
Abstract
Background
Esophageal cancer is a significant global health concern, ranking seventh in incidence and sixth in mortality. It encompasses two pathological types: esophageal squamous cell carcinoma (ESCC) and esophageal adenocarcinoma, with ESCC being more prevalent globally and associated with higher mortality rates. The POU (Pit-Oct-Unc) domain family transcription factors, comprising 15 members, play important roles in embryonic development and organ formation. Aberrant expression of POUs has been observed in several human cancers, influencing cell proliferation, tumor invasion, and drug resistance. However, their specific role in ESCC remains unknown.
Methods
We analyzed TCGA and GEO databases to assess POUs expression in ESCC tissues. Kaplan-Meier and ROC analyses were used to evaluate the prognostic value of POUs. Gene Set Enrichment Analysis and Protein-Protein interaction network were used to explore the potential pathway. Functional assays (Cell Counting Kit-8, EdU Staining assay, and cloning formation assay) and mechanism analyses (RNA-seq, flow cytometry, and Western blot) were conducted to determine the effects of POU4F1 knockdown on ESCC cell phenotypes and signaling pathways.
Results
POU4F1 and POU6F2 were upregulated in various cancer tissues, including ESCC, compared to normal tissues. POU4F1 expression was significantly correlated with patient survival and superior to previous models (AUC = 0.776). Knockdown of POU4F1 inhibited ESCC cell proliferation and affected cell cycle, autophagy, and DNA damage pathways in ESCC cells.
Conclusion
POU4F1 is a novel and promising prognostic and therapeutic target for ESCC patients, providing insights into potential treatment strategies.
Similar content being viewed by others

Introduction
According to the Global Cancer Statistics 2020, esophageal cancer (EC) holds the seventh position in terms of new cases and the sixth position in terms of fatalities worldwide [1]. Due to the economic gain and dietary habits, the incidence of esophageal squamous cell carcinoma (ESCC) is higher in developing countries, usually accounting for more than 90% of all EC cases [1]. The lack of effective diagnosis and treatment is often attributed to the high mortality and incidence rate of ESCC patients. Therefore, it is necessary to identify reliable prognostic biomarkers for the treatment of ESCC.
The POU (Pit-Oct-Unc) domain family genes encode a class of transcription factors that are distinguished by their unique DNA binding structure, known as the POU domain. This structural feature allows these transcription factors to bind to the octameric sequence ATGCAAAT [2]. Recent findings suggest the significant involvement of POUs in tumor initiation and progression. For instance, POU1F1 is associated with the occurrence and progression of tumors [3, 4]. Overexpression of POU5F1 promotes proliferation, migration, invasion, and EMT in colon cancer cells and lung cancer [5, 6]. Furthermore, a recent study published in the journal Advanced Science demonstrates that POU4F1 (also known as Brn3a) regulates G1 transition, thereby affecting the growth and malignant phenotype of breast cancer [7]. However, the precise role and mechanisms of the whole POUs in ESCC progression remain poorly understood.
This study aimed to evaluate the prognostic impact of POUs on ESCC patients and preliminarily explore their functional regulation and mechanisms in ESCC cells. Prognostic significance was assessed through differential analysis, Kaplan-Meier survival analysis, and Cox proportional hazards regression analysis using ESCC data from the TCGA and GEO databases. Functional experiments on ESCC cells and RNA sequencing analysis were conducted to explore the biological function of POUs and their associated pathway mechanisms. The findings are expected to provide valuable insights into the prognostic significance and therapeutic potential of POUs in ESCC.
Materials & methods
Sample and data collection
Public patient data were collected from the GSE53625 dataset, comprising 179 normal and 179 tumor samples. RNA sequencing data from an ESCC cohort, consisting of 11 normal and 81 tumor samples, were retrieved from TCGA. Furthermore, the pan-cancer dataset was sourced from the UCSC database (https://xenabrowser.net/). To streamline the analysis, cancer types with fewer than 3 samples were excluded, resulting in a dataset comprising expression data for 26 distinct cancer types.
Differentially expressed genes (DEGs) analysis
The TCGA database for ESCC employed the “edgeR” package, whereas GSE53625 utilized the “limma” package for differential analysis, applying a filtering criterion of P < 0.05 and |log2 fold change (FC)| > 0.585 to identify differentially expressed genes (DEGs).
Survival analysis of cancer patients
To investigate the potential association between POUs and survival, Kaplan–Meier survival analysis was conducted using the R package “Survival”. P < 0.05 was considered statistically significant.
Forecast model analysis establishment
The prognostic value of POU4F1 was evaluated by drawing the receiver operating characteristic (ROC) curve and calculating the area under curve (AUC) value. AUC is one of the evaluation indices for binary classification tasks (problems), taking values between 0 and 1, with the closer to 1, the higher the prediction accuracy. The Cox proportional hazards regression analysis was performed and a nomogram model, including TNM stage and POU4F1, was established to predict the prognosis of ESCC patients with the help of “nomogram” package.
Pathway analysis
Pathway analysis includes two parts: Gene Set Enrichment Analysis (GSEA) and Protein-Protein interaction (PPI) network. GSEA analysis was carried out between high POU4F1 expression group and low POU4F1 expression group to explore the potential signaling pathway. C5-Gene Ontology gene sets (c5.go.v2023.2.) and C2-Canonical pathway database (c2.cp.v2023.2.) containing KEGG dataset was downloaded as references. The STRING database (http://string.embl.de/) and GeneMANIA database (http://www.genemania.org) were used to construct the PPI network between POU4F1 and its interactional proteins.
Cell culture
ESCC cell line KYSE30 and KYSE70 were obtained from the Cell Bank of Chinese Academy of Sciences (Shanghai, China). Cells were cultured in RPMI1640 medium supplemented with 10% FBS and 1% penicillin / streptomycin at 37 °C with 5% CO2. Mycoplasma testing and STR identification have been performed for KYSE30 and KYSE70.
RNA interference
Small interfering RNAs (siRNA) targeting POU4F1 and a negative control (NC) were synthesized by Shanghai Genechem Co., Ltd. The sequences of the siRNA were listed in Additional file 2:Table S1. Lipofectamine™ RNAiMAX Transfection Reagent (Thermo Fisher Scientific, USA) was used for transiently transfection according to the manufacturer’s instruction.
Reverse transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted by TRIzol reagent (Invitrogen) and was then reverse transcribed to cDNA using a Reverse transcription kit (Takara, Japan). Reverse transcription-quantitative PCR (RT-qPCR) was performed using a SYBR Green qPCR Master Mix (Bimake, USA). The primers were synthesized by Sangon Biotech (Shanghai) Co., Ltd. and listed in Additional file 2:Table S2.
Cell proliferation assay
The influence of si-POU4F1 on the proliferation of ESCC cells was analyzed by CCK8 assay (Beyotime, Shanghai, China). ESCC cells (6000/well) were seeded in the 96-well plate and then were placed at the 37 °C constant temperature cell incubator. After 24 h incubation, si-NC and si-POU4F1 were transfected into the cells for 48 h.
Colony formation assay
KYSE30 and KYSE70 (800/well) were seeded into 12-well plates. After 24 h culture, si-POU4F1 and si-NC were transfected into the cells. After 7 days of culture, the culture medium was aspirated, and the cell clones were fixed by methanol and stained with crystal violet.
EdU staining assay
ESCC cells were seeded in the 96-well plate at a density of 6 × 103 cells per well. After incubation for 24 h, the transfection reagent introduced si-NC and si-POU4F1 into the cells for 48 h. Subsequently, theEdU-594 Kit (Beyotime, Shanghai, China) was conducted to evaluate cell proliferation.
RNA-sequence (RNA-seq) analysis
After transfection with si-NC and si-POU4F1 for 24 h, the cells were sent to the Beijing Genomics Institute. Co., Ltd. (Wuhan, China) for RNA-seq analysis. UMAP analysis using the Sangerbox online tool (http://www.sangerbox.com/tool) was conducted to visualize intra- and inter-group correlations within transcriptome sequencing data [8]. The DEGs were determined using the criteria of |log2 FC| > 0.585 and false discovery rate (FDR) < 0.05, and were highlighted in red (high expressed genes) or blue (low expressed genes) in the volcano plot. Additionally, GSEA analysis were conducted for exploring the potential pathways using C2-Canonical pathway database (c2.cp.v2023.2.) containing KEGG dataset and C5-Gene Ontology gene sets (c5.go.v2023.2.) as a reference.
Western blot analysis
Total protein was extracted using RIPA buffer containing PMSF, protease inhibitors, phosphatase inhibitors and nuclease. The protein was separated by 12% SDS-PAGE and electrotransferred onto polyvinylidene fluoride membranes (Millipore). After blocking with 5% skimmed milk for 2 h, membranes were incubated with primary antibodies at 4 °C overnight. Then, the membranes were incubated with secondary antibodies at room temperature for 1 h and then detected by Invitrogen iBright CL1000 imaging systems (Thermo Fisher, USA). The antibodies used in this paper are listed in Additional file 2:Table S3.
Cell cycle analysis
KYSE30 cells were collected and fixed in 70% ethanol overnight following a 24-hour transfection with si-NC and si-POU4F1. Subsequently, cell cycle analysis was conducted using the Cell Cycle Analysis Kit (C1052, Beyotime, China) in accordance with the provided manual. After staining, the cells were subjected to flow cytometry analysis using the BD FACSCanto™ II system.
Statistical analysis
Results were analyzed by R version 4.1.2 (https://www.r-project.org/), and GraphPad Prism version 8 (GraphPad Software, San Diego, CA, USA). Two-tailed p-values of less than 0.05 were defined as significant differences. Experimental data were presented as mean ± SD of at least three independent experiments and the results were analyzed using an unpaired Student’s t-test, and Two-way ANOVA. The code used in the bioinformatics analysis has been presented in Additional file 2:Table S4.
Result
Expression of POUs differs between ESCC and normal samples
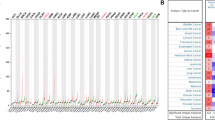
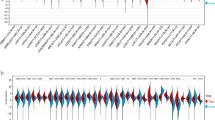
To investigate the expression of POUs in ESCC, we conducted a differential analysis of data from the TCGA and GSE53625 datasets. Boxplots were utilized to visualize the expression patterns of all POUs in these two datasets (Fig. 1A and B). Aberrant mRNA expression was observed in 6 genes in TCGA and 3 genes in GSE53625. Among them, POU6F2 and POU4F1 showed differential expression in both datasets (Fig. 1C). The gene expression profile and differentially expressed genes of POUs in TCGA and GSE53625 were shown in Additional file 2:Table S5-6. Additionally, we conducted a pan-cancer analysis and found that compared to normal tissues, POU6F2 exhibited significantly elevated expression levels across various cancers especially in EC (Fig. 1D), while PUU4F1 also showed significant overexpression in most cancers except for PAAD and renal KICH (Fig. 1E).

Expression of POUs differs between ESCC and normal samples. A-B Boxplots displaying the expression of all POUs in ESCC. Red and blue colors represent tumor and normal tissue expressions, respectively. C Venn diagram of showing the overlap of DEGs between TCGA and GEO datasets. D-E Boxplots illustrating the expression levels of 2 genes between normal and tumor samples across 26 cancer types. *** for p < 0.001, ** for p < 0.01, and * for p < 0.05
Prediction models and a survival nomogram for forecasting survival in ESCC patients
To assess the prognostic value of POUs in ESCC patients, Kaplan-Meier survival analysis was conducted. High POU4F1 expression was significantly correlated with lower survival rates, while POU6F2 did not exhibit significant differences in ESCC patients (Fig. 2A). The survival curves for all POUs are shown in Additional file 1: Fig. S1. To develop a prognostic model for ESCC, the predictive accuracy of POU4F1 was evaluated using the ROC. POU4F1 demonstrated superior predictive ability with 3-Year AUC of 0.707 and 5-Year AUC of 0.776 compared to previously studied biomarkers such as TNM stage, CST1, CD59, and ZNF750 [9,10,11,12] (Fig. 2B). Integration POU4F1 expression and TNM stage into a nomogram resulted in a more accurate prediction model (Fig. 2C), with 3-Year AUC = 0.786 and 5-Year AUC = 0.837 (Fig. 2D). Analysis of the distribution of high- and low-risk patients revealed that those with higher risk scores experienced increased mortality rates compared to those with lower risk scores (Fig. 2E and F). These finding suggests that POU4F1 could serve as a novel, reliable, and independent prognostic marker for ESCC. Furthermore, a prediction model based on POU4F1 and TNM stage may enhance the accuracy of predicting patient survival.

Prediction models and a survival nomogram for forecasting survival in ESCC patients. A Survival analysis of POU4F1 and POU6F2 in TCGA. B The 3 year- and 5 year-ROC curves of POU4F1, Stage, CST1, CD59 and ZNF750. C Nomogram constructed with TNM staging combined with POU4F1. D ROC and survival of the nomogram. E The distribution of patient risk scores, arranged in ascending order and categorized into low-risk (green) and high-risk (red) groups in TCGA. F Presentation of patient survival time and status, organized by increasing risk scores in TCGA. Red and green dots denote deceased and surviving patients, respectively
POU4F1-related signaling pathways based on GSEA and PPI network
Subsequently, our investigation aimed to uncover the possible tumor-associated mechanisms underlying the effects of POU4F1 in ESCC through GSEA. The GSEA result based on the C2-Canonical pathway database (c2.cp.v2023.2.) containing KEGG dataset was included in the Additional file 2:Table S8. According to C5 gene sets, a total of 200 pathways were significantly enriched in the POU4F1-high group (Additional file 2:Table S7). Among these, three pathways are highly ranked in our analysis and well-documented in the literature as being related to tumor occurrence and development, making them readily verifiable through further experiments. These pathways include: GOBP_REGULATION_OF_TRANSCRIPTION_INVOLVED_IN_G1_S_TRANSITION_OF_MITOTIC_CELL_CYCLE (P = 0.000, NES = 2.04), GOBP_GLIAL_CELL_APOPTOTIC_PROCESS (P = 0.008, NES = 1.85), GOBP_REGULATION_OF_INTRINSIC_APOPTOTIC_SIGNALING_PATHWAY_IN_RESPONSE_TO_DNA_DAMAGE (P = 0.014, NES = 1.76) (Fig. 3A). And then, we used the STRING database to construct a PPI network of POU4F1-associated proteins that included proteins such as TP53 and other development related proteins (Fig. 3B) Furthermore, GeneMANIA database analysis showed that POU4F1 was associated with Intrinsic apoptotic signaling pathway-related genes, including BCL2, BAX, ZNF385A, HIPK2, JMY, PMAIP1, POU4F2, DNA damage response, signal transduction by p53 class mediator pathway-related genes, including BAX, ZNF385A, HIPK2, PMAIP1, and Regulation of cell cycle arrest pathway-related genes, including BAX, ZNF385A (Fig. 3C).

POU4F1-related signaling pathways based on GSEA and PPI network. A GSEA analysis of POU4F1-related signaling pathways in TCGA ESCC dataset. B PPI network constructed using the STRING database shows POU4F1 and the POU4F1-interacting proteins. C GeneMANIA analysis of relevant interactive proteins of POU4F1
POU4F1 regulates cell growth and proliferation in ESCC
Based on the results of both differential and prognostic analysis, POU4F1 appears to be the potentially pivotal POUs gene in ESCC. To further substantiate its prognostic and therapeutic value, we performed functional experiments in ESCC cells using siRNA to silence POU4F1 expression. Initially, we identified two siRNAs capable of effectively silencing POU4F1 in KYSE30 and KYSE70 cells (Fig. 4A). Subsequently, we assessed the impact of si-POU4F1 on EC growth using the CCK8 assay, colony formation assay and EdU staining assay. As expected, POU4F1 knockdown in KYSE30 and KYSE70 cells led to significant changes in cell morphology, accompanied by notable decreases in cell number and viability (P < 0.05, Fig. 4B). Additionally, colony formation experiments confirmed a significant reduction in the number of cell colonies after POU4F1 knockdown (Fig. 4C). The EdU assay further demonstrated a significant decrease in the proportion of proliferating ESCC cells after siRNA treatment (Fig. 4D). These results strongly suggest that POU4F1 knockdown exerts a significant inhibitory effect on the growth and proliferation of ESCC cells.

POU4F1 regulates cell growth and proliferation in ESCC. A The interference efficiency of KYSE30 and KYSE70 treated with siRNA. B The evaluation of cellular morphology and viability after a 2-day treatment with siRNA, measured via an optical density of 450 nm. (Scale bar: 100 μm) C Images depicting the colony formation assay results for the three groups within KYSE30 and KYSE70 cells, respectively. Additionally, histograms illustrating the colony numbers for each group in KYSE30 and KYSE70 cells. D Exemplary images from the EdU assays conducted in KYSE30 cells (Scale bar: 200 μm). *P < 0.05, **P < 0.01, ***P < 0.001
POU4F1 knockdown influences cell cycle, autophagy and DNA damage signaling pathways
To investigate the mechanism of POU4F1 knockdown on ESCC cell growth, we conducted RNA-seq analysis following si-POU4F1 treatment in KYSE30 cells. The expression profile in the comparison group was shown in Additional file 2:Table S9. The UMAP analysis revealed a high degree of similarity between samples within the sample group (Fig. 5A). Differential analysis identified 1363 down-regulated and 1157 up-regulated genes, visually represented in a volcano plot (Fig. 5B and Additional file 2:Table S10). GSEA enrichment analysis highlighted many pathways affected by POU4F1 knockdown (Additional file 2: Tables S11-S12). Based on the C2 and C5 gene sets, 162 and 358 pathways, respectively, were significantly enriched, with top-ranked pathways related to cancer cell growth and easily verifiable including SELECTIVE_AUTOPHAGY (P = 0.000, NES = 1.39), DNA_DAMAGE_RECOGNITION (P = 0.000, NES = 1.31), and MITOTIC_G1_PHASE_AND G1_S_TRANSITION (P = 0.000, NES=-1.49) signaling pathway (Fig. 5C). Notably, key genes involved in the cell cycle displayed significant alterations, as depicted in the heatmap (Fig. 5D).

POU4F1 knockdown influences Cell cycle, Autophagy and DNA damage signaling pathways. A UMAP analysis of the samples from RNA sequencing. B Visualization of DEGs from RNA-seq analysis using a volcano plot. C Representative GSEA enrichment analysis. D Heatmap showcasing differential genes associated with Cell Cycle
Experiments validation of RNA sequencing
Additionally, the flow cytometry-based cell cycle assay revealed a significant increase in cells in the G1 phase after si-POU4F1 treatment, suggesting G1 phase cell cycle arrest (Fig. 6A). Western blot analysis suggested that down-regulation of Cyclin D1, CDK4, Cyclin E1, CDK2 and AURKA protein expression, consistent with the RNA-seq results, may contribute to G1 phase cell cycle arrest. Moreover, the increase in LC3B-II levels and decrease in P62 protein levels suggested the induction of autophagy upon POU4F1 knockdown. Furthermore, the elevated γ-H2AX levels and reduced PLK1 protein expression indicated potential DNA damage resulting from POU4F1 knockdown (Fig. 6B and C). The schematic diagram showing the underlying mechanism that POU4F1 may influence ESCC cell proliferation through various pathways, including cell cycle, autophagy and DNA damage signaling (Fig. 7).

Experiments validation of RNA sequencing. A Flow cytometry was used to assess the distribution of cell cycle phases in ESCC cells after POU4F1 knockdown. B-C Western blot detection of protein levels for the indicated proteins

Schematic model. Schematic diagram of the potential mechanism of POU4F1 in the progression of ESCC cells
Discussion
EC, particularly ESCC, imposes a significant global health burden, constituting a majority of cases and presenting challenges in prognosis despite advancements in treatment modalities [13]. Thus, there is a critical need to explore novel prognostic biomarkers [14]. Our study aimed to address this need by investigating in depth the diagnostic and prognostic roles of POUs in ESCC. It’s the first time we’ve used bioinformatics big data mining to explore the diagnostic and prognostic value of the whole POUs in ESCC. Through an analysis of key public databases, we observed elevated expression of POU4F1 in ESCC. Moreover, our analysis revealed that POU4F1 exhibited significantly elevated expression levels across various cancers, suggesting its potential as a diagnostic marker not only in ESCC but also in other malignancies. Furthermore, previous literatures have also indicated the high expression of POU4F1 in other tumors [7].
Our analysis revealed that POU4F1 is associated with lower survival rates in ESCC patients, and demonstrates higher predictive accuracy compared to previously studied biomarkers such as TNM stage, CST1, CD59, and ZNF750 (Fig. 2B), indicating its potential as a prognostic biomarker. Additionally, the integration of POU4F1 expression and TNM stage into a nomogram significantly improves the predictive accuracy for ESCC patient survival, highlighting the clinical relevance of POU4F1 in ESCC prognosis. Furthermore, POU4F1 has been found to significantly influence not only in ESCC development but also the survival and progression of various cancers, including melanoma and head and neck squamous cell carcinoma (HNSCC) [15, 16]. POU4F1 has also been intricately linked to the clinical survival and incorporated into the risk assessment model for predicting clinical outcomes in CRC [17, 18]. Similarly, POU4F1 was identified as a pivotal survival gene and has been specifically chosen for incorporation into the lasso-based Cox model in HNSCC and triple-negative breast cancer [16, 19].
Our study provides a preliminary exploration into the therapeutic potential of targeting POU4F1 in ESCC. Through siRNA-mediated knockdown, we observed a significant inhibition of ESCC cell proliferation and growth, suggesting a potential therapeutic target. These results are consistent with previous findings in other cancer types, where the reduction of POU4F1, combined with trastuzumab, effectively decreased the growth capacity of HER2-positive breast cancer cells resistant to trastuzumab [20]. The suppression of Brn3a through RNA interference significantly diminished the survival rate of melanoma cell lines and lessened the growth of tumors in vivo [15]. However, there is limited research on the mechanistic studies of POU4F1’s impact on tumor cell growth. Our RNA-seq analysis revealed POU4F1’s impact on several ESCC pathways, including the cell cycle, autophagy, and DNA damage. At the same time, GSEA results from TCGA data and PPI network indicate that POU4F1 was associated with pathways involved in tumor development and progression, including cell cycle, apoptotic and DNA damage. These results are consistent with previous studies [15, 21]. GSEA results from RNA-seq data highlighted the significant effect of POU4F1 on the cell cycle, which was validated through subsequent FACS and WB experiments. Our WB experiments results showed that POU4F1 siRNA inhibited the expression of critical checkpoint proteins, cyclin D1/CDK4 and Cyclin E1/CDK2, which are both essential for the G1/S phase transition [22, 23]. We also observed a decrease in AURKA protein expression, which correlates with G0/G1 phase arrest in breast cancer cells [24]. These findings are in line with previous literature, which has shown that silencing POU4F1 via RNA interference significantly reduces melanoma cell viability and inhibits tumor growth by inducing cell cycle arrest and triggering DNA damage signaling [15].
Although there is limited literature available, our RNA-Seq results guided us to explore the effects of knocking down POU4F1 on DNA damage and autophagy. We observed an elevation in γ-H2AX levels within KYSE30 cells. The phosphorylation of the histone H2A variant, H2AX, leads to the formation of γ-H2AX, which is a recognized biomarker for measuring DNA double-strand breaks [25, 26]. Moreover, DNA damage triggers the reduction of PLK1 in an ATM/ATR-dependent manner [27]. Therefore, the persistent rise in γ-H2AX levels coupled with the decrease in PLK1 suggests the potential of POU4F1 suppression to induce DNA damage. Finally, the knockdown of POU4F1 may induce autophagy, as evidenced by the upregulation of LC3BII and the downregulation of p62, both of which are essential markers of autophagy [28]. Thus, apart from its impact on the cell cycle, it is conceivable that POU4F1 may also modulate ESCC progression through mechanisms involving DNA damage and autophagy.
This paper highlights the significance of POU4F1 as both a prognostic and therapeutic target in ESCC, supported by bioinformatics and experimental validation. However, several aspects require further investigation in future studies: [1] We observed a decrease in p21 protein expression following POU4F1 knockdown (Additional file 1: Fig. S2). Previous studies have shown that POU4F1 binds to and activates the p21 promoter, resulting in increased p21 expression [21, 29], which is consistent with our findings. However, since p21 is typically upregulated in response to during anticancer treatment and DNA damage, further exploration is warranted into the role of p21 in POU4F1 knockdown therapy for ESCC [2]. Further elucidation of the detailed role and mechanism of POU4F1 in ESCC requires conducting various experiments, such as overexpression studies and in vivo animal models.
Conclusion
POU4F1 demonstrates significant overexpression in ESCC, showcasing considerable independent prognostic potential. In vitro experiments have underscored POU4F1’s pivotal role in influencing ESCC cell proliferation, cell cycle modulation, DNA damage, and autophagy. Our study highlights POU4F1’s importance as both a therapeutic target and prognostic biomarker in ESCC. Further investigation into the effectiveness of inhibiting POU4F1 in preclinical ESCC models holds promise. Additionally, considering its emerging significance as a prognostic biomarker in ESCC, future clinical studies could evaluate its predictive value regarding treatment response.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- AUC:
-
Area under curve
- DEGs:
-
Differentially expressed genes
- EC:
-
Esophageal cancer
- ESCC:
-
Esophageal squamous cell carcinoma
- FC:
-
Fold change
- FDR:
-
False discovery rate
- GSEA:
-
Gene Set Enrichment Analysis
- HNSCC:
-
Head and neck squamous cell carcinoma
- NC:
-
Negative control
- POU:
-
Pit-Oct-Unc
- PPI:
-
Protein-Protein interaction
- ROC:
-
Receiver operating characteristic
- RT- qPCR:
-
Reverse transcription-quantitative PCR
- siRNA:
-
Small interfering RNAs
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J Clin. 2021;71(3):209–49.
Malik V, Zimmer D, Jauch R. Diversity among POU transcription factors in chromatin recognition and cell fate reprogramming. Cell Mol Life Sci. 2018;75(9):1587–612.
Martinez-Ordonez A, Seoane S, Avila L, Eiro N, Macia M, Arias E, et al. POU1F1 transcription factor induces metabolic reprogramming and breast cancer progression via LDHA regulation. Oncogene. 2021;40(15):2725–40.
Tang C, Lei X, Xiong L, Hu Z, Tang B. HMGA1B/2 transcriptionally activated-POU1F1 facilitates gastric carcinoma metastasis via CXCL12/CXCR4 axis-mediated macrophage polarization. Cell Death Dis. 2021;12(5):422.
Bu X, Liu Y, Wang L, Yan Z, Xin G, Su W. Oct4 promoted proliferation, migration, invasion, and epithelial-mesenchymal transition (EMT) in colon cancer cells by activating the SCF/c-Kit signaling pathway. Cell Cycle. 2023;22(3):291–302.
Lu CS, Shiau AL, Su BH, Hsu TS, Wang CT, Su YC, et al. Oct4 promotes M2 macrophage polarization through upregulation of macrophage colony-stimulating factor in lung cancer. J Hematol Oncol. 2020;13(1):62.
Zhang J, Miao N, Lao L, Deng W, Wang J, Zhu X et al. Activation of bivalent gene POU4F1 promotes and maintains basal-like breast cancer. Adv Sci (Weinh). 2024:e2307660.
Shen W, Song Z, Zhong X, Huang M, Shen D, Gao P, et al. Sangerbox: a comprehensive, interaction-friendly clinical bioinformatics analysis platform. iMeta. 2022;1(3):e36.
Luo FF, Wang J, Zhang ZF, Lin ST, Huang TJ, Liu BQ, et al. SPI1-mediated upregulation of the CST1 gene as an independent poor prognostic factor accelerates metastasis in esophageal squamous cell carcinoma (ESCC) by interacting with MMP2. Front Biosci (Landmark Ed). 2023;28(9):212.
Zhou Y, Chu L, Wang Q, Dai W, Zhang X, Chen J, et al. CD59 is a potential biomarker of esophageal squamous cell carcinoma radioresistance by affecting DNA repair. Cell Death Dis. 2018;9(9):887.
Otsuka R, Akutsu Y, Sakata H, Hanari N, Murakami K, Kano M, et al. ZNF750 expression is a potential prognostic biomarker in esophageal squamous cell carcinoma. Oncology. 2018;94(3):142–8.
Li N, Liu K, Dong S, Ou L, Li J, Lai M, et al. Identification of CHRNB4 as a diagnostic/prognostic indicator and therapeutic target in human esophageal squamous cell carcinoma. Front Oncol. 2020;10:571167.
Lagergren J, Smyth E, Cunningham D, Lagergren P. Oesophageal cancer. Lancet. 2017;390(10110):2383–96.
Rogers JE, Sewastjanow-Silva M, Waters RE, Ajani JA. Esophageal cancer: emerging therapeutics. Expert Opin Ther Targets. 2022;26(2):107–17.
Hohenauer T, Berking C, Schmidt A, Haferkamp S, Senft D, Kammerbauer C, et al. The neural crest transcription factor Brn3a is expressed in melanoma and required for cell cycle progression and survival. EMBO Mol Med. 2013;5(6):919–34.
Dai D, Guo Y, Shui Y, Li J, Jiang B, Wei Q. Combination of radiosensitivity gene signature and PD-L1 status predicts clinical outcome of patients with locally advanced head and neck squamous cell carcinoma: a study based on the cancer genome atlas dataset. Front Mol Biosci. 2021;8:775562.
Fu B, Du C, Wu Z, Li M, Zhao Y, Liu X, et al. Analysis of DNA methylation-driven genes for predicting the prognosis of patients with colorectal cancer. Aging. 2020;12(22):22814–39.
Gong B, Kao Y, Zhang C, Sun F, Gong Z, Chen J. Identification of hub genes related to carcinogenesis and prognosis in colorectal cancer based on integrated bioinformatics. Mediators Inflamm. 2020;2020:5934821.
Qin W, Qi F, Li J, Li P, Zang YS. Prognostic biomarkers on a competitive endogenous RNA network reveals overall survival in triple-negative breast cancer. Front Oncol. 2021;11:681946.
Wu D, Jia HY, Wei N, Li SJ. POU4F1 confers trastuzumab resistance in HER2-positive breast cancer through regulating ERK1/2 signaling pathway. Biochem Biophys Res Commun. 2020;533(3):533–9.
Budram-Mahadeo V, Morris PJ, Latchman DS. The Brn-3a transcription factor inhibits the pro-apoptotic effect of p53 and enhances cell cycle arrest by differentially regulating the activity of the p53 target genes encoding Bax and p21(CIP1/Waf1). Oncogene. 2002;21(39):6123–31.
Qie S, Diehl JA. Cyclin D1, cancer progression, and opportunities in cancer treatment. J Mol Med. 2016;94(12):1313–26.
Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9(3):153–66.
Sharma RK, Chafik A, Bertolin G. Aurora kinase A/AURKA functionally interacts with the mitochondrial ATP synthase to regulate energy metabolism and cell death. Cell Death Discov. 2023;9(1):203.
Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273(10):5858–68.
Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, et al. GammaH2AX and cancer. Nat Rev Cancer. 2008;8(12):957–67.
van Vugt MA, Smits VA, Klompmaker R, Medema RH. Inhibition of Polo-like kinase-1 by DNA damage occurs in an ATM- or ATR-dependent fashion. J Biol Chem. 2001;276(45):41656–60.
Jiang P, Mizushima N. LC3- and p62-based biochemical methods for the analysis of autophagy progression in mammalian cells. Methods. 2015;75:13–8.
Perez-Sanchez C, Budhram-Mahadeo VS, Latchman DS. Distinct promoter elements mediate the co-operative effect of Brn-3a and p53 on the p21 promoter and their antagonism on the Bax promoter. Nucleic Acids Res. 2002;30(22):4872–80.
Acknowledgements
Not applicable.
Funding
This research was funded by the National Natural Science Foundation of China (82073937, 82373126, 32000516), Shenzhen Science and Technology Project (JCYJ20220818102605011, JCYJ20210324093602007, GJHZ20200731095200003 and JCYJ20220531101016035), Shenzhen Key Medical Discipline Construction Fund (SZXK059), China Postdoctoral Science Foundation (No.2022M722213), Guangdong Province Clinical Drug Research Fund (2022JZ14, 2021YXQN07) and Guangdong Basic and Applied Basic Research Foundation (2023A1515110415).
Author information
Authors and Affiliations
Contributions
Nan Li, Siying Chen and Xiao Wang performed the main experiments. Boqing Zhang, Boning Zeng, Chao Sun assisted in some cell experiments. The first draft of the manuscript was written by Nan Li and all authors commented on previous versions of the manuscript. Nan Li, Qiuling Chen analyzed the data. Kai Zheng, Shaoxiang Wang carefully revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, N., Chen, S., Wang, X. et al. Identification of POU4F1 as a novel prognostic biomarker and therapeutic target in esophageal squamous cell carcinoma. Cancer Cell Int 24, 280 (2024). https://doi.org/10.1186/s12935-024-03471-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-024-03471-6