
Abstract
Background
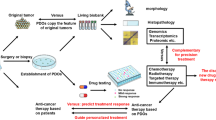
Gastric cancer (GC) encompasses many different histological and molecular subtypes. It is a major driver of cancer mortality because of poor survival and limited treatment options. Personalised medicine in the form of patient-derived organoids (PDOs) represents a promising approach for improving therapeutic outcomes. The goal of this study was to overcome the limitations of current models by ameliorating organoid cultivation.
Methods
Organoids derived from cancer tissue were evaluated by haematoxylin and eosin staining, immunohistochemistry, mRNA, and whole-exome sequencing. Three representative chemotherapy drugs, 5-fluorouracil, docetaxel, and oxaliplatin, were compared for their efficacy against different subtypes of gastric organoids by ATP assay and apoptosis staining. In addition, drug sensitivity screening results from two publicly available databases, the Genomics of Drug Sensitivity in Cancer and Cancer Cell Line Encyclopaedia, were pooled and applied to organoid lines. Once key targeting genes were confirmed, chemotherapy was used in combination with poly (ADP ribose) polymerase (PARP)-targeted therapy.
Results
We successfully constructed GC PDOs surgically resected from GC patient tissue. PDOs closely reflected the histopathological and genomic features of the corresponding primary tumours. Whole-exosome sequencing and mRNA analysis revealed that changes to the original tumour genome were maintained during long-term culture. The drugs caused divergent responses in intestinal, poorly differentiated intestinal, and diffuse gastric cancer organoids, which were confirmed in organoid lines. Poorly differentiated intestinal GC patients benefited from a combination of 5-fluorouracil and veliparib.
Conclusion
The present study demonstrates that combining chemotherapy with PARP targeting may improve the treatment of chemotherapy-resistant tumours.
Similar content being viewed by others

Background
Gastric cancer (GC) is a leading contributor to global cancer incidence and mortality, accounting for 768,793 deaths in 2020 [1, 2]. It is the fifth most commonly diagnosed cancer and the third most common cause of cancer-related deaths [1]. The rates are two-fold higher in men than women, with a strong prevalence in Eastern Asia [2, 3]. China has the largest number of GC cases, deaths, and disability-adjusted life years [4]. Surgical resection remains the primary option for GC [5]; however, GC is highly heterogeneous and most patients are at an advanced stage when diagnosed [6, 7], which limits surgical treatment [3, 8]. Systemic chemotherapy remains the mainstay treatment for advanced or metastatic GC. Currently, two- to three-drug chemotherapy regimens can modestly benefit overall survival, with median overall survival under 12 months [9, 10]. In recent years, molecular targeted therapy has emerged as a novel option, but only two such therapies are currently approved and very few have been clinically validated [11].
There is conflicting evidence regarding the ability of histopathological phenotypes to predict a patient’s response to systemic chemotherapy. Commonly, the Laurén [12] and World Health Organization [13] classifications are used for such purpose. The Laurén classification [5] distinguishes between intestinal GC (IGC) and diffuse GC. Poorly differentiated (PD) IGC is a highly heterogeneous subtype associated with frequent metastasis and chemotherapy resistance. Thus, there is an urgent need for drug susceptibility testing as a clinical strategy for individualised treatment.
Two-dimensional (2D) cell cultures and patient-derived xenografts are used for drug screening. However, predicting the sensitivity of individuals to therapy [7, 8] would be facilitated by a well-characterised model for drug testing. Recently, three-dimensional (3D) “organoids” have been introduced as a new prospect in preclinical personalised therapy testing, including oncology [14,15,16,17]. Organoid technology allows the cultivation of patient-derived GCs [18,19,20]. Compared with conventional 2D cultures and animal models, organoid cultures enable patient specificity, while recapitulating native tissue-like structures and functions in vitro. Organoid cultures are more accessible for manipulation and in-depth studies [21] than animal models, allowing for drug discovery [17, 22], personalised companion diagnostics, and cell therapy [23]. Not surprisingly, they have become the best model for personalised cancer treatment [24].
In this study, we sought to improve the success rate and cultivation timing of organoid lines. We optimised culture conditions by constructing 28 GC organoid lines from tumour tissues (fourteen intestinal GC, nine PDC GC, and five diffuse GC) and verified them by histopathology and whole-exosome and transcriptome sequencing. We demonstrated the utility of gastric organoid lines as preclinical models for drug testing of new targets and their potential applications in personalised treatment. Moreover, we showed the benefit of combining 5-fluorouracil (5-FU) and veliparib to treat patients with PD IGC, thereby creating new therapeutic opportunities.
Results
Patient-derived GC organoids display different morphology and characteristics
Tissue samples with histologically diagnosed gastric adenocarcinoma were obtained from surgically resected specimens (Tables 1 and 2). A flow diagram summarising the study design is shown in Fig. 1a. To boost the initiation of organoid cultures, tissue digestion, Matrigel concentration, and processing of isolated tumour patches were optimised. Using 3D organoid culture technology, the success rate for viable organoids was set to 56% (28/50). These GC organoid lines displayed divergent morphological phenotypes, indicating different molecular genotypes that mimicked in vivo tumours (Fig. 1b).

Construction and histopathological characterizations of GC PDOs. A Schematic overview of GC PDOs isolation, culture and histopathological analysis. B Representative bright-field images and HE staining images of GC PDOs and the primary tumors from which they were derived. These tumors (GO18, GO10, and GO16) represent three histological subtypes. Scale bar, 50 μm
Development of a culture method for the propagation of organoids from gastric tumour patients
To determine Matrigel and growth factor dependency, we designed different culture methods. The response varied markedly across Matrigel concentrations (Fig. 2a) and maximum number of days in culture (Fig. 2b). Expression of stem cell markers identified the number of cultivation days (Fig. 2c); whereas that of GC biomarkers confirmed tumour identity (Fig. 2d). The mRNA expression of gastric stemness markers Prom1, Sox4, and CD44 correlated positively with the culture period for organoids. Expression and immunohistochemistry analysis of typical stemness markers (Fig. 2e, f) indicated significantly shorter culture periods for GC24 and GC25 than for GC22 samples. Continuous observational studies to evaluate organoid growth (Fig. 2g) revealed larger organoid volumes for GC24 and GC25 samples than for the control group under optimal conditions.

Development of a culture method for propagation of organoids from the GC patient tumor. A, B Representative bright-field images and quantification of live-cell yield for organoids from three patients grown from single cells in media supplemented with 70, 80, 90, or 100% of Matrigel matrix (25,000 cells, 1 ml of medium/well) over a 14-day incubation period; scale bars, 200 μm. Data are plotted as mean ± s.d. Statistical significance was attributed to values of p < 0.05 as determined by the Student’s t test. NS, p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001,****p < 0.0001. C Maximum number of days in culture. D Muc5a, Chga, Fabp1, Muc6, Prom1, Sox4 and CD44 mRNA expression of some GC samples. E Prom1, Sox4 and CD44 mRNA expression of GC22, GC24 and GC25 samples. F IHC staining (PCNA and P-P38) showed that GC22, GC24 and GC25. G Representative bright-field images of GO24 and GO25 in Before adjustment and after adjustment conditions
GC organoids maintain the histology of original cancer tissues
To determine whether organoids maintained similar characteristics in culture as the cancers from which they were derived, we performed haematoxylin and eosin (H&E) staining (Fig. 3a), as well as immunohistochemical reactions for typical GC markers, such as muc2, p53, CD133, and EpCAM (Fig. 3b–d). As shown by H&E staining, the organoids reproduced well the overall and immunohistochemical characteristics of the penetrating primary tissue.

Gastric cancer organoids maintain the histology of original cancer tissues. A H&E staining showed that the successfully cultured organoids and primary tumors are consistent in histological characteristics. Scale bars, 50 μm. B–D IHC staining (MUC2、P53、CD133 and EPCam) showed that the successfully cultured organoids and primary tumors are consistent in histological characteristics. Scale bars, 50 μm
GC organoids retain the genetic characteristics of cancer tissues
Next, we investigated whether the organoid models preserved the genomic and transcriptomic characteristics of their corresponding tumours. Transcriptome analysis of GC organoids and corresponding tumour tissues revealed strong agreement between gene expression levels across different histological GC subtypes (Fig. 4a). To study the relationship between the observed results and global transcriptomes, principal component analysis of GC samples and their organoids was performed. Gene expression profiles of GC samples did not differ significantly from those of their organoids in the oncogene I ground state (Fig. 4b).The two most common mutations in GC organoids and tumour tissue samples included nonsense and missense mutations. We used muTect to analyse somatic single-nucleotide variants of the paired sample. The latter was annotated with ANNOVAR to screen the mutation sites and count mutation frequencies of the corresponding genes. We selected the top 50 genes with the highest mutation frequency to construct an oncoplot (Fig. 4c). Interpreting the oncoplot was not difficult. Regardless of whether they were from intestinal, PD intestinal or diffuse cancer, organoids and paired primary lesions had similar single-nucleotide variant mutation characteristics. The base substitutions in all samples included C > T, T > C, C > A, C > G, T > G, and T > A. Notably, C > T or T > C had the highest substitution frequency in GC (Fig. 4d). Whole-exosome sequencing revealed that patient-derived organoids (PDOs) largely recapitulated the primary tumour from which they were derived in terms of copy number (Fig. 4e). KEGG pathway analysis using the DAVID bioinformatics resource pointed to the PDC histological subtype (GO10) being enriched in DNA replication and cell cycle pathways (Fig. 4f). In contrast, the diffuse subtype (GO16) was enriched in Wnt signalling, gastric acid secretion, and cell adhesion molecules (Fig. 4g). Intriguingly, the intestinal histological subtype (GO18) revealed enrichment for immune-related pathways, including Th17 cell differentiation, Th1 and Th2 cell differentiation, as well as B cell receptor signalling (Fig. 4h). These results are consistent with those reported previously [25, 26]. Taken together, the observed findings demonstrated that the established PDOs reflected the genomic profiles of the primary tumour.

Gastric cancer organoids retain the genetic characteristics of cancer tissues. A Pearson correlation coefficients for gastric organoids and their corresponding parental cancers, respectively. B Principle Component Analysis (PCA) for GC RNA sequencing samples and their organoids. C The top 50 mutant genes are obtained by analyzing the SNV of organoids and matched primary tissue. The legend showed the types of mutations. D The mutation types and proportions of all samples. E Heatmap showing CNVs in the primary tumors and the corresponding GC PDOs. The columns represent genomic positions from chromosomes 1–22, and the colors in the plot correspond to the estimated log2 copy ratios of the genomic regions. F Top 30 enriched KEGG analysis in GO10. G Top 30 enriched KEGG analysis in GO16. H Top 30 enriched KEGG analysis in GO18
GC organoids for patient-specific drug trials in vitro
Patients with GC treated with neoadjuvant therapy and conventional chemotherapeutics respond to varying degrees, as documented by their histological regression grade. We investigated whether GC organoids reflected this divergent response. We treated the organoid lines with conventional chemotherapeutics routinely used in GC treatment: 5-FU, oxaliplatin, and docetaxel. Cell viability assays revealed divergent responses to the treatments (Fig. 5a–c). A significant difference in inhibitory concentration at 50% (IC50) was observed among the three chemotherapeutics in the phenotypic organoid lines tested, which consistent with the results of previous studies. Such divergent response was confirmed by annexin V/propidium iodide staining (Fig. 5d). The number of apoptotic cells increased in GO10 organoids upon 5-FU treatment, in GO16 organoids upon docetaxel treatment, and in GO18 organoids upon oxaliplatin treatment.

Human gastric cancer organoids show divergent therapy response to conventional chemotherapeutics. A–C The drug dose–response curve of intestinal(GO18), PDC intestinal (GO10) and diffuse (GO16) after treated with 5-FU, oxaliplatin, docetaxel, and irinotecan; each data point represents three biological replicates, with error bars representing mean ± SD. D Apoptosis assay using annexin V/propidium iodide (PI) flow cytometry (one example of three independent experiments is shown). Data are presented as the mean ± SD. *p < 0.05, **p < 0.01
5-FU and veliparib synergistically induce DNA damage in 5-FU-resistant intestinal GC
We downloaded cell line drug sensitivity data from the Cancer Cell Line Encyclopaedia and Genomics of Drug Sensitivity in Cancer databases. Differential gene expression analysis was performed for 5-FU sensitivity (SNU5) or resistance (SNU16) in a cell line from the PDC Intestinal cell subtype (Fig. 6a; Supplementary Fig. S1a, b). Analysis of KEGG pathways across SNU16 cells (Fig. 6b; Supplementary Fig. S1c) revealed significant enrichment in DNA replication and cell cycle signalling. To confirm these observations in 2D SNU16 cell lines, we selected three PDC Intestinal samples with elevated expression of the PARP1, ANAPC13, and CDT1 genes associated with DNA replication. The mRNA expression of PARP1 was higher in the GC30 and GC33 groups than in the GC10 group; whereas that of ANAPC13 and CDT1 was higher in GC33 than in GC10 (Fig. 6c). Western blotting revealed that protein expression of PARP1, ANAPC13, and CDT1 was significantly higher in the GC30 and GC33 groups than in the GC10 group (Fig. 6d). These results were also confirmed by immunohistochemistry (Fig. 6e). We therefore focused on the PARP1 gene encoding the poly (ADP ribose) polymerase. Veliparib, a PARP inhibitor developed by AbbVie (Chicago, IL, USA), exerts an anticancer effect by inducing DNA damage [27].

PDC intestinal carcinoma can increase chemotherapy sensitivity through combination therapy. A Venn diagram showing the number of genes that are uniquely expressed within each cell line group, green = SNU5, blue = SNU16. B KEGG pathway analysis of SNU16 uniquely expressed genes. C Relative mRNA expression of PARP1, ANAPC13 and CDT1 in GC10, GC30 and GC33 samples. D Western blot analysis of PARP1, ANAPC13 and CDT1 in GC10, GC30 and GC33 samples. E IHC staining of PARP1, ANAPC13 and CDT1 in GC10, GC30 and GC33 samples. Scale bars: 50 μm. F Dose effect curve of 5-FU, Veliparib and combination on GO30 organoid cells. G Combination index of 5-FU and Veliparib co-treatment on GO30 organoid cells. H Dose effect curve of 5-FU, Veliparib and combination on GO33 organoid cells. I Combination index of 5-FU and Veliparib co-treatment on GO33 organoid cells. J, K The relative protein levels of apoptosis-related proteins and DNA damage were measured via western blotting in GO30 organoid cells. Statistical analysis was performed using two-tailed Student’s t-tests. *p < 0.05; **p < 0.01; ***p < 0.001. vs GC10 or control
We hypothesised that integrating existing chemotherapy agents with targeted therapies could represent a novel strategy for patients who have developed resistance to conventional chemotherapy. To test this hypothesis, we treated the intestinal organoid with 5-FU and veliparib alone or in combination at a constant ratio of 1:2, then assessed cell viability 48 h after drug treatment. As shown in Fig. 6f, h, the combination of 10 μM 5-FU and 20 μM veliparib had a more severe inhibitory effect on the proliferation of GO30 and GO33 than 5-FU or veliparib alone. To test the synergistic cell-killing effect of 5-FU and veliparib co-administration, we calculated the combination index for the IC50 in GO30 and GO33 organoid samples (Fig. 6g, i; Supplementary Fig. S1d). PARP-1 is responsible for nearly 90% of the universal PAR synthsis following DNA strand breakage [28]. To further define the role of veliparib could improve the sensitivity of cancer cells to chemotherapeutic drugs by apoptosis, we tested the pro-apoptotic proteins, Bax and caspase-3, and the anti-apoptotic protein, Bcl-2, were examined via western blotting. GO30 organoid Cells were classified in four groups: control group, treated with 10 μM 5-Fu, treated with 10 μM Veliparib and the combination group receiving both treatments.The results demonstrated that Bax and cleaved caspase-3 expression increased highest in the combination, respectively, the expression of Bcl-2 decreased highest in the combination (Fig. 6j, k). Our research revealed that veliparib enhances the cytotoxic effect on 5-FU-resistant GC organoids. This is achieved by inducing a slowdown in the DNA damage repair process, leading to sustained higher levels of γH2AX, which in turn, promotes cell apoptosis (Fig. 6j, k).
Discussion
Here, we tested the use of human GC organoids in the study of different pathological types and their drug sensitivity. The study included 28 intestinal, 14 PD intestinal, and five diffuse GC lines obtained from tumour tissue (Fig. 1b). Failure to culture 10 of these lines may have been caused by tissue contamination and was related to the collection of materials. Failure to passage in the other 16 cases may have been related to the pathological characteristics of the patients or the limitations of culture technology.
The growth of organoids requires an environment similar to the extracellular matrix, which allows intercellular or matrix-cellular crosstalk. Currently used matrices rely on animal tumour-derived basement membrane extracts (e.g. Matrigel), which limits application in human regenerative medicine [29, 30]. Although patient-derived GC cells can be cultured in vitro as organoids in Matrigel, the optimal concentration of Matrigel remains unclear. To make efficient use of organoid technology, we compared the effects of different Matrigel concentrations on organoid proliferation (Fig. 2a). We demonstrated for the first time that a 100% concentration of Matrigel was the best condition for gastric organoid cultivation (Fig. 2b).
An organoid is a self-organised 3D tissue, whose functional, structural, and biological complexity resemble that of an organ [31,32,33]. As in other tissues, gastric stem cells are used as a source for GC. Thus, we examined the expression of the stem cell markers Prom1, Sox4, and CD44. Prom1 is a cell surface membrane protein and a homolog to the CD133 protein in humans [34]. It is expressed in both rod and cone photoreceptors of the eye. Prom1 has been used as a cancer stem cell marker alone or in combination with other markers such as CD44 [35]. Sox proteins leave their footprint in every cell lineage and are involved in stem cell biology, developmental biology, pathology, and human genetics [36]. The maximum number of days in culture correlated with the mRNA expression of Prom1, Sox4, and CD44 (Fig. 2c–e). Mitogen-activated protein kinase p38 is essential for the long-term expansion of human intestinal organoids. Therefore, we compared PCNA and phosphorylated-p38 protein levels in the three samples (Fig. 2f). Fgf10 is expressed in mesenchymal and epithelial tissues and is an epithelial-mesenchymal signalling molecule. It also plays a crucial role in the stomach [37]. The concentrations of Fgf10 and SB202190 (a P38 inhibitor) were adjusted to increase the proliferation and long-term passaging of gastrointestinal organoids (Table 1, Fig. 2g). Based on the above steps, we established a new approach for culturing organoids derived from human GC.
One of the major challenges in the culture of GC is a tough stroma with sparse cancer cells and frequent contamination by non-cancerous cells, leading to overgrowth by the latter. Both these factors contributed to the failure of culturing some samples, as determined by immunohistochemical analysis of the tumours from which they were derived (Fig. 3). Next, whole-exosome and mRNA sequencing of the corresponding parental tumours were performed to determine the histological architecture, cancer driver gene mutations, copy number variations, and single-nucleotide variants after in vitro culture (Fig. 4). PDOs offer exciting novel opportunities for drug screening and testing [38]. In the present study, gastric organoids varied in their responses to drugs. Using the transcriptome sequencing of organoid samples from the three pathological subtypes, we enriched pathways and screened for three commonly used chemotherapy drugs (5-FU,oxaliplatin and docetaxel) in clinical practice, which were then validated (Fig. 4f–h). The diffuse type (GO16) was highly sensitive to docetaxel (Fig. 5a). The intestinal type (GO18) was highly sensitive to oxaliplatin (Fig. 5b). Finally, the PD intestinal type (GO10) was sensitive to 5-FU (Fig. 5c). Through bioinformatic mining, along with transcriptome and whole-exosome sequencing, we predicted and confirmed that PARP inhibitors could increase chemotherapy sensitivity in resistant PDC intestinalGC samples (Fig. 6).
Although the proposed method successfully predicted chemotherapy outcomes, lack of tumour microenvironment components and vasculature factors, such as stromal cells and immune cells, are major disadvantages of organoid models. Thus, future studies should focus on mimicking the tumour microenvironment in gastric organoids [39, 40]. Second, this study was constrained by a small sample size and results should be verified in a larger sample. Third, it is important to evaluate the tumorigenicity of gastric organoids by translating them into immunodeficient mice, where they can assume the morphology and histological characteristics of primary tumour samples.
In this study, we undertook a systematic effort to create a biobank of GC cells in the form of organoids and performed exosome and transcriptome sequencing to gain insight into their molecular characteristics. In addition, we investigated the long-term stability of the organoids in culture.
Conclusions
Our results show that the proposed organoid culture method successfully captured both early- and advanced-stage cancers. Importantly, we observed that molecular changes in the organoids closely resembled those in tumour samples obtained directly from patients. This suggested that our organoids faithfully recreated in vitro the genetic and molecular features of in vivo GC.
Furthermore, we tested the sensitivity of GC organoids to various therapeutic agents. GC organoids were particularly sensitive to a combination of 5-FU (a chemotherapeutic agent) and veliparib (a PARP inhibitor). This finding opens up new therapeutic opportunities for the treatment of GC, specifically for patients with PDC. In summary, we established a reliable culture method for generating tumour organoids from human GC cells. Our study highlights the potential of veliparib as a promising addition to chemotherapy regimens for the treatment of PDC and personalised strategies.
Availability of data and materials
Not applicable.
Abbreviations
- 2D:
-
Two-dimensional
- 3D:
-
Three-dimensional
- 5-FU:
-
5-Fluorouracil
- GC:
-
Gastric cancer
- H&E:
-
Haematoxylin and eosin
- IGC:
-
Intestinal gastric cancer
- PARP:
-
Poly (ADP ribose) polymerase
- PDC:
-
Poorly differentiated
- PDO:
-
Patient-derived organoid
References
Smyth EC, Nilsson M, Grabsch HI, van Grieken NC, Lordick F. Gastric cancer. Lancet. 2020;396:635–48.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Yeoh KG, Tan P. Mapping the genomic diaspora of gastric cancer. Nat Rev Cancer. 2022;22:71–84.
Fitzmaurice C, Abate D, Abbasi N, Abbastabar H, Abd-Allah F, Abdel-Rahman O, Abdelalim A, Abdoli A, Abdollahpour I, Abdulle ASM, Abebe ND, Abraha HN, Abu-Raddad LJ, Abualhasan A, Adedeji IA, Advani SM, Afarideh M, Afshari M, Aghaali M, Agius D, Agrawal S, Ahmadi A, Ahmadian E, Ahmadpour E, Ahmed MB, Akbari ME, Akinyemiju T, Al-Aly Z, AlAbdulKader AM, Alahdab F, Alam T, Alamene GM, Alemnew BTT, Alene KA, Alinia C, Alipour V, Aljunid SM, Bakeshei FA, Almadi MAH, Almasi-Hashiani A, Alsharif U, Alsowaidi S, Alvis-Guzman N, Amini E, Amini S, Amoako YA, Anbari Z, Anber NH, Andrei CL, Anjomshoa M, Ansari F, Ansariadi A, Appiah SCY, Arab-Zozani M, Arabloo J, Arefi Z, Aremu O, Areri HA, Artaman A, Asayesh H, Asfaw ET, Ashagre AF, Assadi R, Ataeinia B, Atalay HT, Ataro Z, Atique S, Ausloos M, Avila-Burgos L, Avokpaho E, Awasthi A, Awoke N, Ayala Quintanilla BP, Ayanore MA, Ayele HT, Babaee E, Bacha U, Badawi A, Bagherzadeh M, Bagli E, Balakrishnan S, Balouchi A, Bärnighausen TW, Battista RJ, Behzadifar M, Behzadifar M, Bekele BB, Belay YB, Belayneh YM, Berfield KKS, Berhane A, Bernabe E, Beuran M, Bhakta N, Bhattacharyya K, Biadgo B, Bijani A, Bin Sayeed MS, Birungi C, Bisignano C, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 29 cancer groups, 1990 to 2017: a systematic analysis for the global burden of disease study. JAMA Oncol. 2019;5:1749–68.
Seidlitz T, Merker SR, Rothe A, Zakrzewski F, von Neubeck C, Grützmann K, Sommer U, Schweitzer C, Schölch S, Uhlemann H, Gaebler AM, Werner K, Krause M, Baretton GB, Welsch T, Koo BK, Aust DE, Klink B, Weitz J, Stange DE. Human gastric cancer modelling using organoids. Gut. 2019;68:207–17.
Allemani C, Matsuda T, Di Carlo V, Harewood R, Matz M, Nikšić M, Bonaventure A, Valkov M, Johnson CJ, Estève J, Ogunbiyi OJ, Azevedo ESG, Chen WQ, Eser S, Engholm G, Stiller CA, Monnereau A, Woods RR, Visser O, Lim GH, Aitken J, Weir HK, Coleman MP. Global surveillance of trends in cancer survival 2000–14 (CONCORD-3): analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet. 2018;391:1023–75.
Hunt RH, Camilleri M, Crowe SE, El-Omar EM, Fox JG, Kuipers EJ, Malfertheiner P, McColl KE, Pritchard DM, Rugge M, Sonnenberg A, Sugano K, Tack J. The stomach in health and disease. Gut. 2015;64:1650–68.
Takahashi T, Saikawa Y, Kitagawa Y. Gastric cancer: current status of diagnosis and treatment. Cancers. 2013;5:48–63.
Wagner AD, Syn NL, Moehler M, Grothe W, Yong WP, Tai BC, Ho J, Unverzagt S. Chemotherapy for advanced gastric cancer. Cochrane Database Syst Rev. 2017;8:004064.
Ter Veer E, Haj Mohammad N, van Valkenhoef G, Ngai LL, Mali RMA, Anderegg MC, van Oijen MGH, van Laarhoven HWM. The efficacy and safety of first-line chemotherapy in advanced esophagogastric cancer: a network meta-analysis. J Natl Cancer Inst. 2016. https://doi.org/10.18632/oncotarget.13216.
Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical development success rates for investigational drugs. Nat Biotechnol. 2014;32:40–51.
Lauren P. The two histological main types of gastric carcinoma: diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classification. Acta Pathol Microbiol Scand. 1965;64:31–49.
J.F. Fl´ejou, WHO Classification of digestive tumors: the fourth edition. Ann Pathol. 2011;S27–31. https://doi.org/10.1016/j.annpat.2011.08.001
Kopper O, de Witte CJ, Lõhmussaar K, Valle-Inclan JE, Hami N, Kester L, Balgobind AV, Korving J, Proost N, Begthel H, van Wijk LM, Revilla SA, Theeuwsen R, van de Ven M, van Roosmalen MJ, Ponsioen B, Ho VWH, Neel BG, Bosse T, Gaarenstroom KN, Vrieling H, Vreeswijk MPG, van Diest PJ, Witteveen PO, Jonges T, Bos JL, van Oudenaarden A, Zweemer RP, Snippert HJG, Kloosterman WP, Clevers H. An organoid platform for ovarian cancer captures intra- and interpatient heterogeneity. Nat Med. 2019;25:838–49.
Pasch CA, Favreau PF, Yueh AE, Babiarz CP, Gillette AA, Sharick JT, Karim MR, Nickel KP, DeZeeuw AK, Sprackling CM, Emmerich PB, DeStefanis RA, Pitera RT, Payne SN, Korkos DP, Clipson L, Walsh CM, Miller D, Carchman EH, Burkard ME, Lemmon KK, Matkowskyj KA, Newton MA, Ong IM, Bassetti MF, Kimple RJ, Skala MC, Deming DA. Patient-derived cancer organoid cultures to predict sensitivity to chemotherapy and radiation. Clin Cancer Res. 2019;25:5376–87.
Yao Y, Xu X, Yang L, Zhu J, Wan J, Shen L, Xia F, Fu G, Deng Y, Pan M, Guo Q, Gao X, Li Y, Rao X, Zhou Y, Liang L, Wang Y, Zhang J, Zhang H, Li G, Zhang L, Peng J, Cai S, Hu C, Gao J, Clevers H, Zhang Z, Hua G. Patient-derived organoids predict chemoradiation responses of locally advanced rectal cancer. Cell Stem Cell. 2020;26:17-26.e6.
Kim M, Mun H, Sung CO, Cho EJ, Jeon HJ, Chun SM, Jung DJ, Shin TH, Jeong GS, Kim DK, Choi EK, Jeong SY, Taylor AM, Jain S, Meyerson M, Jang SJ. Patient-derived lung cancer organoids as in vitro cancer models for therapeutic screening. Nat Commun. 2019;10:3991.
Barker N, Huch M, Kujala P, van de Wetering M, Snippert HJ, van Es JH, Sato T, Stange DE, Begthel H, van den Born M, Danenberg E, van den Brink S, Korving J, Abo A, Peters PJ, Wright N, Poulsom R, Clevers H. Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell. 2010;6:25–36.
Bartfeld S, Bayram T, van de Wetering M, Huch M, Begthel H, Kujala P, Vries R, Peters PJ, Clevers H. In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology. 2015;148:126-136.e6.
Nanki K, Toshimitsu K, Takano A, Fujii M, Shimokawa M, Ohta Y, Matano M, Seino T, Nishikori S, Ishikawa K, Kawasaki K, Togasaki K, Takahashi S, Sukawa Y, Ishida H, Sugimoto S, Kawakubo H, Kim J, Kitagawa Y, Sekine S, Koo BK, Kanai T, Sato T. Divergent routes toward Wnt and R-spondin niche independency during human gastric carcinogenesis. Cell. 2018;174:856-869.e17.
Clevers H. Modeling development and disease with organoids. Cell. 2016;165:1586–97.
Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernández-Mateos J, Khan K, Lampis A, Eason K, Huntingford I, Burke R, Rata M, Koh DM, Tunariu N, Collins D, Hulkki-Wilson S, Ragulan C, Spiteri I, Moorcraft SY, Chau I, Rao S, Watkins D, Fotiadis N, Bali M, Darvish-Damavandi M, Lote H, Eltahir Z, Smyth EC, Begum R, Clarke PA, Hahne JC, Dowsett M, de Bono J, Workman P, Sadanandam A, Fassan M, Sansom OJ, Eccles S, Starling N, Braconi C, Sottoriva A, Robinson SP, Cunningham D, Valeri N. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science. 2018;359:920–6.
Al Shihabi A, Davarifar A, Nguyen HTL, Tavanaie N, Nelson SD, Yanagawa J, Federman N, Bernthal N, Hornicek F, Soragni A. Personalized chordoma organoids for drug discovery studies. Sci Adv. 2022;8:3674.
Drost J, Clevers H. Organoids in cancer research. Nat Rev Cancer. 2018;18:407–18.
Wang J, Qin D, Tao Z, Wang B, Xie Y, Wang Y, Li B, Cao J, Qiao X, Zhong S, Hu X. Identification of cuproptosis-related subtypes, construction of a prognosis model, and tumor microenvironment landscape in gastric cancer. Front Immunol. 2022;13:1056932.
Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513(7517):202–9. https://doi.org/10.1038/nature13480.
Wagner LM. Profile of veliparib and its potential in the treatment of solid tumors. Onco Targets Ther. 2015;8:1931–9.
Talhaoui I, Lebedeva NA, Zarkovic G, Saint-Pierre C, Kutuzov MM, Sukhanova MV, Matkarimov BT, Gasparutto D, Saparbaev MK, Lavrik OI, Ishchenko AA. Poly(ADP-ribose) polymerases covalently modify strand break termini in DNA fragments in vitro. Nucleic Acids Res. 2016;44:9279–95.
Steele NG, Chakrabarti J, Wang J, Biesiada J, Holokai L, Chang J, Nowacki LM, Hawkins J, Mahe M, Sundaram N, Shroyer N, Medvedovic M, Helmrath M, Ahmad S, Zavros Y. An organoid-based preclinical model of human gastric cancer. Cell Mol Gastroenterol Hepatol. 2019;7:161–84.
Yang R, Yu Y. Patient-derived organoids in translational oncology and drug screening. Cancer Lett. 2023;562:216180.
Simian M, Bissell MJ. Organoids: a historical perspective of thinking in three dimensions. J Cell Biol. 2017;216:31–40.
Reis RL. 2nd Consensus conference on definitions on biomaterials science. J Tissue Eng Regen Med. 2020;14:561–2.
Z. Zhao, X. Chen, A.M. Dowbaj, A. Sljukic, K. Bratlie, L. Lin, E.L.S. Fong, G.M. Balachander, Z. Chen, A. Soragni, M. Huch, Y.A. Zeng, Q. Wang, and H. Yu, Organoids. Nat Rev Methods Primers. 2022; 2.
Taïeb N, Maresca M, Guo XJ, Garmy N, Fantini J, Yahi N. The first extracellular domain of the tumour stem cell marker CD133 contains an antigenic ganglioside-binding motif. Cancer Lett. 2009;278:164–73.
Sahlberg SH, Spiegelberg D, Glimelius B, Stenerlöw B, Nestor M. Evaluation of cancer stem cell markers CD133, CD44, CD24: association with AKT isoforms and radiation resistance in colon cancer cells. PLoS ONE. 2014;9:e94621.
Harley V, Lefebvre V. Twenty Sox, twenty years. Int J Biochem Cell Biol. 2010;42:376–7.
Itoh N, Ohta H. Fgf10: a paracrine-signaling molecule in development, disease, and regenerative medicine. Curr Mol Med. 2014;14:504–9.
Weeber F, Ooft SN, Dijkstra KK, Voest EE. Tumor organoids as a pre-clinical cancer model for drug discovery. Cell Chem Biol. 2017;24:1092–100.
Yuki K, Cheng N, Nakano M, Kuo CJ. Organoid models of tumor immunology. Trends Immunol. 2020;41:652–64.
Bar-Ephraim YE, Kretzschmar K, Clevers H. Organoids in immunological research. Nat Rev Immunol. 2020;20:279–93.
Acknowledgements
We would like to thank all the members of the Gastroenterology Department of Xijing Hospital, Air Force Medical University, Xi’an, China for kind support and Editage (www.editage.cn) for English language editing.
Funding
This research was funded by the Key R&D Program of Ningxia Province (Project Number: 2021BEG03037), the Medical Enhancement Program (Project Number: 2020SWAQ06), the Technology Development Incubation Fund (Project Number: 2023YJY-15) and the State Key Laboratory of Cancer Biology Fund (Project Number: CBSKL2022ZZ51.
Author information
Authors and Affiliations
Contributions
G. Chen: Conceptualization, data curation, formal analysis, visualization, writing the original draft. Ruidong Han: Conceptualization, resources, writing original draft. Li Wang: Validation. Wen Ma: Data curation, validation. Wenli Zhang: Data curation, validation. Z. Lu: Conceptualization, resources, supervision, funding acquisition, writing–review and editing. L. Wang: Conceptualization, data curation, supervision, funding acquisition.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Medical Research Ethics Review Committee of General Hospital of Ningxia Medical University (No. 2020–835).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, G., Han, R., Wang, L. et al. Establishment of patient-derived organoids and a characterization based drug discovery platform for treatment of gastric cancer. Cancer Cell Int 24, 288 (2024). https://doi.org/10.1186/s12935-024-03460-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-024-03460-9