sagara I, Doumbo O, Wu Y. Blood stage vaccines for Plasmodium falciparum: current status and the way forward. Hum Vaccin. 2010;6:627–34.
Sagara I, Dicko A, Ellis RD, Fay MP, Diawara SI, Assadou MH, et al. A randomized controlled phase 2 trial of the blood stage AMA1-C1/Alhydrogel malaria vaccine in children in Mali. Vaccine. 2009;27:3090–8.
Spring MD, Cummings JF, Ockenhouse CF, Dutta S, Reidler R, Angov E, et al. Phase 1/2a study of the malaria vaccine candidate apical membrane antigen-1 (AMA-1) administered in adjuvant system AS01B or AS02A. PLoS ONE. 2009;4:e5254.
Thera MA, Doumbo OK, Coulibaly D, Laurens MB, Ouattara A, Kone AK, et al. A field trial to assess a blood-stage malaria vaccine. N Engl J Med. 2011;365:1004–13.
Ogutu BR, Apollo OJ, McKinney D, Okoth W, Siangla J, Dubovsky F, et al. Blood stage malaria vaccine eliciting high antigen-specific antibody concentrations confers no protection to young children in Western Kenya. PLoS ONE. 2009;4:e4708.
Schwartz L, Brown GV, Genton B, Moorthy VS. A review of malaria vaccine clinical projects based on the WHO rainbow table. Malar J. 2012;11:11.
Seder RA, Chang LJ, Enama ME, Zephir KL, Sarwar UN, Gordon IJ, et al. Protection against malaria by intravenous immunization with a nonreplicating sporozoite vaccine. Science. 2013;341:1359–65.
Epstein JE, Richie TL. The whole parasite, pre-erythrocytic stage approach to malaria vaccine development: a review. Curr Opin Infect Dis. 2013;26:420–8.
Spring M, Murphy J, Nielsen R, Dowler M, Bennett JW, Zarling S, et al. First-in-human evaluation of genetically attenuated Plasmodium falciparum sporozoites administered by bite of Anopheles mosquitoes to adult volunteers. Vaccine. 2013;31:4975–83.
Vaughan AM, Wang R, Kappe SH. Genetically engineered, attenuated whole-cell vaccine approaches for malaria. Hum Vaccin. 2010;6:107–13.
Bijker EM, Bastiaens GJ, Teirlinck AC, van Gemert GJ, Graumans W, van de Vegte-Bolmer M, et al. Protection against malaria after immunization by chloroquine prophylaxis and sporozoites is mediated by preerythrocytic immunity. Proc Natl Acad Sci USA. 2013;110:7862–7.
Roestenberg M, McCall M, Hopman J, Wiersma J, Luty AJ, van Gemert GJ, et al. Protection against a malaria challenge by sporozoite inoculation. N Engl J Med. 2009;361:468–77.
McCarthy JS, Good MF. Whole parasite blood stage malaria vaccines: a convergence of evidence. Hum Vaccin. 2010;6:114–23.
Good MF, Reiman JM, Rodriguez IB, Ito K, Yanow SK, El-Deeb IM, et al. Cross-species malaria immunity induced by chemically attenuated parasites. J Clin Invest. 2013;123:3353–62.
Shi Q, Lynch MM, Romero M, Burns JM Jr. Enhanced protection against malaria by a chimeric merozoite surface protein vaccine. Infect Immun. 2007;75:1349–58.
Stowers AW, Kennedy MC, Keegan BP, Saul A, Long CA, Miller LH. Vaccination of monkeys with recombinant Plasmodium falciparum apical membrane antigen 1 confers protection against blood-stage malaria. Infect Immun. 2002;70:6961–7.
Sedegah M, Charoenvit Y, Minh L, Belmonte M, Majam VF, Abot S, et al. Reduced immunogenicity of DNA vaccine plasmids in mixtures. Gene Ther. 2004;11:448–56.
Sedegah M, Charoenvit Y, Aguiar J, Sacci J, Hedstrom R, Kumar S, et al. Effect on antibody and T-cell responses of mixing five GMP-produced DNA plasmids and administration with plasmid expressing GM-CSF. Genes Immun. 2004;5:553–61.
Pichyangkul S, Tongtawe P, Kum-Arb U, Yongvanitchit K, Gettayacamin M, Hollingdale MR, et al. Evaluation of the safety and immunogenicity of Plasmodium falciparum apical membrane antigen 1, merozoite surface protein 1 or RTS, S vaccines with adjuvant system AS02A administered alone or concurrently in rhesus monkeys. Vaccine. 2009;28:452–62.
Forbes EK, Biswas S, Collins KA, Gilbert SC, Hill AV, Draper SJ. Combining liver- and blood-stage malaria viral-vectored vaccines: investigating mechanisms of CD8+ T cell interference. J Immunol. 2011;187:3738–50.
Alaro JR, Lynch MM, Burns JM Jr. Protective immune responses elicited by immunization with a chimeric blood-stage malaria vaccine persist but are not boosted by Plasmodium yoelii challenge infection. Vaccine. 2010;28:6876–84.
Alaro JR, Angov E, Lopez AM, Zhou H, Long CA, Burns JM Jr. Evaluation of the immunogenicity and vaccine potential of recombinant Plasmodium falciparum merozoite surface protein 8. Infect Immun. 2012;80:2473–84.
Alaro JR, Partridge A, Miura K, Diouf A, Lopez AM, Angov E, et al. A chimeric Plasmodium falciparum merozoite surface protein vaccine induces high titers of parasite growth inhibitory antibodies. Infect Immun. 2013;81:3843–54.
Black CG, Wu T, Wang L, Hibbs AR, Coppel RL. Merozoite surface protein 8 of Plasmodium falciparum contains two epidermal growth factor-like domains. Mol Biochem Parasitol. 2001;114:217–26.
Angov E, Hillier CJ, Kincaid RL, Lyon JA. Heterologous protein expression is enhanced by harmonizing the codon usage frequencies of the target gene with those of the expression host. PLoS ONE. 2008;3:e2189.
Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Meth. 2012;9:671–5.
Lyon JA, Angov E, Fay MP, Sullivan JS, Girourd AS, Robinson SJ, et al. Protection induced by Plasmodium falciparum MSP1(42) is strain-specific, antigen and adjuvant dependent, and correlates with antibody responses. PLoS ONE. 2008;3:e2830.
Miura K, Zhou H, Moretz SE, Diouf A, Thera MA, Dolo A, et al. Comparison of biological activity of human anti-apical membrane antigen-1 antibodies induced by natural infection and vaccination. J Immunol. 2008;181:8776–83.
Drew DR, O’Donnell RA, Smith BJ, Crabb BS. A common cross-species function for the double epidermal growth factor-like modules of the highly divergent plasmodium surface proteins MSP-1 and MSP-8. J Biol Chem. 2004;279:20147–53.
Drew DR, Sanders PR, Crabb BS. Plasmodium falciparum merozoite surface protein 8 is a ring-stage membrane protein that localizes to the parasitophorous vacuole of infected erythrocytes. Infect Immun. 2005;73:3912–22.
Vita R, Zarebski L, Greenbaum JA, Emami H, Hoof I, Salimi N, et al. The immune epitope database 2.0. Nucleic Acids Res. 2010;38:D854–62.
Authors’ contributions
JMB, CAL and JWB conceived and designed the experiments. JMB, KM and JS performed the experiments. JMB, KM, CAL, and JWB analysed the data. JMB and KM wrote the paper. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by NIH-NIAID Grants AI035661 (JMB) and AI114292 (JMB), the Department of Microbiology and Immunology, Drexel University College of Medicine (JMB) and the Intramural Program of NIH-NIAID (CAL). The GIA Reference Center where the GIA assay was performed was supported by the PATH Malaria Vaccine Initiative. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Competing interests
JMB is an inventor listed on US Patent No. 7931,908 entitled “Chimeric MSP-Based Malaria Vaccine”.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Burns, J.M., Miura, K., Sullivan, J. et al. Immunogenicity of a chimeric Plasmodium falciparum merozoite surface protein vaccine in Aotus monkeys. Malar J 15, 159 (2016). https://doi.org/10.1186/s12936-016-1226-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-016-1226-5