
Abstract
Introduction
Acute myeloid leukemia (AML) is a complex hematologic malignancy characterized by uncontrolled proliferation of myeloid precursor cells within bone marrow. Despite advances in understanding of its molecular underpinnings, AML remains a therapeutic challenge due to its high relapse rate and clonal evolution.
Methods
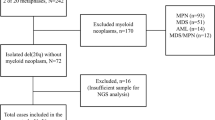
In this retrospective study, we analyzed data from 24 AML patients diagnosed at a single institution between January 2017 and August 2023. Comprehensive genetic analyses, including chromosomal karyotyping, next-generation sequencing, and gene fusion assays, were performed on bone marrow samples obtained at initial diagnosis and relapse. Clinical data, treatment regimens, and patient outcomes were also documented.
Results
Mutations in core genes of FLT3, NPM1, DNMT3A, and IDH2 were frequently discovered in diagnostic sample and remained in relapse sample. FLT3-ITD, TP53, KIT, RUNX1, and WT1 mutation were acquired at relapse in one patient each. Gene fusion assays revealed stable patterns, while chromosomal karyotype analyses indicated a greater diversity of mutations in relapsed patients. Clonal evolution patterns varied, with some cases showing linear or branching evolution and others exhibiting no substantial change in core mutations between diagnosis and relapse.
Conclusions
Our study integrates karyotype, gene rearrangements, and gene mutation results to provide a further understanding of AML heterogeneity and evolution. We demonstrate the clinical relevance of specific mutations and clonal evolution patterns, emphasizing the need for personalized therapies and measurable residual disease monitoring in AML management. By bridging the gap between genetics and clinical outcome, we move closer to tailored AML therapies and improved patient prognoses.
Similar content being viewed by others


Introduction
Acute myeloid leukemia (AML) is a heterogeneous and aggressive hematologic malignancy characterized by uncontrolled proliferation of myeloid precursor cells within the bone marrow [1]. Despite advancements in the understanding of its molecular pathogenesis, AML remains challenging to manage, with a high propensity for relapse following initial therapy [2]. Relapsed AML often exhibits genetic and clonal evolution, further complicating its diagnosis and treatment [3]. Like other cancers, relapsed AML has a poor prognosis, and treatment options are also challenging.
To identify genetic mutations in AML, various methods have been developed. The historic approach is chromosomal karyotyping [4], through which it is possible to determine if there are recurrent mutations and to identify chromosome gains or losses [5]. From chromosomal karyotyping, typical abnormalities such as deletion 5, deletion 7, inversion 16, and t(15;17) can be detected. To examine mutations at the gene level, next-generation sequencing (NGS) has emerged as a prominent method [6]. Clinical laboratories use panels targeting specific genes for testing [7], while research studies may involve whole exome sequencing or whole genome sequencing [8]. Through methods such as NGS, mutations in genes such as FLT3, NPM1, and DNMT3A can be detected. Since it determines whether there is a mutation in specific codons, it is advantageous for patient-specific minimal residual disease follow-up. NGS allows the identification of gene amplifications, deletions, as well as chromosomal gains and losses but may fall short in identifying gene breakpoints [9]. Therefore, RNA-based gene fusion assays have been developed [10]. In most clinical laboratories, tests are conducted for genes with common known breakpoints in acute leukemia, and these results can be used for precise diagnosis [11]. RNA-based sequencing is also being actively researched recently, and it is suitable for detecting various types of gene fusions in recent AML diagnostic guidelines. Recently, optical mapping methods have been employed to perform comprehensive testing for all genes [12].
Historically, the prognosis for acute myeloid leukemia has been quite poor, and there have been extensive discussions regarding relapse and post-relapse treatment [13]. Research using NGS methods has focused on identifying driver mutations and understanding how clonal evolution progresses based on these drivers [14]. Recent studies have suggested the existence of linear evolution and branching evolution when specific AML clones survive during remission to later contribute to relapse [15]. Linear evolution refers to the gradual accumulation of individual mutations, while branching evolution is characterized by the elimination of the predominant clone, succeeded by the emergence and expansion of a subclone.
Furthermore, while several studies have reported NGS results based on samples collected at AML diagnosis and relapse, our comprehensive analysis revealed certain limitations in their findings. First, they did not present RNA gene fusion results, which can yield positivity rates as high as 60% [16]. Second, there was a lack of chromosome results considered significant in the diagnostic criteria for AML. Third, these studies did not provide information regarding the treatment regimens employed for AML patients and the associated outcomes. In that regard, we believe it is necessary to integrate and analyze multiple tests for relapsed AML patients.
In this study, we aimed to broaden our knowledge of relapsed AML by examining patients who underwent NGS diagnosis and treatment at a single institution, encompassing treatment progress, prognosis, and various other tests. We sought to understand how clonal evolution occurs in these patients as well as which mutations persist through relapse to provide a more comprehensive understanding of AML.
Methods
Patients and data
This retrospective study focused on patients diagnosed with AML at a single institution from January 1, 2017, to August 31, 2023, who underwent NGS analysis on bone marrow samples at the time of initial diagnosis and again when experiencing relapse. Clinical data, laboratory test results, and genetic analysis results were collected and analyzed. The data included bone marrow aspiration results and flow cytometry findings at the time of AML diagnosis, as well as details of the treatments received. The initial diagnosis was determined according to WHO guidelines [17], considering flow cytometry, chromosome karyotyping, gene fusion, and NGS results. In cases of AML relapse, information was collected regarding the duration to relapse, clinical data and laboratory test results at the time of relapse, treatments received, and current prognosis. Patient outcomes were categorized as alive, dead, or follow-up loss. This study protocol received approval from the Institutional Review Board of Yonsei University Health System (4-2023-0930).
Cytogenetic and molecular genetic analyses
The G-banding karyotyping procedure followed standard protocols and was performed on heparinized bone marrow aspirate. A minimum of 20 metaphases was assessed, and the karyotype was described in accordance with the International System for Human Cytogenetic Nomenclature.
During the initial diagnosis, bone marrow aspirate samples were collected in EDTA vials. The QIAamp RNA Blood Mini Kit (Qiagen, Hilden, Germany) was used to extract total RNA for NGS testing. The Agilent 4200 TapeStation System (Agilent Technologies Inc, Santa Clara, CA) was used to measure the quantity and quality of RNA. The Archer FusionPlex Pan-Heme kit (ArcherDX, Boulder, CO) manufacturer’s instructions were followed to prepare a target-enriched cDNA library. Starting with 100 ng of RNA, random primers were used to provide random start sites for the initial cDNA synthesis. The end-repaired cDNA molecules were then ligated with sample-specific indices and specific molecular barcode adapters at both ends. A total of 199 target genes was covered by gene-specific primers used in two rounds of low-cycle PCR. On the NextSeq 550Dx instrument (Illumina, San Diego, CA), the finished products underwent sequencing, producing roughly three million reads per sample over 151 cycles. Next, default parameters were used with Archer Analysis Software (version 5.1, ArcherDX) to process and analyze the raw data, including extraction and evaluation of gene fusions, oncogenic variations, oncogenic isoforms, and mRNA expression data.
For targeted NGS, a customized set of probes (Dxome Co. Ltd., Gyeonggi-do, Korea) targeting 213, 497, or 531 genes associated with hematologic neoplasms was utilized (Supplementary Table S1). Genomic DNA extracted from the diagnostic bone marrow aspirate was used for library construction. These libraries underwent hybridization with capture probes and were sequenced using Illumina’s NextSeq 550Dx (San Diego, CA, USA), following the manufacturer’s guidelines. NGS data analysis was carried out using the DxSeq analyzer (Dxome). Single-nucleotide variants, small insertions and deletions, and copy number variants were detected using established methods [18, 19]. Variant allele frequency (VAF) represents the proportion of sequence reads that align with a particular DNA variant, divided by the total coverage observed at that specific genomic location. Validation of germline variants was conducted by comparing NGS results with buccal swabbing or skin fibroblast analysis. The identified variants were classified into four tiers based on the Association for Molecular Pathology’s guidelines, which incorporate input from the American College of Medical Genetics, the American Society of Clinical Oncology, and the College of American Pathologists [20]. During classification, web databases such as OncoKB [21] and cBioPortal [22] were consulted. The accuracy of all variants was visually confirmed using the Integrated Genomics Viewer.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Results
Patient demographics
A total of 24 patients, 10 male and 14 female, was enrolled in this study (Table 1). The ages of the patients ranged from 2 to 72 years. The initial diagnosis was determined by integrating the results of flow cytometry and bone marrow aspiration, resulting in 8 different AML diagnoses: acute megakaryoblastic leukemia, acute monocytic leukemia, acute myeloid leukemia with maturation, acute myeloid leukemia with mutated NPM1, acute myeloid leukemia with t(16;16)(p13.1;q22), acute monocytic leukemia, acute myeloid leukemia with t(8;21)(q22;q22.1), and acute myelomonocytic leukemia. Among the patients, 15 received hematopoietic stem cell transplantation (HSCT) during their initial treatment, while 9 underwent only chemotherapy. The average time to relapse was 14 months (ranging from 5 months to 2 years and 7 months). Among the relapsed patients, 11 received HSCT, 10 underwent chemotherapy only, and 3 did not receive any treatment. Among the HSCT-treated patients, 7 of 11 survived, while 4 succumbed to the disease. Among the patients who underwent chemotherapy alone, 8 died, and 2 were lost to follow-up. Among the 3 patients who did not receive treatment, 2 died, and 1 was lost to follow-up.
Oncogenic mutations
To identify oncogenic mutations in the patients, three main tests were conducted for cytogenetics and molecular genetics: NGS, gene fusion analysis, and chromosome karyotyping. The average depth for NGS was 1346.3x (from 407.4x to 3648.0x). The results of these tests were correlated with treatment and prognosis (Fig. 1). Among the core gene mutations identified by NGS, the most frequent genes were FLT3, NPM1, DNMT3A, and IDH2, in that order. In the case of FLT3 mutations, 7 patients had mutations at diagnosis, and all 7 maintained the mutations at relapse; the 8th patient presented with the mutation at relapse. The core gene mutations identified as tier 1/2 occurred on 26 genes, present in panels ranging from the smallest 213 gene panel to the largest 531 gene panel.

Oncoplot of the case distribution of relapsed acute myeloid leukemia patients
Twelve patients underwent a fusion test both at diagnosis and relapse and seven patients were fusion positive. Gene fusions detected included RUNX::RUNX1T1, KMT2A::MLLT10, NUP98::HOXA9, NUP214::ABL1, CBFB::MYH11, and CBFA2T3::GLIS2 fusions. Among them, RUNX1::RUNX1T1 fusion was detected in 4 patients, while the other gene fusions were detected in 1 patient each. All gene fusions remained stable upon relapse detection, with no new fusions gained or lost. Chromosome karyotype analysis revealed various types of abnormalities, with relapsed patients often exhibiting a greater diversity of abnormalities compared to their diagnostic samples.
Analyses of 24 patients regarding the correspondence of tier 1/2 core mutations to evolutionary models are described in Table 2. Five patients experienced linear evolution, 4 with branching evolution, and 15 were classified as unclear. Among the 15 unclear cases, 11 exhibited an exact match in core mutations between diagnosis and relapse. Among the 24 patients and the 26 core mutation genes, 9 were categorized as linear or branching evolution. The 4 most frequent core genes (FLT3, NPM1, DNMT3A, IDH2) were compared in terms of VAF between diagnosis and prognosis (Fig. 2). Additionally, graphs were generated for the variation in VAF across tiers 1, 2, and 3 for patients classified under linear evolution and branching evolution. Linear evolution was characterized by a high occurrence of new mutations, while the VAF of pre-existing mutations remained relatively consistent or slightly decreased. In contrast, branching evolution often featured decreased VAF for pre-existing mutations, with fewer new mutations compared to linear evolution. All 4 core genes that were present at diagnosis were also present at relapse, except for 1 patient who acquired an FLT3 mutation at relapse and 1 who acquired an NPM1 mutation at relapse. FLT3 mutations exhibited increased VAF at relapse in all patients except 1, while a consensus on VAF changes for NPM1, DNMT3A, and IDH2 was not observed. All variants identified by NGS panels are provided in Supplementary Table S2.

The changes of variant allele frequencies in (A) linear evolution patients’ tier 1,2 and 3 variants and (B) branching evolution patients’ tier 1,2 and 3 variants for (C) FLT3, (D) NPM1, (E) DNMT3A, and (F) IDH2.
Discussion
In this study, we present comprehensive results of a cohort of 24 patients diagnosed with AML. Through this study, we were able to integrate chromosomal karyotyping, gene fusion results, and NGS findings. The findings from this study contribute to our understanding of AML heterogeneity and evolution, providing valuable insights for future therapeutic strategies.
Oncogenic mutations play a pivotal role in AML pathogenesis and progression [23]. Our investigation into the mutational landscape through cytogenetic and molecular genetic analyses revealed several significant insights. The most frequently observed core mutations, FLT3, NPM1, DNMT3A, and IDH2, mostly remained consistent between diagnosis and relapse stages. Mutations in DNMT3A, ASXL1, and RUNX1 genes and FLT3-ITD were known to be frequently acquired at relapse [15]. In our study, FLT3-ITD, TP53, KIT, RUNX1, and WT1 mutation were acquired at relapse in one patient each. These genes are associated with a poor prognosis of AML [24,25,26,27] and mutations in these genes have been reported to be AML relapse-associated mutations in previous studies [28,29,30]. Therefore, they are presumed to be associated with the progression and treatment resistance of AML.
In our study, fusion results showed that mutations present at diagnosis remained unchanged at relapse. These findings suggest that the gene rearrangements were early events in these cases. However, performing a gene fusion test at diagnosis and relapse might be helpful when referring to existing reports that BCR::ABL1 can be required in AML relapse [31]. Unlike gene fusions, chromosomal karyotyping revealed various alterations, including gains, losses, and translocations, acquired by the chromosomes at relapse. Our finding supports that chromosomal instability acts as a mechanism for AML relapse, as previously suggested [32].
In our research, the patient cohort was highly diverse and was classified into four groups: pediatric (under 15 years old), adolescents and young adults (from 15 to 29 years old), adults (30 to 64 years old), and elderly (65 years and older), which consisted of 3, 1, 18, and 2 patients, respectively. The adult group had the largest number of patients, and survivors were distributed only among the adolescents and young adults and adult groups. Notably, four patients belonging to the pediatric or adolescents and young adults groups showed a characteristic feature of gene rearrangement detection in targeted RNA sequencing. This observation is in agreement with recent diagnostic trends, where the presence of specific gene rearrangements can lead to a change in the diagnosis of AML and plays a significant role in prognosis [27]. Branching evolution was found to occur in the higher age bracket of those aged 55 to 73 years. indicating that branching evolution occurs more frequently in the elderly population. These patients exhibited a pattern of certain remaining clones after anticancer treatment. If relapse is predicted after chemotherapy due to the persistence of specific clones, alternative chemotherapy regimens may be necessary.
The importance of MRD is being emphasized, and an ideal MRD technique in AML must be established [33]. In our study, four patients were followed up with reverse-transcription PCR (RT-PCR). RT-PCR detected morphological relapse at three months (P6) and four months (P10) before morphological relapse or continued to be RT-PCR positive during morphological complete remission (P8 and P19), suggesting the usefulness of MRD monitoring using RT-PCR. However, gene fusion was not detected in more than half of our patients. Therefore, NGS-based MRD might be helpful in these patients. In addition, considering that 50% of the total 24 patients showed clonal evolution with emerging or vanished mutations, our results suggest that the agnostic panel approaches might be more valuable than the patient-specific panel among NGS-based MRD strategies in these patients.
This study has several limitations. First, it is a retrospective study conducted in a single institution, so sample size is small, and there might be potential biases in patient selection. However, our findings highlight the complex interplay of genetic and clonal factors in AML, emphasizing the need for larger studies to validate these observations. Second, our study could only identify some of the molecular patterns that appear in the diagnosis and relapse of AML because we used targeted panel sequencing. Previous studies have used whole exome or whole genome sequencing to determine molecular profiles associated with AML relapse [3, 28]. Moreover, recent studies used single-cell analysis to reveal the pathogenic mechanisms of AML relapse [34, 35]. With the advancement of genomic techniques, more information has been available to determine the precise mechanisms of chemoresistance and relapse of AML. However, our study suggests that the gene panel sequencing currently available in routine diagnostics without using complex genomic techniques may help identify changes in genomic profiles associated with the recurrence of AML in a clinical setting. Third, we included only samples of diagnosis and relapse in this study. Studies including complete remission samples need to determine how much earlier the AML relapse can be predicted through molecular studies [36]. Whether the mutations found at diagnosis were cleared at complete remission is also important in predicting the patient’s prognosis [37].
Conclusions
We tried to identify the molecular feature associated with the progression of AML by comparing chromosome abnormalities, gene rearrangements, and gene mutation results at diagnosis and relapse. Patients showed branching and linear evolution patterns; some patients were not clearly classified. Acquired mutations at relapse are likely to play a role in the chemoresistance of AML. Moreover, we could suggest evidence on the optimal MRD monitoring strategy of AML through our study result. Since mutations at the time of diagnosis are not maintained at relapse and disappear or newly emerge in about half of patients, gene panel-based MRD monitoring would be helpful for AML patients.
While our findings contribute to understanding AML heterogeneity, acknowledging limitations like the small sample size, future research should focus on refining MRD measurement methods and exploring comprehensive diagnostic approaches for more effective therapeutic strategies. Overall, our study lays the groundwork for further investigations to enhance the management of relapsed AML patients.
Data availability
All data generated or analysed during this study are included in this published article and its supplementary information files.
References
Grove CS, Vassiliou GS. Acute myeloid leukaemia: a paradigm for the clonal evolution of cancer? Dis Models Mech. 2014;7(8):941–51.
Reville PK, Kadia TM. Maintenance therapy in AML. Front Oncol. 2020;10:619085.
Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506–10.
Kumar CC. Genetic abnormalities and challenges in the treatment of acute myeloid leukemia. Genes cancer. 2011;2(2):95–107.
Mrózek K. Cytogenetic, molecular genetic, and clinical characteristics of acute myeloid leukemia with a complex karyotype. Semin Oncol. 2008;35(4):365–77.
Haferlach T. Advancing leukemia diagnostics: role of Next Generation sequencing (NGS) in acute myeloid leukemia. Hematol Rep. 2020;12(Suppl 1):8957.
Zhong Y, Xu F, Wu J, Schubert J, Li MM. Application of Next Generation sequencing in Laboratory Medicine. Annals Lab Med. 2021;41(1):25–43.
Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, Dooling D, Dunford-Shore BH, McGrath S, Hickenbotham M, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456(7218):66–72.
Zeng X, Lin D, Liang D, Huang J, Yi J, Lin D, Zhang Z. Gene sequencing and result analysis of balanced translocation carriers by third-generation gene sequencing technology. Sci Rep. 2023;13(1):7004.
Heydt C, Wölwer CB, Velazquez Camacho O, Wagener-Ryczek S, Pappesch R, Siemanowski J, Rehker J, Haller F, Agaimy A, Worm K, et al. Detection of gene fusions using targeted next-generation sequencing: a comparative evaluation. BMC Med Genom. 2021;14(1):62.
Engvall M, Cahill N, Jonsson BI, Höglund M, Hallböök H, Cavelier L. Detection of leukemia gene fusions by targeted RNA-sequencing in routine diagnostics. BMC Med Genom. 2020;13(1):106.
Tembrink M, Gerding WM, Wieczorek S, Mika T, Schroers R, Nguyen HP, Vangala DB, Nilius-Eliliwi V. Novel NUP98::ASH1L Gene Fusion in Acute myeloid leukemia detected by Optical Genome Mapping. Cancers 2023, 15(11).
Thol F, Ganser A. Treatment of relapsed Acute myeloid leukemia. Curr Treat Options Oncol. 2020;21(8):66.
Pannuti A, Filipovic A, Hicks C, Lefkowitz E, Ptacek T, Stebbing J, Miele L. Novel putative drivers revealed by targeted exome sequencing of advanced solid tumors. PLoS ONE. 2018;13(3):e0194790.
Vosberg S, Greif PA. Clonal evolution of acute myeloid leukemia from diagnosis to relapse. Genes Chromosomes Cancer. 2019;58(12):839–49.
Heyer EE, Deveson IW, Wooi D, Selinger CI, Lyons RJ, Hayes VM, O’Toole SA, Ballinger ML, Gill D, Thomas DM, et al. Diagnosis of fusion genes using targeted RNA sequencing. Nat Commun. 2019;10(1):1388.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–405.
Kook HW, Kim JJ, Park MR, Jang JE, Min YH, Lee ST, Shin S, Cheong JW. Therapy-related Acute Lymphoblastic Leukaemia has a Unique Genetic Profile Compared to De Novo Acute Lymphoblastic Leukaemia. J Cancer. 2022;13(12):3326–32.
Kim H, Shim Y, Lee TG, Won D, Choi JR, Shin S, Lee ST. Copy-number analysis by base-level normalization: an intuitive visualization tool for evaluating copy number variations. Clin Genet. 2023;103(1):35–44.
Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, Tsimberidou AM, Vnencak-Jones CL, Wolff DJ, Younes A, et al. Standards and guidelines for the interpretation and reporting of sequence variants in Cancer: a Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagnostics: JMD. 2017;19(1):4–23.
Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, Rudolph JE, Yaeger R, Soumerai T, Nissan MH et al. OncoKB: A Precision Oncology Knowledge Base. JCO precision oncology 2017, 2017.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4.
Goldman SL, Hassan C, Khunte M, Soldatenko A, Jong Y, Afshinnekoo E, Mason CE. Epigenetic modifications in Acute myeloid leukemia: prognosis, treatment, and heterogeneity. Front Genet. 2019;10:133.
Wakita S, Yamaguchi H, Miyake K, Mitamura Y, Kosaka F, Dan K, Inokuchi K. Importance of c-kit mutation detection method sensitivity in prognostic analyses of t(8;21)(q22;q22) acute myeloid leukemia. Leukemia. 2011;25(9):1423–32.
Gaidzik VI, Teleanu V, Papaemmanuil E, Weber D, Paschka P, Hahn J, Wallrabenstein T, Kolbinger B, Köhne CH, Horst HA, et al. RUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic features. Leukemia. 2016;30(11):2160–8.
Rampal R, Figueroa ME. Wilms tumor 1 mutations in the pathogenesis of acute myeloid leukemia. Haematologica. 2016;101(6):672–9.
Döhner H, Wei AH, Appelbaum FR, Craddock C, DiNardo CD, Dombret H, Ebert BL, Fenaux P, Godley LA, Hasserjian RP, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood. 2022;140(12):1345–77.
Greif PA, Hartmann L, Vosberg S, Stief SM, Mattes R, Hellmann I, Metzeler KH, Herold T, Bamopoulos SA, Kerbs P, et al. Evolution of cytogenetically normal Acute Myeloid Leukemia during Therapy and Relapse: an Exome sequencing study of 50 patients. Clin Cancer Res. 2018;24(7):1716–26.
Höllein A, Meggendorfer M, Dicker F, Jeromin S, Nadarajah N, Kern W, Haferlach C, Haferlach T. NPM1 mutated AML can relapse with wild-type NPM1: persistent clonal hematopoiesis can drive relapse. Blood Adv. 2018;2(22):3118–25.
Höllein A, Nadarajah N, Meggendorfer M, Jeromin S, Kern W, Haferlach C, Haferlach T. Molecular characterization of AML with RUNX1-RUNX1T1 at diagnosis and relapse reveals net loss of co-mutations. Hemasphere. 2019;3(1):e178.
Alotaibi AS, Yilmaz M, Loghavi S, DiNardo C, Borthakur G, Kadia TM, Thakral B, Pemmaraju N, Issa GC, Konopleva M, et al. Emergence of BCR-ABL1 Fusion in AML Post-FLT3 inhibitor-based therapy: a potentially targetable mechanism of Resistance - A Case Series. Front Oncol. 2020;10:588876.
de Oliveira Lisboa M, Brofman PRS, Schmid-Braz AT, Rangel-Pozzo A, Mai S. Chromosomal instability in Acute myeloid leukemia. Cancers 2021, 13(11).
Heuser M, Freeman SD, Ossenkoppele GJ, Buccisano F, Hourigan CS, Ngai LL, Tettero JM, Bachas C, Baer C, Béné MC, et al. 2021 Update on MRD in acute myeloid leukemia: a consensus document from the European LeukemiaNet MRD Working Party. Blood. 2021;138(26):2753–67.
Mumme H, Thomas BE, Bhasin SS, Krishnan U, Dwivedi B, Perumalla P, Sarkar D, Ulukaya GB, Sabnis HS, Park SI, et al. Single-cell analysis reveals altered tumor microenvironments of relapse- and remission-associated pediatric acute myeloid leukemia. Nat Commun. 2023;14(1):6209.
Li K, Du Y, Cai Y, Liu W, Lv Y, Huang B, Zhang L, Wang Z, Liu P, Sun Q, et al. Single-cell analysis reveals the chemotherapy-induced cellular reprogramming and novel therapeutic targets in relapsed/refractory acute myeloid leukemia. Leukemia. 2023;37(2):308–25.
Dunlap JB, Leonard J, Rosenberg M, Cook R, Press R, Fan G, Raess PW, Druker BJ, Traer E. The combination of NPM1, DNMT3A, and IDH1/2 mutations leads to inferior overall survival in AML. Am J Hematol. 2019;94(8):913–20.
Jongen-Lavrencic M, Grob T, Hanekamp D, Kavelaars FG, Al Hinai A, Zeilemaker A, Erpelinck-Verschueren CAJ, Gradowska PL, Meijer R, Cloos J, et al. Molecular minimal residual disease in Acute myeloid leukemia. N Engl J Med. 2018;378(13):1189–99.
Acknowledgements
None.
Funding
This work was supported by a grant from the National Research Foundation of Korea (NRF-2021R1I1A1A01045980) and a faculty research grant of Yonsei University College of Medicine (6-2022-0050).
Author information
Authors and Affiliations
Contributions
Conceptualization, N.K., S.S., and J.C; methodology, S.L. and J.R.C.; investigation, C.J.L. and J.C.; resources, C.J.L. and J.C.; data curation, N.K. and S.S.; writing—original draft preparation, S.H. and J.C.; writing—review and editing, Y.J.C., H.C., H.C., and J.E.J.; supervision, S.S. and J.C. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study protocol received approval from the Institutional Review Board of Yonsei University Health System (4-2023-0930).
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kim, N., Hahn, S., Choi, Y.J. et al. Comprehensive insights into AML relapse: genetic mutations, clonal evolution, and clinical outcomes. Cancer Cell Int 24, 174 (2024). https://doi.org/10.1186/s12935-024-03368-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-024-03368-4