
Abstract
Immune checkpoint blockade (ICB) has become a promising strategy in treating advanced cancers, providing significant survival benefits for patients with various cancer types. However, among the vast population of cancer patients, only a small fraction are able to respond to and derive benefits from ICB therapy. Numerous factors contribute to the diminished efficacy of ICB, with the complex tumor microenvironment (TME) playing an important role. Therefore, comprehensively understanding the intricate composition of the TME is critical for elucidating the mechanisms that underlie distinct responses to ICB in patients. Single-cell RNA sequencing (scRNA-seq) is a novel technique that reveals gene expression profiles of individual cells, facilitating the investigation of TME heterogeneity at a high resolution and the identification of key cell subsets participating in the response to ICB. This review emphasizes the importance of scRNA-seq in studying ICB and summarizes recent findings in the discovery of biomarkers that predict ICB response and novel potential therapeutic targets for immunotherapy. These findings suggest future directions for the clinical implementation of cancer immunotherapy, facilitating further advancements in precision medicine.
Similar content being viewed by others
Background
The immune system monitors the human body and eradicates abnormal cells. This intricate surveillance system operates through a complex network of immune cells and signaling molecules to identify and eliminate foreign invaders. Immune checkpoints, located on the surface of immune cells, play an important role in preventing their overactivation and maintaining immune homeostasis. Despite the defense mechanisms, cancer cells can evade immune surveillance by multiple mechanisms, such as binding to the immune checkpoints, thereby suppressing the anti-tumor response.
Treatment selection for cancer patients depends on the clinical stage, usually assessed using the TNM classification system. For patients with unresectable tumors or those in advanced stages, systemic treatment is necessary to control the progression of the disease. Immunotherapy, emerging after chemotherapy and targeted therapy, represents a promising avenue for improving patient survival. Compared with other treatments, immunotherapy shows the advantages of long-lasting anti-tumor effects and reduced side effects, making it a significant modality for treating cancers [1, 2]. Immunotherapy activates the immune system to kill cancer cells. Immune checkpoint inhibitors (ICIs), which inhibit programmed cell death 1 (PD-1) or programmed cell death 1 ligand 1 (PD-L1) and cytotoxic T lymphocyte-associated protein 4 (CTLA-4), have shown success in treating many cancers. Nonetheless, most patients receiving ICIs do not experience survival benefits. While biomarkers such as PD-L1 expression [3], mismatch repair deficiency (dMMR) or microsatellite instability-high (MSI-H) [4], and tumor mutational burden (TMB) [5] are routinely utilized to identify people who may benefit from immune checkpoint blockade (ICB), these methods are not always accurate in predicting response to ICB in clinical practice. Therefore, it is essential to identify novel biomarkers that better predict response to ICB.
Current immunotherapy strategies mainly focus on activating CD8+ T cells, the major cytotoxic immune cells that kill cancer cells via the Fas-FasL pathway and the perforin/granzyme mechanism [6]. Nonetheless, the tumor microenvironment (TME) is composed of heterogeneous cells, extracellular matrix (ECM) components, blood vessels, and biomolecules [7], and the complex network formed by direct and indirect interactions between these heterogeneous cells can contribute to an immunosuppressive pattern in the TME and result in ICB failure. Therefore, it is also essential to search for new therapeutic targets in the TME that can synergize with current ICIs to suppress tumors.
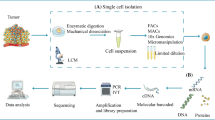
Single-cell technologies have the capacity to unravel the phenotypes of individual cells within a tissue, revolutionizing our understanding of cancer biology. Single-cell RNA sequencing (scRNA-seq), involving the main steps of single-cell isolation, cell lysis for mRNA capture, reverse transcription, cDNA amplification, and sequencing library construction, enables the researchers to decipher the cell heterogeneity. Unlike bulk RNA sequencing (RNA-seq), which quantifies the average expression of genes in a variety of cells, scRNA-seq allows for the detection of transcriptomic heterogeneity by measuring RNA expression levels of individual cells [8]. This technology can detect heterogeneity in various samples, including cancer and peritumoral tissues, as well as peripheral blood, enabling the discovery of new cell subsets with unique gene expression profiles and the studies of intercellular crosstalk, cellular differentiation trajectories, and gene regulation networks within the TME [9,10,11]. Therefore, scRNA-seq is a potent tool for unraveling why certain patients respond to ICB while others do not. By revealing differences in cellular components between responders and non-responders, and by discovering pivotal cell subsets that contribute to the response to ICB, scRNA-seq can identify novel biomarkers that have the potential to predict ICB responsiveness and new molecules that can be therapeutically targeted. In this review, we will first introduce the present status of ICB therapy and briefly overview the immunosuppressive cellular components in the TME. We will then review recent scRNA-seq studies that reveal the dynamic changes in cellular components during ICB treatment, discover novel biomarkers, and identify new therapeutic targets (Fig. 1).

The schematic diagram of the application of scRNA-seq in ICB. Tumor or peripheral blood mononuclear cells are obtained from animal models or human patients and are first dissociated into single-cell suspensions. The cells are then lysed for reverse transcription and amplification to construct the cDNA library. The resulting scRNA-seq data can be applied to study the dynamic changes in the TME or peripheral blood during ICB therapy, discover novel biomarkers predictive of ICB response, and search for potential therapeutic targets. scRNA-seq, single-cell RNA sequencing; ICB, immune checkpoint blockade. Created with Figdraw (www.figdraw.com)
Immune checkpoint blockade and predictive biomarkers in cancer treatment
In current clinical practice, cancer staging is essential for disease assessment and treatment selection. The TNM system is the most widely used classification method which evaluates the primary tumor (T), regional lymph nodes (N), and distant metastasis (M) [12]. In addition, many cancers also have their unique staging systems, such as the Barcelona Clinic Liver Cancer (BCLC) staging system in hepatocellular carcinoma (HCC) [13]. Regrettably, many patients are diagnosed at advanced stages and fail to undergo surgery, which is the only radical treatment method. Consequently, other therapeutic approaches, including radiotherapy, chemotherapy, targeted therapy, and immunotherapy, have become necessary for the management of advanced-stage tumors. Among these methods, radiotherapy is a local treatment modality that employs high-energy radiation to eradicate cancer cells within localized tumors. It has limited impacts on distant lesions, and it can cause damage to healthy tissues, leading to toxicity [14]. Chemotherapy is a first-line treatment for many cancers, functioning by inhibiting the proliferation of cancer cells. However, it cannot effectively differentiate between cancer cells and normal cells and can cause various side effects [15]. Targeted therapy precisely targets specific proteins or signal transduction pathways in cancer cells, taking effect quickly with low toxicity. However, it is effective mainly for patients with specific genetic mutations [16].
Immunotherapy, mainly including immune checkpoint inhibitors, cancer vaccines, and CAR-T cell therapy, is the latest emerging therapeutic approach that has demonstrated promising clinical efficacy [17]. Unlike other treatment modalities, immunotherapy activates the patient’s immune system instead of directly targeting cancer cells, thereby leading to long-lasting clinical benefits.
Currently used immune checkpoints and their inhibitors
In the cancer immune cycle, antigen-presenting cells (APCs) capture tumor antigens and then present them to naïve T cells when migrating to the lymph nodes. Then the T cells are primed and activated, obtaining the ability to eliminate cancer cells [18]. However, during this cycle, immune checkpoints function as a brake to suppress T cell activation, thereby enabling the immune escape of cancer cells. The immunoinhibitory receptors PD-1 and CTLA-4 are the most-studied immune checkpoints regulating the activation of T cells. Targeting these receptors with antibodies can activate effector T cells in suppressed states and enhance the immune response, which has yielded encouraging outcomes in cancer treatment.
CTLA-4 and PD-1/PD-L1
CTLA-4 is present on the surface of T cells and binds B7-1 (CD80) and B7-2 (CD86), competing with the costimulatory receptor CD28. CTLA-4 inhibition was shown to induce potent immune responses and tumor-killing activity of T cells in vivo [19]. Ipilimumab, the first immune checkpoint inhibitor targeting CTLA-4, was approved by the US Food and Drug Administration (FDA) in 2011 after showing improved survival benefits in patients with metastatic melanoma [20]. PD-1 is another immune checkpoint that binds to the ligands PD-L1 and PD-L2, inducing the suppression of T cell receptor (TCR)-mediated proliferation and the CD28-mediated costimulation pathway [21, 22]. Inhibiting the interaction between PD-1 and PD-L1 can enhance the anti-tumor responses of T cells. The first anti-PD-1 antibody, nivolumab, and the first anti-PD-L1 antibody, atezolizumab, demonstrated their anti-tumor efficacy in clinical trials and were approved by the FDA in 2014 and 2016, respectively [23, 24].
Currently, the FDA has approved several anti-PD-1/PD-L1 and anti-CTLA-4 drugs for the treatment of various cancers. These ICIs have shown anti-tumor efficacy in treating cancers, including HCC, melanoma, renal cell carcinoma (RCC), triple-negative breast cancer (TNBC), non-small cell lung carcinoma (NSCLC), head and neck squamous cell carcinomas (HNSCC), MSI-H/dMMR colorectal cancer (CRC), urothelial cancer, and Merkel cell carcinoma (MCC), as well as nonsolid tumors, such as diffuse large B-cell lymphoma and classic Hodgkin lymphoma [25]. However, the clinical efficacy varies greatly among patients receiving ICIs, and only a fraction of patients receiving ICB achieve objective responses, with a response rate of only 20-30% on average [26]. Some patients may respond well to ICB and even achieve a complete response, while more patients fail to benefit from treatment or only show a temporary response. In addition, the response rate to ICB varies across different cancer types, with an objective response rate of 87.1% in patients with classical Hodgkin lymphoma [27] and no more than 30% in patients with other cancers, such as melanoma, RCC, NSCLC, and HCC [28, 29].
Other immune checkpoints
In addition to the canonical immune checkpoints PD-1 and CTLA-4, other immune checkpoint molecules have been discovered, such as lymphocyte-activation gene 3 (LAG-3), T-cell immunoglobulin and mucin-domain 3 (TIM-3), and T cell immunoreceptor with Ig and ITIM domains (TIGIT). LAG-3 is primarily expressed on activated T cells and CD4+ regulatory T cells (Tregs) and can bind to stable peptide-MHC class II complexes, downregulating T-cell activation [30]. LAG-3 can inhibit the immune response synergistically with PD-1, acting as a nonredundant immune checkpoint molecule [31]. TIM-3 is expressed on activated CD8+ T cells, CD4+ T helper 1 (Th1) cells, Tregs, monocytes, and natural killer (NK) cells [32]. By binding to ligands such as galectin-9, TIM-3 triggers apoptosis of CD8+ T cells, negatively regulating the anti-tumor activity [33]. And a negative correlation has been found between TIM-3 expression on PD-1+ CD8+ T cells and clinical outcomes in cancer patients [34]. TIGIT is another immune checkpoint protein showing expression on activated T cells, Tregs, and NK cells [35]. It competes with the costimulatory receptor CD226 for the binding of CD155, which results in T and NK cell inhibition, similar to the CD28/CTLA-4 axis [35, 36]. Additionally, TIGIT can bind to CD226 directly and prevent CD155/CD226 signaling by disrupting CD226 homodimerization [35, 36]. Numerous novel drugs targeting these immune checkpoints are currently undergoing clinical trials, either alone or combined with PD-1/PD-L1 inhibitors. Some trials have reported promising results, suggesting that these drugs might enhance existing immunotherapy.
Biomarkers used in current clinical practice for ICB
The application of ICIs in cancer treatment has shown substantial success. However, there is significant variability in outcomes among patients treated with ICB, and the overall response rate remains relatively low [26]. To prevent non-responsive patients from undergoing ineffective treatments and incurring high costs, it is imperative to find reliable biomarkers to identify patients who may benefit from ICB. Three biomarkers are often used to identify potential ICB responders in current clinical practice, including PD-L1, TMB, and MSI-H/dMMR, and they have been found to be associated with the efficacy of ICB in clinical practice [3,4,5]. However, these biomarkers are not perfect, as they face inevitable challenges in practical applications. Their predictive abilities vary across different cancer types, and standardized thresholds to discriminate patients are lacking [37]. Furthermore, these biomarkers cannot reflect the heterogeneity of the patient’s TME, which directly influences the anti-tumor response. Arbitrarily relying on these biomarkers for discrimination may deprive certain patients of the opportunity to benefit from ICB.
PD-L1
PD-L1 is often expressed on the surface of cancer cells or APCs under conditions of T cell infiltration and interferon-γ production, which indicates the preexistence of T cell response and predicts the response when receiving ICIs [38]. PD-L1 is the first FDA-approved biomarker for ICB, and its expression in tumor tissues is primarily detected by immunohistochemistry in clinical practice. A high tumor PD-L1 expression level predicts enhanced ICB efficacy [3], and higher expression of PD-L1 is associated with a more substantial improvement [39]. However, some patients with low PD-L1 expression can also achieve survival benefits from ICB [40, 41]. Conversely, some patients with high PD-L1 expression may not respond to ICB, as the PD-1/PD-L1 interaction is more correlated with ICB response compared with PD-L1 expression levels [42]. PD-L1 expression exhibits temporospatial dynamics as it is heterogeneous across different tissue sample sites and at different time points, which may also compromise its predictive accuracy [43, 44]. Moreover, researchers have discovered that specific somatic molecular alterations were also correlated with ICB outcome, which could also differentially impact the predictive power of PD-L1 [43]. In addition, the predictive ability of PD-L1 is diminished when ICB is combined with other therapies such as chemotherapy [45]. These limitations indicate that PD-L1 expression is an imperfect biomarker, emphasizing the urgent need for other biomarkers.
TMB
TMB represents the number of non-synonymous somatic mutations per million bases of a tested genomic sequence, which is often determined by whole-exome sequencing or targeted panel sequencing [46]. A higher TMB may lead to a greater mutational load, increase the generation of immunogenic neoantigens, and activate more CD8+ T cells, thus enhancing the response to ICB. The KEYNOTE-158 study showed significantly enhanced ICB outcomes in TMB-high patients compared with TMB-low patients with advanced solid tumors [47]. Patients with high TMB tend to benefit from ICB irrespective of PD-L1 expression, indicating TMB as a nonredundant biomarker [48]. However, a higher TMB does not necessarily mean a better response to ICB. Only a small fraction of somatic mutations can be transcribed and translated into neoantigens that are presented by major histocompatibility complex (MHC) molecules on the cell surface. And even fewer neoantigens can be recognized by the immune system, thereby leading to limited recruitment of CD8+ T cells [49]. A pan-cancer study [50] found that high TMB did not accurately predict ICB response in certain cancers that showed no correlation between the levels of CD8+ T cells and neoantigen loads, and some TMB-high patients even exhibited worse ICB outcomes in these cancer types. In addition, the diversity of human leukocyte antigen class I (HLA-I) genotypes is also associated with ICB response, which influences the presentation of neoantigens to CD8+ T cells [51]. Therefore, a high TMB does not ensure the production of adequate immunogenic neoantigens recognized by CD8+ T cells and may not always predict the response to ICB.
MSI-H/dMMR
Loss-of-function mutations or transcriptional silencing of mismatch repair genes like MLH1, MSH2, MSH6, and PMS2 can alter the size of DNA microsatellite regions and is defined as microsatellite instability (MSI) [52]. MSI-H is a specific type of TMB-high, as most MSI-H tumors show high TMB while only a small fraction of TMB-high tumors are MSI-H [53]. MSI-H or dMMR can increase the TMB and generate immunogenic neoantigens, which can serve as a useful biomarker predicting a favorable ICB response [4]. However, the frequency of MSI varies significantly across different types of cancers. MSI-H/dMMR is more common in endometrial cancer, gastric cancer, and colorectal cancer, while it is rare in other cancers, such as lung adenocarcinoma, limiting its utility as a pan-cancer biomarker [54, 55].
Other biomarkers
While the biomarkers discussed above have varying degrees of limitations, it is imperative to unravel the underlying mechanisms and develop robust predictive models to better stratify patients. Recent studies have identified other factors that may predict the response to ICB, including specific infiltrating immune cells in the TME [56], specific gene mutations such as BRCA2 [57], somatic copy number alterations [58], circulating tumor DNA (ctDNA) [59], and the gut microbiome [60]. Owing to the heterogeneity of the TME, the predictive accuracy of biomarkers may be impacted by specific cell subclusters. Recent advances in single-cell technologies such as scRNA-seq have provided a detailed investigation of cellular components in the TME and peripheral blood to discover critical cell subsets that correlate with the response to ICB.
Cellular components of the tumor microenvironment
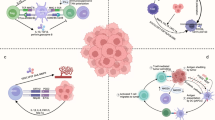
Mobilizing immune cells to induce a durable anti-tumor immune response is the primary goal of ICB. The TME is a heterogeneous microenvironment including cancer cells and stromal cells that can either promote or suppress anti-tumor effects and influence cancer cell growth, metabolism, proliferation, and metastasis (Fig. 2). Innate immune cells, including dendritic cells (DCs), NK cells, macrophages, natural killer T (NKT) cells, and gamma delta T (γδ T) cells, participate in killing cancer cells in a nonspecific manner [61]. Anti-tumor adaptive immune cells include B cells and T cells, with the latter comprising CD8+ cytotoxic T cells and CD4+ helper T cells. Despite the existence of anti-tumor immune cells, the TME overall exhibits a pro-tumor pattern due to the presence of immunosuppressive cells, including Tregs, myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), tumor-associated neutrophils (TANs), regulatory B cells (Bregs), and cancer-associated fibroblasts (CAFs) [62, 63]. These immunosuppressive components of the TME may impede the activation of anti-tumor immunity and contribute to ICB failure.

The schematic presentation of the TME composition. The tumor microenvironment (TME) contains both cellular and non-cellular components. Multiple cell types are present in the TME, including cancer cells and stromal cells. The heterogeneous cells contain both immunostimulatory and immunosuppressive subsets that exert anti-tumor and pro-tumor effects, respectively. And the preponderance of the pro-tumor cell subsets contributes to the immunosuppressive TME, leading to cancer progression and the failure of ICB. The application of single-cell RNA sequencing (scRNA-seq) provides insights into TME heterogeneity. Th1 cell, CD4+ T helper 1 cell; NK cell, natural killer cell; TAM, tumor-associated macrophage; TAN, tumor-associated neutrophil; Th2 cell, CD4+ T helper 2 cell; CAF, cancer-associated fibroblast; Treg, CD4+ regulatory T cell; MDSC, myeloid-derived suppressor cell; ECM, extracellular matrix. Created with Figdraw (www.figdraw.com)
CD8+ T cells
Within T cells, CD8+ T cells are the primary effector cells to kill cancer cells, and CD4+ T cells, including Tregs and helper T cells, modulate the function of CD8+ T cells. CD8+ T cells exert tumor-killing cytotoxicity primarily through two mechanisms: the Fas-FasL pathway and the perforin-granzyme pathway [6]. In the Fas-FasL pathway, CD8+ T cells can secrete vesicles containing the Fas ligand (FasL) that bind to the Fas molecule on cancer cells, activating intracellular caspase 8 and inducing cell apoptosis. In the perforin-granzyme pathway, perforin released by CD8+ T cells can open channels on the surface of cancer cells, allowing granzymes to enter and lead to apoptosis.
Tregs
Tregs are a subset of immunosuppressive CD4+ T cells that express forkhead box protein P3 (FOXP3) and participate in maintaining self-tolerance [64]. Tregs are more abundant in tumor tissues than in peripheral blood and peritumoral tissues, suggesting their pro-tumor function [65]. FOXP3+ CD4+ T cells can be classified into effector Treg cells (eTregs), naïve Treg cells, and non-Treg cells based on specific markers. Of these subsets, eTregs (CD4+ CD25high FOXP3high CD45RA−) are the predominant subset of Tregs that can differentiate from naïve Treg cells upon TCR stimulation and exert potent immunosuppressive effects [66]. Tregs facilitate immunosuppression via multiple mechanisms, including consumption of interleukin-2 (IL-2), suppression of APC function, production of immunosuppressive molecules such as IL-10 and transforming growth factor-β (TGF-β), and destruction of effector cells [66]. 4-1BB is specifically expressed on activated intratumoral Tregs, enhancing their suppressive function [67].
Macrophages
Macrophages exhibit different developmental trajectories depending on the context and are commonly classified into two classes: classically activated macrophages (M1) and alternatively activated macrophages (M2). M1 macrophages express CD80, CD86, and HLA-DR surface markers and have anti-tumor effects by producing molecules such as IL-12, reactive oxygen species (ROS), and nitric oxide synthase (iNOS). In contrast, M2 macrophages express CD163, CD206, and CD204 surface markers and exert pro-tumor effects by producing IL-10, vascular endothelial growth factor (VEGF), and matrix metalloprotein (MMP) to promote angiogenesis, matrix remodeling, and immunosuppression [68, 69]. The TAM population is heterogeneous and primarily exhibits an M2-like immunosuppressive pattern. Nonetheless, the binary M1/M2 division of macrophages is imprecise, as recent scRNA-seq studies have identified specific TAM clusters that express both M1 and M2 signatures [70, 71] and discovered TAM clusters that do not fall within the conventional M1/M2 classification [72], highlighting the complexity of TAMs and the need for further investigation. Additionally, scRNA-seq can identify novel TAM subsets with crucial effects. FOLR2+ macrophages, a subset of tissue-resident macrophages, can prime CD8+ T cells, leading to a better prognosis [73]. Conversely, TAMs enriched for the scavenger receptor MARCO in gliomas are associated with the mesenchymal signature and hypoxia, contributing to tumor progression [74]. TAMs highly expressing SPP1 can drive malignancy and are correlated with unfavorable outcomes and metastasis in CRC [75].
Neutrophils
Neutrophils present in the TME exhibit heterogeneity and can be classified into anti-tumor N1-TANs and pro-tumor N2-TANs, similar to TAMs [76]. N2-polarized TANs constitute the major part of TANs. Cytokines such as TGF-β can facilitate the transition of TANs from the N1 to the N2 phenotype, promoting tumor cell proliferation, matrix remodeling, and angiogenesis [76]. Recent studies using scRNA-seq have further investigated the heterogeneity within TAN subtypes. In pancreatic ductal adenocarcinoma (PDAC), a terminally differentiated TAN subcluster named TAN-1 highly expresses VEGFA and exhibits hyperactivated glycolytic activity, which is correlated with poor prognosis [77]. In liver tumors, scRNA-seq analysis identified three pro-tumor TAN subsets (CCL4+ TANs, IFIT1+ TANs, and SPP1+ TANs) and one anti-tumor subset (APOA2+ TANs) [78]. CCL4+ TANs secrete CCL4 protein and recruit macrophages via the CCL4-CCR5 axis, leading to immunosuppression in the TME. IFIT1+ TANs highly express CD274 (encoding PD-L1) and directly suppress the function of CD8+ T cells.
CAFs
CAFs are another important constituent of the TME and are particularly abundant in tumors enriched for tumor reactive stroma, such as cholangiocarcinoma, pancreatic cancer, and breast cancer [79]. CAFs originate primarily from tissue-resident fibroblasts, and they are also derived from fibrocytes, epithelial cells, mesenchymal stem cells, and endothelial cells. In some rare conditions, CAFs may also be derived from the transdifferentiation of adipocytes, pericytes, and smooth muscle cells [80]. CAFs participate in ECM remodeling by producing ECM components or matrix metalloproteinases (MMPs). They also engage in crosstalk with other cells and contribute to angiogenesis and cancer cell metastasis by secreting multiple cytokines, including VEGF, TGF-β, IL-6, C-X-C motif chemokine ligand 12 (CXCL12), and C-C motif chemokine ligand 2 (CCL2) [80, 81]. However, CAFs are a heterogeneous population including both pro- and anti-tumor subsets. Two important subsets of CAFs have been identified: inflammatory CAFs (iCAFs) and myofibroblastic CAFs (myCAFs), which are characterized by cytokine secretion and stromal remodeling, respectively [82]. Some scRNA-seq studies have confirmed the existence of these two CAF subsets and identified other novel CAF subclusters in various cancer types [83,84,85,86].
Endothelial cells
Endothelial cells (ECs) of blood vessels in the tumor control the recruitment of immune cells by producing angiocrine factors [87]. ECs also control the intravasation and extravasation of cancer cells, resulting in tumor progression and metastasis [87]. One scRNA-seq study identified a tumor-specific PLVAP+ EC subset that could be activated by VEGF signaling and contribute to the immunosuppressive TME [88]. However, ECs can also exert anti-tumor effects, as a study found that MECA-79+ tumor-associated HEV endothelial cells could mediate lymphocyte extravasation during ICB and enhance anti-tumor immunity [89].
Dynamic changes during ICB therapy revealed by scRNA-seq
Reprogramming of the TME is a key outcome of ICB therapy. To understand the mechanisms underlying ICB function, it is essential to capture the dynamic changes in cellular components of the TME or peripheral blood before and after treatment.
As CD8+ T cells are the predominant cells responsible for tumor eradication that experience a series of lineage transitions and transcriptional changes during the anti-tumor response, it is especially important to study the changes in CD8+ T cells. CD8+ T cells are heterogeneous and multiple states along the development trajectory have been defined by recent studies. Some scRNA-seq studies have identified a CD8+ T cell development trajectory starting with LEF1+ naïve cells, transitioning through GZMK+ transitory cells, and then bifurcating into two branches with two distinct final states: CX3CR1+ terminally differentiated effector memory or effector T cells (Temra) and LAYN+ exhausted T cells (Tex) [90,91,92]. Notably, exhaustion is a gradual process, and there are different subsets of exhausted CD8+ T cells. Two subsets, progenitor exhausted T cells (Texprog) and terminally exhausted T cells (Texterm), have different expression profiles and transcriptional changes during ICB [93]. Transcription factor 1 (TCF1), encoded by TCF7, is a crucial transcription factor that maintains stem-like functions of CD8+ progenitor cells, and the absence of Tcf1 indicates progressive differentiation into exhausted CD8+ T cells [94]. TCF-1+ PD-1+ Texprog cells show stem-like signatures, have relatively lower levels of cytotoxicity, and can differentiate into Texterm cells. In contrast, TCF-1− PD-1+ Texterm cells are more exhausted, express higher levels of immune checkpoints, and transiently exhibit robust tumor-killing activity by producing more cytotoxic cytokines before rapidly becoming dysfunctional [93, 95, 96]. Of the two Tex subsets, Texprog cells are ICB-responsive, and Texterm cells are not [93, 97]. This is because Texprog cells can both self-renew and differentiate into Texterm cells to temporarily fight against cancer cells, while Texterm cells cannot proliferate. These findings reveal that CD8+ T cells contain multiple subsets along the activation and exhaustion trajectory. Understanding the dynamic subsets and states of CD8+ T cells is crucial for elucidating the mechanisms underlying studying immunotherapeutic strategies against cancer.
Use of scRNA-seq can capture the changes among different CD8+ T cell subsets during ICB. Integration of TCR sequencing (TCR-seq) with scRNA-seq provides insights into CD8+ T cell clonality, diversity, and dynamics in both human and animal models [98].
ScRNA-seq studies have found that CD8+ T cells transitioned from a naïve or stem state to an active or effector state during ICB. Zhou et al. [99] found four major CD8+ T cell subclusters in murine oral carcinoma, including Tcf7+ Pd1− naïve/memory cells, Tcf7+ Pd1+ progenitor cells, Tcf7− Pd1+ effector cells, and Mki67+ proliferative cells. They demonstrated the transition from naïve/memory and progenitor cells to effector cells and the acquisition of a more activated/exhausted state during ICB. Kurtulus et al. [100] studied PD-1− CD8+ T cells from mice bearing colon carcinoma and observed a reduction in naïve-like cells along with the expansion of memory-precursor- and effector-like cells after anti-PD-1 and anti-TIM-3 blockade. Similarly, Khojandi et al. [101] also observed a significant decrease in naïve-like gene expression in peripheral CD8+ T cells after ICB, such as TCF7, SELL, IL7R, and CCR7. Collectively, scRNA-seq reveals that CD8+ T cells can shift from the naïve state to a more activated state during ICB, characterized by the expansion of effector cells and increased cytotoxic activities against cancer cells.
The dynamic changes in different subsets of exhausted CD8+ T cells during ICB vary among patients with different cancer types and TME compositions. Some scRNA-seq studies have reported the accumulation of progenitor exhausted T cells after ICB, while others have found the accumulation of terminally exhausted T cells. Liu et al. [102] found that GZMK+ precursor exhausted T (Texp) cells upregulated the expression of CXCL13 and significantly accumulated after ICB in patients with NSCLC. Khan et al. [103] found that after PD-1 inhibition, Texprog cells differentiated into intermediate Tex (Texinter) cells in anti-PD-1 responsive GL261 glioblastoma models. Kwon et al. [104] collected tumor tissues from patients with MSI-H gastric cancer who received ICB and conducted scRNA-seq analysis. They found an increase in the proportions of transitory and terminally exhausted CD8+ T cells after ICB, which contributed to the enhancement of cytotoxic function. Bi et al. [105] investigated a human RCC cohort and found that a subset of CD8+ T cells with low expression of the activation marker 4-1BB (4-1BB-Lo) had high progenitor exhausted signatures at baseline. These 4-1BB-Lo cells were activated by ICB and converted from a progenitor exhausted state to a more terminally exhausted state after ICB. The above studies revealed different distribution patterns of exhausted CD8+ T cell subsets after ICB, including progenitor, intermediate, and terminally exhausted cells. Liu et al. [106] found that this might be explained by different levels of immunosuppression in different TMEs. Terminally exhausted CD8+ T cells are more likely to accumulate in tumors with higher levels of immunosuppression after ICB, such as squamous cell carcinoma (SCC), basal cell carcinoma (BCC), and RCC, while precursor-like CD8+ T cells tend to accumulate in tumors with lower levels of immunosuppression, such as NSCLC and melanoma [102, 106].
Apart from CD8+ T cells, the application of scRNA-seq also reveals the dynamic changes in other cell types during ICB. For CD4+ T cells, Liu et al. [106] observed an increase in Th1-like CXCL13+ CD4+ T cells following effective ICB treatment across multiple cancer types, indicating their role as tumor-reactive cells. Liu et al. [102] found a slight decrease in Tregs after responding to ICB in NSCLC, consistent with the immunosuppressive role of Tregs. Li et al. [107] also observed a decrease in Tregs and an increase in CD40+ B cells after responding to ICB in patients with dMMR/MSI-H CRC. Additionally, Bi et al. [105] studied macrophages in RCC patients and observed a significant increase in M1-like proinflammatory patterns after ICB, which also upregulated some immune checkpoint and anti-inflammatory genes, potentially leading to transient effects and the ultimate failure of ICB.
In summary, scRNA-seq can identify dynamic changes in cell numbers and transcriptional patterns of cells in the TME, especially CD8+ T cells, in animal models or human patients at different time points during ICB treatment. Studying the changes in these cells can enhance our understanding of the impact of ICB on the TME, elucidating the process of patients’ responses to ICB and revealing the mechanisms underlying ICB function.
Discovering novel biomarkers of ICB with scRNA-seq
ScRNA-seq is a potent technology for investigating crucial cell clusters with varying expression profiles between responders and non-responders to cancer immunotherapy. Differentially expressed genes or cell subsets displaying high expression differences between responders and non-responders can be used as biomarkers. Recent research has aimed to identify new biomarkers to predict the efficacy of ICB using scRNA-seq, shedding light on specific cell populations that might be indicative of ICB responsiveness in both the tumor and peripheral blood (Table 1).
Biomarkers in the tumor
T cells
CD8+ T cells can directly kill cancer cells, and their expression patterns can determine the effectiveness of ICB therapy. Sade-Feldman et al. [108] identified two CD8+ T cell states in melanoma by scRNA-seq. One cluster, CD8_G, was enriched for genes related to memory and activation, including TCF7 and IL7R, and was more abundant in ICB responders. They examined the expression levels of TCF7 in CD8+ T cells and found more tumor-infiltrating TCF7+ CD8+ T cells in ICB responders and more TCF7− CD8+ T cells in non-responders, demonstrating the predictive ability of TCF7 expression of CD8+ T cells. Consistent with this study, other scRNA-seq studies also found that responders had higher levels of stem or progenitor T cells at baseline, whereas non-responders tended to accumulate more terminally exhausted T cells. Zhang et al. [109] applied scRNA-seq in TNBC tumors and discovered two clusters of CXCL13+ T cells (CD8-CXCL13 and CD4-CXCL13) with high expression of CXCL13 and exhaustion genes PDCD1, TIGIT, and CTLA-4. Compared with non-responders, CD8-CXCL13 and CD4-CXCL13 were enriched at baseline and expanded after ICB in responders, indicating their tumor-reactive roles. They discovered a strong interaction between CXCL13+ T cells and follicular B cells via the CXCL13-CXCR5 axis, which enhanced the anti-tumor activity, consistent with other scRNA-seq studies [110, 111]. In addition, another study showed that CXCL13 was a B cell chemoattractant and was associated with the formation of tertiary lymphoid structures (TLSs), the ectopic lymphoid tissues that form around tumors and participate in anti-tumor immune responses [112]. Liu et al. [106] demonstrated that CXCL13 was a marker of tumor-reactive T cells. Measuring the abundance of CXCL13+ CD8+ and CXCL13+ CD4+ T cells together in tumor samples is a powerful biomarker of response to ICB in multiple cohorts, with higher predictive accuracy than TMB [106]. Kim et al. [113] classified CD8+ T cells into PDCD1-high and PDCD1-low subsets based on PDCD1 expression levels from scRNA-seq datasets of both melanoma and NSCLC. They found that the transcription factor TOX was a crucial promoter regulating T cell exhaustion. Additionally, low baseline expression of TOX in tumor-infiltrating T cells was associated with a positive ICB response, suggesting the predictive ability of TOX. These studies highlight the importance of baseline accumulation of stem/memory-like or progenitor exhausted cells for a favorable response to ICB, as opposed to terminally exhausted CD8+ T cells. A pan-cancer scRNA-seq study [114] discovered a novel T-cell state characterized by high expression of HSPA1A and HSPA1B as well as a stress response signature. This CD8+/CD4+ TSTR group was found to be predictive of an unfavorable response to ICB across multiple cancer types, providing a new T-cell biomarker different from T-cell exhaustion.
B cells
B cells within the TME are another group of immune cells crucial for robust anti-tumor immunity. Patil et al. [115] clustered B cells from scRNA-seq data of NSCLC patients and discovered a plasma cell subcluster with signature genes, including MZB1, DERL3, JSRP1, and IGHG2. They demonstrated that a high plasma cell signature in the TME could predict a favorable response to ICB in multiple cohorts. Cabrita et al. [116] studied B cells using a melanoma scRNA-seq dataset and noticed that baseline CD69+ B cells and IGLL5− CD69+ B cells were both more abundant in responders. They also discovered that B-cell-rich tumors had a higher proportion of TCF7- or IL7R-expressing naïve or memory-like CD8+ and CD4+ T cells than B-cell-poor tumors, which suggested that a higher level of tumor-infiltrating B cells could recruit more progenitor T cells to TLSs and potentially lead to a better response to ICB.
TAMs
TAMs can serve as another type of biomarker for predicting ICB response, as M1- and M2-type macrophages play distinct roles in the TME. Xiong et al. [117] identified a macrophage subset primarily in melanoma non-responders with high expression of TREM2, M2-like signatures, and genes associated with the complement system and complement cascade activation. The complement system associated with this TREM2high macrophage subset could alter the composition of the TME and contribute to ICB resistance. Consistent with Xiong et al. [117], a study by Zhang et al. [118] also found a TREM2+ TAM subset from scRNA-seq data of NSCLC and demonstrated its immunosuppressive role in the TME. This TREM2+ cluster was enriched for genes linked to pro-tumor pathways and could strongly interact with FOXP3+ Tregs through the IL1B/IL1R interaction, suppressing the anti-tumor effects. These studies revealed that high infiltration of TREM2+ TAMs might predict a poor response to ICB in various cancers. In addition, Hao et al. [119] identified a specific TAM subset expressing APOC1, FABP1, and C1QC in HCC patients. They discovered that APOC1 exhibited significantly higher expression levels in TAMs of HCC tissues than adjacent tissues or peripheral blood and was negatively correlated with PD-1 and PD-L1 expression. Therefore, high expression of APOC1 in TAMs could predict an unfavorable ICB response in HCC patients. In the study by Zhang et al. [109], MMP9+ macrophages with high expression of PLA2G2D, IL2RG, and CXCL9 were enriched in responders at baseline, suggesting their predictive value in TNBC patients receiving ICB therapy.
CAFs
CAFs interact with cancer cells and immune cells in the TME, impacting the responsiveness to ICB therapy. Dominguez et al. [85] analyzed stromal cells expressing podoplanin from the pancreases of normal mice and mice with PDAC. They identified a subcluster of myCAFs that showed enrichment for TGFβ gene signatures with significantly higher expression of LRRC15 than natural fibroblasts. They demonstrated that high LRRC15+ CAF signatures could predict poor responses to atezolizumab in several types of human cancers. Similar to this finding, Kieffer et al. [86] discovered two subsets of myCAFs from breast cancer scRNA-seq data: ecm-myCAFs, which exhibited high expression of LRRC15 and were associated with ECM remodeling, and TGFβ-myCAFs, which had high expression of TGFβ1 and were associated with the TGF-β signaling pathway. These two subclusters might be linked to an immunosuppressive TME enriched for Tregs and deficient in CD8+ T cells. Non-responders with melanoma or NSCLC who received ICB had higher baseline expression levels of ecm-myCAFs and TGFβ-myCAFs. Wang et al. [120] identified a novel CAF subcluster termed meCAF that had a highly activated metabolic state with the marker genes PLA2G2A and CRABP2 based on scRNA-seq data from PDAC. In loose-type PDAC with low desmoplasia, meCAFs were enriched and were associated with high metastatic potential and poor prognosis. However, patients with high meCAF infiltration at baseline showed a dramatic response to anti-PD-1 therapy, indicating that meCAFs could predict favorable responses to PDAC immunotherapy. Obradovic et al. [121] conducted scRNA-seq on HNSCC samples and identified five distinct CAF subclusters. In anti-PD-1 responders, the gene signatures of two CAF subsets (HNCAF-0 and HNCAF-3) were enriched at baseline and expanded after treatment. The researchers also demonstrated that HNCAF-0/3 could inhibit the exhaustion of CD8+ T cells and enhance their memory phenotypes and cytolytic function, as evidenced by functional assays. Thus, the gene signatures of HNCAF-0/3 were designated as T cell-stimulating CAF (tsCAF), which could predict the responsiveness to ICB. These studies provide additional evidence that CAFs in the TME are heterogeneous, comprising both pro- and anti-tumor subsets that can differentially regulate anti-tumor immunity and ultimately modulate the effects of ICB [82].
NK cells
As innate immune cells, NK cells also play an essential role in the anti-tumor response, and their heterogeneity has been revealed through single-cell technologies. In a pan-cancer scRNA-seq study [122], CD56dimCD16hi c6-DNAJB1 NK cells were designated as tumor-associated NK (TaNK) cells, which were enriched in tumors and exhibited impaired cytotoxicity, indicative of a terminal state. The signatures of TaNK cells were more pronounced in non-responders than responders, implying their potential as an ICB biomarker.
Cancer cells
In addition to cells in the cancer stroma, the RNA expression patterns of cancer cells themselves may be indicative of ICB outcome. For example, Zhou et al. [123] analyzed several melanoma scRNA-seq datasets and revealed that melanoma cells had elevated expression levels of signal regulatory protein α (SIRPα, encoded by SIRPA) and could strongly interact with CD8+ T cells via the SIRPα/CD47 axis, thus enhancing the cytotoxicity of CD8+ T cells. ICB responders had higher SIRPA expression in melanoma cells than non-responders, demonstrating the predictive potential of SIRPA expression in melanoma cells.
Biomarkers in peripheral blood
Although cellular components in the TME directly reflect factors that influence cancer cell growth, metabolism, and metastasis, obtaining these samples from patients requires invasive procedures. Therefore, it is essential to investigate circulating biomarkers to minimize the harm caused by biopsies. Watson et al. [124] used scRNA-seq and single-cell V(D)J sequencing to analyze peripheral CD8+ T cells collected before and after ICB therapy from melanoma patients. They found that patients with low cytotoxicity and small expanded clone numbers in their peripheral CD8+ T cells at baseline had poor clinical outcomes following ICB. De Biasi et al. [125] conducted scRNA-seq on circulating CD8+ T cells from patients with metastatic melanoma and found that mucosal-associated invariant T (MAIT) cells were present in a higher proportion and had higher activation signatures before and after ICB in responders. Moreover, CXCR4 expression on peripheral MAIT cells could mediate their migration to the site of metastatic tumors and exert their proinflammatory and cytotoxic functions, further supporting the predictive value of peripheral MAIT cells for melanoma ICB therapy. Li et al. [126] analyzed scRNA-seq data from melanoma patients and identified a subset of CD8+ T cells that exhibited high levels of oxidative phosphorylation (OXPHOS) metabolism and expression of exhaustion and cytotoxic signatures. This unique CD8+ T cell cluster was present in both tumors and peripheral blood and was termed CD8+ TOXPHOS cells. Additionally, the researchers found that melanoma patients who showed resistance to ICB had higher levels of OXPHOS in CD8+ TOXPHOS cells in both tumors and peripheral blood.
In addition to CD8+ T cells, other immune cells in peripheral blood have been shown to carry predictive potential in other studies using scRNA-seq. Pour et al. [127] analyzed peripheral blood mononuclear cells (PBMCs) from melanoma patients before and during nivolumab treatment. They identified a monocyte cluster expressing significantly higher levels of S100A9 and other S100A proteins in non-responders than in responders. Further validation showed that S100A9+ monocytes were significantly more abundant in non-responders in a melanoma cohort, suggesting its predictive potential for ICB. Sui et al. [128] analyzed scRNA-seq data from MSI-H CRC patients and discovered that neutrophils interacted with CD8+ T cells in the tumor via the CD80/CD86-CTLA4 axis, contributing to local inflammation that predicted an unfavorable response to ICB. Moreover, the inflammatory response was correlated with peripheral leukocytes, and a high neutrophil-to-lymphocyte ratio (NLR) could also predict a poor response to ICB.
Applying scRNA-seq to identify novel therapeutic targets for immunotherapy
Due to the low response rate to ICB in cancer treatment, identifying novel therapeutic targets to combine with CTLA-4 or PD-1/PD-L1 inhibitors is essential for enhancing anti-tumor effects. Applying scRNA-seq enables the identification of critical molecules or cell subtypes that contribute to immunosuppressive tumor microenvironments and the failure of ICB therapy. To identify new treatment targets for immunotherapy, scRNA-seq can be employed to discover crucial genes expressed by cancer cells as well as other immunosuppressive cells, compare the expression of genes between responders and non-responders, evaluate changes in the TME after targeting therapeutic targets, and identify immunosuppressive intercellular interactions in the TME. Moreover, to strengthen the credibility of scRNA-seq results, it is necessary to confirm the efficacy of proposed therapeutic targets through functional assays, such as cell lines and animal models. The studies applying scRNA-seq to identify novel treatment targets for immunotherapy are summarized in Table 2.
By utilizing scRNA-seq, it is possible to identify immune-evasion genes expressed by cancer cells that can lead to the failure of ICB. Targeting these genes represents a potential approach to enhancing the efficacy of ICB treatment. Jerby-Arnon et al. [132] studied malignant cells from melanoma patients using scRNA-seq and identified an immune resistance program including cyclin-dependent kinase 4 (CDK4). Combining CDK4/6 inhibitors and ICB enhanced the response to therapy. Cappellesso et al. [133] performed scRNA-seq on PDAC patients and identified SLC4A4 as the most abundantly expressed bicarbonate transporter gene of PDAC epithelial cells. Inhibition of SLC4A4 reduced the acidification of the TME and repressed tumor growth when combined with ICB, confirming its potential as a therapeutic target.
Some genes expressed by immunosuppressive cells in the TME revealed by scRNA-seq can also be therapeutic targets. Abdelfattah et al. [134] observed high expression of S100A4 in exhausted T cells, Tregs, and pro-tumor myeloid cells by scRNA-seq of human gliomas and found that it was associated with a poor prognosis. Further functional assays confirmed the immunosuppressive role of S100A4, indicating its potential as a therapeutic target. Alvisi et al. [135] found increased expression of MEOX1 in tumor-infiltrating Tregs by scRNA-seq data of ICC and demonstrated that the transcription factor MEOX1 could reprogram peripheral Tregs toward a tumor-infiltrating immunosuppressive signature.
Genes showing differential expression between responders and non-responders, as discovered by scRNA-seq, may serve as potential treatment targets to improve ICB efficacy. Yuen et al. [136] found that baseline IL8 (CXCL8) expression in peripheral myeloid cells was higher in non-responders, according to scRNA-seq data from PBMCs of patients with metastatic urothelial carcinoma and metastatic renal cell carcinoma. IL8-high cells downregulated genes related to antigen presentation, contributing to immunosuppression. Therefore, blocking CXCR2, the receptor of IL-8, could be an effective strategy to sensitize tumors to PD-1 inhibitors. Wen et al. [137] analyzed peripheral CD8+ T cells from melanoma patients receiving ICB and found that NKG7 was upregulated in responders and downregulated in non-responders after ICB treatment. The researchers confirmed the cytolytic function of NKG7 through functional assays and demonstrated that combining NKG7 mRNA therapy and ICB enhanced the cytotoxicity of CD8+ T cells.
Using scRNA-seq to study changes in the TME after targeting certain genes or proteins can evaluate their potential as therapeutic targets. Molgora et al. [138] observed that in mouse models, the combination of anti-TREM2 treatment of TAMs and anti-PD-1 treatment remodeled the landscape of macrophages in the TME, with a decrease in MRC1+ (CD206+) macrophages and an increase in immunostimulatory iNOS+ macrophages. The researchers highlighted the crucial role of TREM2 in immunotherapy resistance, consistent with the findings of Xiong et al. [117] and Zhang et al. [118]. Hui et al. [139] performed scRNA-seq on tumor-bearing mice deficient in APOE and noticed a decrease in M2-like C1QC+ and CCR2+ TAMs, suggesting that targeting APOE+ macrophages could be a novel strategy to combine with ICB for cancer treatment. Kim et al. [140] conducted scRNA-seq analysis to compare the effects of CD73 inhibitors and PD-1 inhibitors on tumor-bearing mice with CRC. They discovered that CD73 inhibition improved the anti-tumor functions of Tregs and exhausted CD8+ T cells distinctly from PD-1 inhibition, showing that the combination of CD73 inhibitors and PD-1 inhibitors could synergistically kill cancer cells in CRC treatment. Liu et al. [141] showed that inhibiting CD39 in bladder cancer increased tumor-infiltrating NK cells and conventional type 1 dendritic cells (cDC1) and enhanced the cytotoxicity of CD8+ T cells. They also found that CD39 inhibition had synergistic effects with cisplatin but not with PD-1 inhibitors, indicating its potential for combination with chemotherapy rather than immunotherapy.
ScRNA-seq analysis can also effectively capture cellular interactions within the TME, revealing the ligand-receptor pairs that may serve as promising targets for immunotherapy interventions. Zhang et al. [84] applied scRNA-seq in ICC samples and identified an interaction between CD146+ vascular CAFs (vCAFs) and malignant cells via the IL-6/IL-6R axis, which could promote tumorigenesis and cancer stemness. Inhibiting the IL-6/IL-6R axis reduced tumor progression, indicating its potential as a treatment target. Heidegger et al. [142] focused on tumor endothelial cells (TECs) in prostate cancer patients using scRNA-seq data. They identified an interaction between arterial TECs and tip TECs mediated by the CXCL12/CXCR4 axis, which could promote angiogenesis. The CXCR4 inhibitor AMD3100 could target this ligand-receptor pair and reduce the number and density of vessels. Gong et al. [143] analyzed scRNA-seq data and found that nasopharyngeal carcinoma cells interacted with Tregs via the CD70-CD27 axis, thereby enhancing the proliferation and suppressive function of Tregs. CD70 blockade could be synergistic with PD-1 inhibition to enhance the anti-tumor response of T cells.
Conclusions and further perspectives
Over the past decade, the use of ICB in cancer treatment has proven successful in delaying tumor progression and prolonging overall survival in some patients. However, this therapy only benefits a small proportion of individuals with specific cancer types. The intratumoral and intertumoral heterogeneity of the TME is a key factor that contributes to differences in responsiveness to ICB. As an emerging high-throughput technology, scRNA-seq is increasingly being used to study the heterogeneous transcriptomic information of individual cells in the TME and identify crucial cell subsets involved in ICB responsiveness from a large amount of bioinformatic data. In recent years, many studies have employed scRNA-seq to investigate the mechanisms underlying responsiveness to ICB in multiple cancers, in both animal models and humans. By applying scRNA-seq to tumor samples and PBMCs, researchers can capture the dynamic changes in crucial cell subsets during ICB, identify biomarkers that can predict response to ICB, and discover novel therapeutic targets in the TME for immunotherapy.
To precisely capture the dynamic alterations of cell subsets and obtain convincing results, it may be helpful to take tumor samples from patients in a large cohort at multiple time points during ICB. However, the recruitment of enough patients for scRNA-seq is difficult, and the repeated procedures necessary to obtain multiple samples can cause harm to patients. To solve this problem, some studies have conducted scRNA-seq on animal models and validated the conclusions in human cohorts. In addition, investigating key cell subclusters in peripheral blood, which is noninvasive to patients, provides an alternative approach to understanding the mechanisms of ICB response or resistance.
However, the reliability of scRNA-seq analysis is limited not only by the number of cells collected from patients but also by the inherent shortcomings of the technology, such as single-cell isolation, library amplification, technical noise, and sequencing depth and quality [166]. In addition, since scRNA-seq can only detect RNA expression levels and cannot directly capture protein expression, it is often used in combination with other technologies, such as flow cytometry, mass spectrometry, and cytometry by time-of-flight (CyToF) to obtain transcriptomic and proteomic data of individual cells. Another single-cell technology, named cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq), has been applied in many studies. It combines scRNA-seq with cell surface protein detection, enabling the simultaneous profiling of the single-cell transcriptome and cell surface proteome [167]. Furthermore, although single-cell technologies capture biological information at the single-cell level, they do not preserve the spatial information of cells’ original locations since cells are dissociated from tissues. By integrating single-cell technologies with spatial omics technologies, such as spatial transcriptomes or spatial proteomics, we can both map the cellular phenotypes at a high resolution and reflect the architecture of the tissue microenvironment, allowing us to have a deep understanding of the TME heterogeneity [168]. In future studies, it would be beneficial to combine scRNA-seq with these techniques to more precisely characterize cellular profiles in large cohorts of cancer patients. Additionally, confirming scRNA-seq findings through functional assays would provide greater confidence in the results. These insights potentially inform clinical practice for precision medicine by identifying critical cell subsets involved in ICB responsiveness, improving patient stratification strategies, and searching for novel therapeutic targets.
Data availability
Not applicable.
Abbreviations
- ICB:
-
Immune checkpoint blockade
- TME:
-
Tumor microenvironment
- scRNA-seq:
-
Single-cell RNA sequencing
- ICIs:
-
Immune checkpoint inhibitors
- PD-1:
-
Programmed cell death 1
- PD-L1:
-
Programmed cell death 1 ligand 1
- CTLA-4:
-
Cytotoxic T lymphocyte-associated protein 4
- dMMR:
-
Mismatch repair deficiency
- MSI-H:
-
Microsatellite instability-high
- TMB:
-
Tumor mutational burden
- ECM:
-
Extracellular matrix
- RNA-seq:
-
RNA sequencing
- BCLC:
-
Barcelona Clinic Liver Cancer
- HCC:
-
Hepatocellular carcinoma
- APCs:
-
Antigen-presenting cells
- FDA:
-
Food and Drug Administration
- TCR:
-
T cell receptor
- RCC:
-
Renal cell carcinoma
- TNBC:
-
Triple-negative breast cancer
- NSCLC:
-
Non-small cell lung carcinoma
- HNSCC:
-
Head and neck squamous cell carcinomas
- MCC:
-
Merkel cell carcinoma
- LAG-3:
-
Lymphocyte-activation gene 3
- TIM-3:
-
T-cell immunoglobulin and mucin-domain 3
- TIGIT:
-
T cell immunoreceptor with Ig and ITIM domains
- Tregs:
-
Regulatory T cells
- Th1 cells:
-
T helper 1 cells
- NK cells:
-
Natural killer cells
- MHC:
-
Major histocompatibility complex
- HLA-I:
-
Human leukocyte antigen class I
- ctDNA:
-
Circulating tumor DNA
- DCs:
-
Dendritic cells
- NKT cells:
-
Natural killer T cells
- γδT cells:
-
gamma delta T cells
- MDSCs:
-
Myeloid-derived suppressor cells
- TAMs:
-
Tumor-associated macrophages
- TANs:
-
Tumor-associated neutrophils
- Bregs:
-
Regulatory B cells
- CAFs:
-
Cancer-associated fibroblasts
- IL-2:
-
Interleukin-2
- TGF-β:
-
Transforming growth factor-β
- ROS:
-
Reactive oxygen species
- iNOS:
-
Nitric oxide synthase
- VEGF:
-
Vascular endothelial growth factor
- MMP:
-
Matrix metalloprotein
- PDAC:
-
Pancreatic ductal adenocarcinoma
- MMPs:
-
Matrix metalloproteinases
- CXCL12:
-
C-X-C motif chemokine ligand 12
- CCL2:
-
C-C motif chemokine ligand 2
- iCAFs:
-
inflammatory CAFs
- myCAFs:
-
myofibroblastic CAFs
- ECs:
-
Endothelial cells
- TCF1:
-
Transcription factor 1
- TCR-seq:
-
TCR sequencing
- SCC:
-
Squamous cell carcinoma
- BCC:
-
Basal cell carcinoma
- TLSs:
-
Tertiary lymphoid structures
- MAIT cells:
-
Mucosal-associated invariant T cells
- OXPHOS:
-
Oxidative phosphorylation
- PBMCs:
-
Peripheral blood mononuclear cells
- NLR:
-
Neutrophil-to-lymphocyte ratio
- CDK4:
-
Cyclin-dependent kinase 4
- cDC1:
-
Conventional type 1 dendritic cells
- TECs:
-
Tumor endothelial cells
- vCAFs:
-
vascular CAFs
- CyToF:
-
Cytometry by time-of-flight
- CITE-seq:
-
Cellular indexing of transcriptomes and epitopes by sequencing
References
Yousefi H, Yuan J, Keshavarz-Fathi M, Murphy JF, Rezaei N. Immunotherapy of cancers comes of age. Expert Rev Clin Immunol. 2017;13(10):1001–15.
Brower V. Checkpoint blockade immunotherapy for cancer comes of age. J Natl Cancer Inst. 2015;107(3).
Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell Lung cancer. N Engl J Med. 2015;372(21):2018–28.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372(26):2509–20.
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell Lung cancer. Science. 2015;348(6230):124–8.
Golstein P, Griffiths GM. An early history of T cell-mediated cytotoxicity. Nat Rev Immunol. 2018;18(8):527–35.
Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the Tumor microenvironment. Science. 2015;348(6230):74–80.
Wang D, Gu J. VASC: dimension reduction and visualization of single-cell RNA-seq data by Deep Variational Autoencoder. Genom Proteom Bioinf. 2018;16(5):320–31.
Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat Protoc. 2020;15(4):1484–506.
Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32(4):381–6.
Aibar S, Gonzalez-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods. 2017;14(11):1083–6.
Ogata D, Namikawa K, Takahashi A, Yamazaki N. A review of the AJCC Melanoma staging system in the TNM classification (eighth edition). Jpn J Clin Oncol. 2021;51(5):671–4.
Reig M, Forner A, Rimola J, Ferrer-Fabrega J, Burrel M, Garcia-Criado A, et al. BCLC strategy for prognosis prediction and treatment recommendation: the 2022 update. J Hepatol. 2022;76(3):681–93.
De Ruysscher D, Niedermann G, Burnet NG, Siva S, Lee AWM, Hegi-Johnson F. Radiotherapy toxicity. Nat Rev Dis Primers. 2019;5(1):13.
Salas-Benito D, Perez-Gracia JL, Ponz-Sarvise M, Rodriguez-Ruiz ME, Martinez-Forero I, Castanon E, et al. Paradigms on Immunotherapy combinations with Chemotherapy. Cancer Discov. 2021;11(6):1353–67.
Llovet JM, Villanueva A, Lachenmayer A, Finn RS. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat Rev Clin Oncol. 2015;12(7):408–24.
Kamrani A, Hosseinzadeh R, Shomali N, Heris JA, Shahabi P, Mohammadinasab R, et al. New immunotherapeutic approaches for cancer treatment. Pathol Res Pract. 2023;248:154632.
Li Y, Liu X, Zhang X, Pan W, Li N, Tang B. Immune cycle-based strategies for Cancer Immunotherapy. Adv Funct Mater. 2021;31(50).
Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–6.
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic Melanoma. N Engl J Med. 2010;363(8):711–23.
Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–34.
Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2(3):261–8.
Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28(19):3167–75.
Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic Bladder cancer. Nature. 2014;515(7528):558–62.
Naimi A, Mohammed RN, Raji A, Chupradit S, Yumashev AV, Suksatan W, et al. Tumor immunotherapies by immune checkpoint inhibitors (ICIs); the pros and cons. Cell Commun Signal. 2022;20(1):44.
Jiang Y, Zhao X, Fu J, Wang H. Progress and challenges in Precise Treatment of Tumors with PD-1/PD-L1 blockade. Front Immunol. 2020;11:339.
Song Y, Gao Q, Zhang H, Fan L, Zhou J, Zou D, et al. Treatment of relapsed or refractory classical Hodgkin Lymphoma with the anti-PD-1, tislelizumab: results of a phase 2, single-arm, multicenter study. Leukemia. 2020;34(2):533–42.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Five-year survival and correlates among patients with Advanced Melanoma, Renal Cell Carcinoma, or Non-small Cell Lung Cancer treated with Nivolumab. JAMA Oncol. 2019;5(10):1411–20.
Yau T, Hsu C, Kim TY, Choo SP, Kang YK, Hou MM, et al. Nivolumab in advanced hepatocellular carcinoma: Sorafenib-experienced Asian cohort analysis. J Hepatol. 2019;71(3):543–52.
Maruhashi T, Sugiura D, Okazaki IM, Shimizu K, Maeda TK, Ikubo J, et al. Binding of LAG-3 to stable peptide-MHC class II limits T cell function and suppresses autoimmunity and anti-cancer immunity. Immunity. 2022;55(5):912–24. e8.
Maruhashi T, Sugiura D, Okazaki IM, Okazaki T. LAG-3: from molecular functions to clinical applications. J Immunother Cancer. 2020;8(2):e001014.
Tang R, Rangachari M, Kuchroo VK. Tim-3: a co-receptor with diverse roles in T cell exhaustion and tolerance. Semin Immunol. 2019;42:101302.
Kang CW, Dutta A, Chang LY, Mahalingam J, Lin YC, Chiang JM, et al. Apoptosis of Tumor infiltrating effector TIM-3 + CD8 + T cells in colon Cancer. Sci Rep. 2015;5:15659.
Granier C, Dariane C, Combe P, Verkarre V, Urien S, Badoual C, et al. Tim-3 expression on Tumor-infiltrating PD-1 + CD8 + T cells correlates with poor clinical outcome in renal cell carcinoma. Cancer Res. 2017;77(5):1075–82.
Chauvin JM, Zarour HM. TIGIT in cancer immunotherapy. J Immunother Cancer. 2020;8(2):e000957.
Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev. 2017;276(1):112–20.
Shum B, Larkin J, Turajlic S. Predictive biomarkers for response to immune checkpoint inhibition. Semin Cancer Biol. 2022;79:4–17.
Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350–5.
Mok TSK, Wu YL, Kudaba I, Kowalski DM, Cho BC, Turna HZ, et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell Lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet. 2019;393(10183):1819–30.
Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, et al. Atezolizumab versus Docetaxel in patients with previously treated non-small-cell Lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389(10066):255–65.
Borghaei H, Langer CJ, Paz-Ares L, Rodriguez-Abreu D, Halmos B, Garassino MC, et al. Pembrolizumab plus chemotherapy versus chemotherapy alone in patients with advanced non-small cell Lung cancer without Tumor PD-L1 expression: a pooled analysis of 3 randomized controlled trials. Cancer. 2020;126(22):4867–77.
Sanchez-Magraner L, Gumuzio J, Miles J, Quimi N, Del Martinez P, Abad-Villar MT, et al. Functional Engagement of the PD-1/PD-L1 Complex but Not PD-L1 expression is highly predictive of patient response to Immunotherapy in Non-small-cell Lung Cancer. J Clin Oncol. 2023;41(14):2561–70.
Schoenfeld AJ, Rizvi H, Bandlamudi C, Sauter JL, Travis WD, Rekhtman N, et al. Clinical and molecular correlates of PD-L1 expression in patients with lung adenocarcinomas. Ann Oncol. 2020;31(5):599–608.
Mansfield AS, Aubry MC, Moser JC, Harrington SM, Dronca RS, Park SS, et al. Temporal and spatial discordance of programmed cell death-ligand 1 expression and lymphocyte Tumor infiltration between paired primary lesions and brain metastases in Lung cancer. Ann Oncol. 2016;27(10):1953–8.
Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gumus M, Mazieres J, et al. Pembrolizumab plus Chemotherapy for squamous non-small-cell Lung Cancer. N Engl J Med. 2018;379(21):2040–51.
Merino DM, McShane LM, Fabrizio D, Funari V, Chen SJ, White JR, et al. Establishing guidelines to harmonize Tumor mutational burden (TMB): in silico assessment of variation in TMB quantification across diagnostic platforms: phase I of the friends of Cancer Research TMB Harmonization Project. J Immunother Cancer. 2020;8(1):e000147.
Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020;21(10):1353–65.
Hellmann MD, Ciuleanu TE, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus Ipilimumab in Lung Cancer with a high Tumor Mutational Burden. N Engl J Med. 2018;378(22):2093–104.
Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, et al. Development of Tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol. 2019;30(1):44–56.
McGrail DJ, Pilie PG, Rashid NU, Voorwerk L, Slagter M, Kok M, et al. High Tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann Oncol. 2021;32(5):661–72.
Chowell D, Morris LGT, Grigg CM, Weber JK, Samstein RM, Makarov V, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science. 2018;359(6375):582–7.
Iacopetta B, Grieu F, Amanuel B. Microsatellite instability in Colorectal cancer. Asia Pac J Clin Oncol. 2010;6(4):260–9.
Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of Tumor mutational burden. Genome Med. 2017;9(1):34.
Takamochi K, Takahashi F, Suehara Y, Sato E, Kohsaka S, Hayashi T, et al. DNA mismatch repair deficiency in surgically resected lung adenocarcinoma: microsatellite instability analysis using the Promega panel. Lung Cancer. 2017;110:26–31.
Dudley JC, Lin MT, Le DT, Eshleman JR. Microsatellite instability as a biomarker for PD-1 blockade. Clin Cancer Res. 2016;22(4):813–20.
Stenzel PJ, Schindeldecker M, Tagscherer KE, Foersch S, Herpel E, Hohenfellner M, et al. Prognostic and predictive value of Tumor-infiltrating leukocytes and of Immune Checkpoint molecules PD1 and PDL1 in Clear Cell Renal Cell Carcinoma. Transl Oncol. 2020;13(2):336–45.
Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to Anti-PD-1 therapy in metastatic Melanoma. Cell. 2016;165(1):35–44.
Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science. 2017;355(6322):eaaf8399.
Zhang Q, Luo J, Wu S, Si H, Gao C, Xu W, et al. Prognostic and predictive impact of circulating Tumor DNA in patients with Advanced Cancers treated with Immune Checkpoint Blockade. Cancer Discov. 2020;10(12):1842–53.
Katayama Y, Yamada T, Shimamoto T, Iwasaku M, Kaneko Y, Uchino J, et al. The role of the gut microbiome on the efficacy of immune checkpoint inhibitors in Japanese responder patients with advanced non-small cell Lung cancer. Transl Lung Cancer Res. 2019;8(6):847–53.
Sun H, Sun C, Xiao W, Sun R. Tissue-resident lymphocytes: from adaptive to innate immunity. Cell Mol Immunol. 2019;16(3):205–15.
Murakami Y, Saito H, Shimizu S, Kono Y, Shishido Y, Miyatani K, et al. Increased regulatory B cells are involved in immune evasion in patients with gastric cancer. Sci Rep. 2019;9(1):13083.
Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z, Chen EB, et al. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory cells to promote progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology. 2016;150(7):1646–58e17.
Oleinika K, Nibbs RJ, Graham GJ, Fraser AR. Suppression, subversion and Escape: the role of regulatory T cells in cancer progression. Clin Exp Immunol. 2013;171(1):36–45.
Goda N, Sasada S, Shigematsu H, Masumoto N, Arihiro K, Nishikawa H, et al. The ratio of CD8 + lymphocytes to tumor-infiltrating suppressive FOXP3 + effector regulatory T cells is associated with treatment response in invasive Breast cancer. Discov Oncol. 2022;13(1):27.
Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol. 2019;16(6):356–71.
Freeman ZT, Nirschl TR, Hovelson DH, Johnston RJ, Engelhardt JJ, Selby MJ, et al. A conserved intratumoral regulatory T cell signature identifies 4-1BB as a pan-cancer target. J Clin Invest. 2020;130(3):1405–16.
Cutolo M, Campitiello R, Gotelli E, Soldano S. The role of M1/M2 macrophage polarization in Rheumatoid Arthritis Synovitis. Front Immunol. 2022;13:867260.
Pan Y, Yu Y, Wang X, Zhang T. Tumor-Associated macrophages in Tumor Immunity. Front Immunol. 2020;11:583084.
Zhang Q, He Y, Luo N, Patel SJ, Han Y, Gao R, et al. Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell. 2019;179(4):829–45. e20.
Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, et al. Single-cell map of Diverse Immune Phenotypes in the breast Tumor Microenvironment. Cell. 2018;174(5):1293–308. e36.
Wu SZ, Al-Eryani G, Roden DL, Junankar S, Harvey K, Andersson A, et al. A single-cell and spatially resolved atlas of human breast cancers. Nat Genet. 2021;53(9):1334–47.
Nalio Ramos R, Missolo-Koussou Y, Gerber-Ferder Y, Bromley CP, Bugatti M, Nunez NG, et al. Tissue-resident FOLR2 + macrophages associate with CD8 + T cell infiltration in human Breast cancer. Cell. 2022;185(7):1189–207. e25.
Chen AX, Gartrell RD, Zhao J, Upadhyayula PS, Zhao W, Yuan J, et al. Single-cell characterization of macrophages in glioblastoma reveals MARCO as a mesenchymal pro-tumor marker. Genome Med. 2021;13(1):88.
Liu Y, Zhang Q, Xing B, Luo N, Gao R, Yu K, et al. Immune phenotypic linkage between Colorectal cancer and liver Metastasis. Cancer Cell. 2022;40(4):424–37. e5.
Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: N1 versus N2 TAN. Cancer Cell. 2009;16(3):183–94.
Wang L, Liu Y, Dai Y, Tang X, Yin T, Wang C, et al. Single-cell RNA-seq analysis reveals BHLHE40-driven pro-tumour neutrophils with hyperactivated glycolysis in pancreatic tumour microenvironment. Gut. 2023;72(5):958–71.
Xue R, Zhang Q, Cao Q, Kong R, Xiang X, Liu H, et al. Liver tumour immune microenvironment subtypes and neutrophil heterogeneity. Nature. 2022;612(7938):141–7.
Brivio S, Cadamuro M, Strazzabosco M, Fabris L. Tumor reactive stroma in cholangiocarcinoma: the fuel behind cancer aggressiveness. World J Hepatol. 2017;9(9):455–68.
Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discov. 2019;18(2):99–115.
Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. 2020;20(3):174–86.
Ohlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in Pancreatic cancer. J Exp Med. 2017;214(3):579–96.
Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals Antigen-Presenting Cancer-Associated fibroblasts. Cancer Discov. 2019;9(8):1102–23.
Zhang M, Yang H, Wan L, Wang Z, Wang H, Ge C, et al. Single-cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J Hepatol. 2020;73(5):1118–30.
Dominguez CX, Muller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, et al. Single-cell RNA sequencing reveals stromal evolution into LRRC15 + myofibroblasts as a determinant of patient response to Cancer Immunotherapy. Cancer Discov. 2020;10(2):232–53.
Kieffer Y, Hocine HR, Gentric G, Pelon F, Bernard C, Bourachot B, et al. Single-cell analysis reveals fibroblast clusters linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020;10(9):1330–51.
Maishi N, Annan DA, Kikuchi H, Hida Y, Hida K. Tumor endothelial heterogeneity in Cancer Progression. Cancers (Basel). 2019;11(10):1511.
Sharma A, Seow JJW, Dutertre CA, Pai R, Bleriot C, Mishra A, et al. Onco-fetal reprogramming of endothelial cells drives immunosuppressive macrophages in Hepatocellular Carcinoma. Cell. 2020;183(2):377–94. e21.
Asrir A, Tardiveau C, Coudert J, Laffont R, Blanchard L, Bellard E, et al. Tumor-associated high endothelial venules mediate lymphocyte entry into tumors and predict response to PD-1 plus CTLA-4 combination immunotherapy. Cancer Cell. 2022;40(3):318–34. e9.
Zheng L, Qin S, Si W, Wang A, Xing B, Gao R, et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science. 2021;374(6574):abe6474.
Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, et al. Global characterization of T cells in non-small-cell Lung cancer by single-cell sequencing. Nat Med. 2018;24(7):978–85.
Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, et al. Landscape of infiltrating T cells in Liver Cancer revealed by single-cell sequencing. Cell. 2017;169(7):1342–56e16.
Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted CD8 + T cells differentially mediate Tumor control and respond to checkpoint blockade. Nat Immunol. 2019;20(3):326–36.
Kim C, Jin J, Weyand CM, Goronzy JJ. The transcription factor TCF1 in T cell differentiation and aging. Int J Mol Sci. 2020;21:18.
Wang D, Fang J, Wen S, Li Q, Wang J, Yang L, et al. A comprehensive profile of TCF1 + progenitor and TCF1- terminally exhausted PD-1 + CD8 + T cells in head and neck squamous cell carcinoma: implications for prognosis and immunotherapy. Int J Oral Sci. 2022;14(1):8.
Jiang W, He Y, He W, Wu G, Zhou X, Sheng Q, et al. Exhausted CD8 + T cells in the Tumor Immune Microenvironment: New pathways to Therapy. Front Immunol. 2020;11:622509.
Guo Y, Xie YQ, Gao M, Zhao Y, Franco F, Wenes M, et al. Metabolic reprogramming of terminally exhausted CD8 + T cells by IL-10 enhances anti-tumor immunity. Nat Immunol. 2021;22(6):746–56.
De Simone M, Rossetti G, Pagani M. Single cell T cell receptor sequencing: techniques and Future challenges. Front Immunol. 2018;9:1638.
Zhou L, Zeng Z, Egloff AM, Zhang F, Guo F, Campbell KM, et al. Checkpoint blockade-induced CD8 + T cell differentiation in Head and Neck cancer responders. J Immunother Cancer. 2022;10(1):e004034.
Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, et al. Checkpoint blockade immunotherapy induces dynamic changes in PD-1–CD8 + tumor-infiltrating T cells. Immunity. 2019;50(1):181–94. e6.
Khojandi N, Connelly L, Piening A, Hoft SG, Pherson M, Donlin MJ, et al. Single-cell analysis of peripheral CD8 + T cell responses in patients receiving checkpoint blockade immunotherapy for cancer. Cancer Immunol Immunother. 2023;72(2):397–408.
Liu B, Hu X, Feng K, Gao R, Xue Z, Zhang S, et al. Temporal single-cell tracing reveals clonal revival and expansion of precursor exhausted T cells during anti-PD-1 therapy in Lung cancer. Nat Cancer. 2022;3(1):108–21.
Khan SM, Desai R, Coxon A, Livingstone A, Dunn GP, Petti A, et al. Impact of CD4 T cells on intratumoral CD8 T-cell exhaustion and responsiveness to PD-1 blockade therapy in mouse brain tumors. J Immunother Cancer. 2022;10(12):e005293.
Kwon M, An M, Klempner SJ, Lee H, Kim KM, Sa JK, et al. Determinants of response and intrinsic resistance to PD-1 blockade in microsatellite instability-high gastric Cancer. Cancer Discov. 2021;11(9):2168–85.
Bi K, He MX, Bakouny Z, Kanodia A, Napolitano S, Wu J, et al. Tumor and immune reprogramming during immunotherapy in advanced renal cell carcinoma. Cancer Cell. 2021;39(5):649–61. e5.
Liu B, Zhang Y, Wang D, Hu X, Zhang Z. Single-cell meta-analyses reveal responses of tumor-reactive CXCL13 + T cells to immune-checkpoint blockade. Nat Cancer. 2022;3(9):1123–36.
Li J, Wu C, Hu H, Qin G, Wu X, Bai F, et al. Remodeling of the immune and stromal cell compartment by PD-1 blockade in mismatch repair-deficient Colorectal cancer. Cancer Cell. 2023;41(6):1152–69. e7.
Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T Cell States Associated with response to Checkpoint Immunotherapy in Melanoma. Cell. 2018;175(4):998–1013e20.
Zhang Y, Chen H, Mo H, Hu X, Gao R, Zhao Y, et al. Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative Breast cancer. Cancer Cell. 2021;39(12):1578–93e8.
Leader AM, Grout JA, Maier BB, Nabet BY, Park MD, Tabachnikova A, et al. Single-cell analysis of human non-small cell Lung cancer lesions refines Tumor classification and patient stratification. Cancer Cell. 2021;39(12):1594–609. e12.
Chen J, Liu K, Luo Y, Kang M, Wang J, Chen G, et al. Single-cell profiling of Tumor Immune Microenvironment reveals Immune Irresponsiveness in gastric Signet-Ring Cell Carcinoma. Gastroenterology. 2023;165(1):88–103.
Henry RA, Kendall PL. CXCL13 blockade disrupts B lymphocyte organization in tertiary lymphoid structures without altering B cell receptor bias or preventing Diabetes in nonobese diabetic mice. J Immunol. 2010;185(3):1460–5.
Kim K, Park S, Park SY, Kim G, Park SM, Cho J-W et al. Single-cell transcriptome analysis reveals TOX as a promoting factor for T cell exhaustion and a predictor for anti-PD-1 responses in human cancer. Genome Med. 2020;12(1).
Chu Y, Dai E, Li Y, Han G, Pei G, Ingram DR, et al. Pan-cancer T cell atlas links a cellular stress response state to immunotherapy resistance. Nat Med. 2023;29(6):1550–62.
Patil NS, Nabet BY, Muller S, Koeppen H, Zou W, Giltnane J, et al. Intratumoral plasma cells predict outcomes to PD-L1 blockade in non-small cell Lung cancer. Cancer Cell. 2022;40(3):289–300. e4.
Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, et al. Tertiary lymphoid structures improve immunotherapy and survival in Melanoma. Nature. 2020;577(7791):561–5.
Xiong D, Wang Y, You M. A gene expression signature of TREM2hi macrophages and γδ T cells predicts immunotherapy response. Nat Commun. 2020;11(1):5084.
Zhang H, Liu Z, Wen H, Guo Y, Xu F, Zhu Q, et al. Immunosuppressive TREM2 + macrophages are associated with undesirable prognosis and responses to anti-PD-1 immunotherapy in non-small cell Lung cancer. Cancer Immunol Immunother. 2022;71(10):2511–22.
Hao X, Zheng Z, Liu H, Zhang Y, Kang J, Kong X, et al. Inhibition of APOC1 promotes the transformation of M2 into M1 macrophages via the ferroptosis pathway and enhances anti-PD1 immunotherapy in hepatocellular carcinoma based on single-cell RNA sequencing. Redox Biol. 2022;56:102463.
Wang Y, Liang Y, Xu H, Zhang X, Mao T, Cui J, et al. Single-cell analysis of pancreatic ductal adenocarcinoma identifies a novel fibroblast subtype associated with poor prognosis but better immunotherapy response. Cell Discov. 2021;7(1):36.
Obradovic A, Graves D, Korrer M, Wang Y, Roy S, Naveed A, et al. Immunostimulatory Cancer-Associated fibroblast subpopulations can predict Immunotherapy Response in Head and Neck Cancer. Clin Cancer Res. 2022;28(10):2094–109.
Tang F, Li J, Qi L, Liu D, Bo Y, Qin S et al. A pan-cancer single-cell panorama of human natural killer cells. Cell. 2023.
Zhou Z, Chen MM, Luo Y, Mojumdar K, Peng X, Chen H, et al. Tumor-intrinsic SIRPA promotes sensitivity to checkpoint inhibition immunotherapy in Melanoma. Cancer Cell. 2022;40(11):1324–40. e8.
Watson RA, Tong O, Cooper R, Taylor CA, Sharma PK, de Los Aires AV, et al. Immune checkpoint blockade sensitivity and progression-free survival associates with baseline CD8 + T cell clone size and cytotoxicity. Sci Immunol. 2021;6(64):eabj8825.
De Biasi S, Gibellini L, Lo Tartaro D, Puccio S, Rabacchi C, Mazza EMC, et al. Circulating mucosal-associated invariant T cells identify patients responding to anti-PD-1 therapy. Nat Commun. 2021;12(1):1669.
Li C, Phoon YP, Karlinsey K, Tian YF, Thapaliya S, Thongkum A, et al. A high OXPHOS CD8 T cell subset is predictive of immunotherapy resistance in Melanoma patients. J Exp Med. 2022;219(1):e20202084.
Rad Pour S, Pico de Coana Y, Demorentin XM, Melief J, Thimma M, Wolodarski M, et al. Predicting anti-PD-1 responders in malignant Melanoma from the frequency of S100A9 + monocytes in the blood. J Immunother Cancer. 2021;9(5):e002171.
Sui Q, Zhang X, Chen C, Tang J, Yu J, Li W, et al. Inflammation promotes resistance to immune checkpoint inhibitors in high microsatellite instability Colorectal cancer. Nat Commun. 2022;13(1):7316.
Cho JW, Park S, Kim G, Han H, Shim HS, Shin S, et al. Dysregulation of TFH-B-TRM lymphocyte cooperation is associated with unfavorable anti-PD-1 responses in EGFR-mutant Lung cancer. Nat Commun. 2021;12(1):6068.
Oh DY, Kwek SS, Raju SS, Li T, McCarthy E, Chow E, et al. Intratumoral CD4 + T cells mediate anti-tumor cytotoxicity in human Bladder Cancer. Cell. 2020;181(7):1612–25. e13.
Hu J, Zhang L, Xia H, Yan Y, Zhu X, Sun F, et al. Tumor microenvironment remodeling after neoadjuvant immunotherapy in non-small cell Lung cancer revealed by single-cell RNA sequencing. Genome Med. 2023;15(1):14.
Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, et al. A Cancer Cell Program promotes T cell exclusion and resistance to checkpoint blockade. Cell. 2018;175(4):984–97. e24.
Cappellesso F, Orban MP, Shirgaonkar N, Berardi E, Serneels J, Neveu MA, et al. Targeting the bicarbonate transporter SLC4A4 overcomes immunosuppression and immunotherapy resistance in Pancreatic cancer. Nat Cancer. 2022;3(12):1464–83.
Abdelfattah N, Kumar P, Wang C, Leu JS, Flynn WF, Gao R, et al. Single-cell analysis of human glioma and immune cells identifies S100A4 as an immunotherapy target. Nat Commun. 2022;13(1):767.
Alvisi G, Termanini A, Soldani C, Portale F, Carriero R, Pilipow K, et al. Multimodal single-cell profiling of intrahepatic cholangiocarcinoma defines hyperactivated Tregs as a potential therapeutic target. J Hepatol. 2022;77(5):1359–72.
Yuen KC, Liu LF, Gupta V, Madireddi S, Keerthivasan S, Li C, et al. High systemic and tumor-associated IL-8 correlates with reduced clinical benefit of PD-L1 blockade. Nat Med. 2020;26(5):693–8.
Wen T, Barham W, Li Y, Zhang H, Gicobi JK, Hirdler JB, et al. NKG7 is a T-cell-intrinsic therapeutic target for improving antitumor cytotoxicity and Cancer immunotherapy. Cancer Immunol Res. 2022;10(2):162–81.
Molgora M, Esaulova E, Vermi W, Hou J, Chen Y, Luo J, et al. TREM2 modulation remodels the Tumor Myeloid Landscape enhancing Anti-PD-1 immunotherapy. Cell. 2020;182(4):886–900. e17.
Hui B, Lu C, Li H, Hao X, Liu H, Zhuo D, et al. Inhibition of APOE potentiates immune checkpoint therapy for cancer. Int J Biol Sci. 2022;18(14):5230–40.
Kim M, Min YK, Jang J, Park H, Lee S, Lee CH. Single-cell RNA sequencing reveals distinct cellular factors for response to immunotherapy targeting CD73 and PD-1 in Colorectal cancer. J Immunother Cancer. 2021;9(7):e002503.
Liu L, Hou Y, Deng C, Tao Z, Chen Z, Hu J, et al. Single cell sequencing reveals that CD39 inhibition mediates changes to the Tumor microenvironment. Nat Commun. 2022;13(1):6740.
Heidegger I, Fotakis G, Offermann A, Goveia J, Daum S, Salcher S, et al. Comprehensive characterization of the prostate Tumor microenvironment identifies CXCR4/CXCL12 crosstalk as a novel antiangiogenic therapeutic target in Prostate cancer. Mol Cancer. 2022;21(1):132.
Gong L, Luo J, Zhang Y, Yang Y, Li S, Fang X et al. Nasopharyngeal carcinoma cells promote regulatory T cell development and suppressive activity via CD70-CD27 interaction. Nat Commun. 2023;14(1).
Yamashita N, Long M, Fushimi A, Yamamoto M, Hata T, Hagiwara M, et al. MUC1-C integrates activation of the IFN-gamma pathway with suppression of the Tumor immune microenvironment in triple-negative Breast cancer. J Immunother Cancer. 2021;9(1):e002115.
Wang B, Zhang W, Jankovic V, Golubov J, Poon P, Oswald EM, et al. Combination cancer immunotherapy targeting PD-1 and GITR can rescue CD8 + T cell dysfunction and maintain memory phenotype. Sci Immunol. 2018;3(29):eaat7061.
Salmon AJ, Shavkunov AS, Miao Q, Jarjour NN, Keshari S, Esaulova E, et al. BHLHE40 regulates the T-Cell effector function required for Tumor Microenvironment Remodeling and Immune Checkpoint Therapy Efficacy. Cancer Immunol Res. 2022;10(5):597–611.
Mathewson ND, Ashenberg O, Tirosh I, Gritsch S, Perez EM, Marx S, et al. Inhibitory CD161 receptor identified in glioma-infiltrating T cells by single-cell analysis. Cell. 2021;184(5):1281–98. e26.
Aoki T, Chong LC, Takata K, Milne K, Marshall A, Chavez EA, et al. Single-cell profiling reveals the importance of CXCL13/CXCR5 axis biology in lymphocyte-rich classic Hodgkin Lymphoma. Proc Natl Acad Sci U S A. 2021;118(41):e2105822118.
Wan C, Keany MP, Dong H, Al-Alem LF, Pandya UM, Lazo S, et al. Enhanced efficacy of simultaneous PD-1 and PD-L1 Immune Checkpoint Blockade in High-Grade Serous Ovarian Cancer. Cancer Res. 2021;81(1):158–73.
Ni J, Wang X, Stojanovic A, Zhang Q, Wincher M, Buhler L, et al. Single-cell RNA sequencing of Tumor-infiltrating NK cells reveals that inhibition of transcription factor HIF-1alpha unleashes NK Cell activity. Immunity. 2020;52(6):1075–87. e8.
Li X, Chen M, Wan Y, Zhong L, Han X, Chen X, et al. Single-cell transcriptome profiling reveals the key role of ZNF683 in natural killer cell exhaustion in Multiple Myeloma. Clin Transl Med. 2022;12(10):e1065.
Van Damme H, Dombrecht B, Kiss M, Roose H, Allen E, Van Overmeire E, et al. Therapeutic depletion of CCR8 + tumor-infiltrating regulatory T cells elicits antitumor immunity and synergizes with anti-PD-1 therapy. J Immunother Cancer. 2021;9(2):e001749.
Kim MC, Borcherding N, Ahmed KK, Voigt AP, Vishwakarma A, Kolb R, et al. CD177 modulates the function and homeostasis of tumor-infiltrating regulatory T cells. Nat Commun. 2021;12(1):5764.
Chen K, Wang Y, Hou Y, Wang Q, Long D, Liu X, et al. Single cell RNA-seq reveals the CCL5/SDC1 receptor-ligand interaction between T cells and Tumor cells in Pancreatic cancer. Cancer Lett. 2022;545:215834.
Liu Y, Xun Z, Ma K, Liang S, Li X, Zhou S, et al. Identification of a tumour immune barrier in the HCC microenvironment that determines the efficacy of immunotherapy. J Hepatol. 2023;78(4):770–82.
Weng J, Liu S, Zhou Q, Xu W, Xu M, Gao D et al. Intratumoral PPT1-positive macrophages determine immunosuppressive contexture and immunotherapy response in hepatocellular carcinoma. J Immunother Cancer. 2023;11(6).
Mei Y, Wang X, Zhang J, Liu D, He J, Huang C et al. Siglec-9 acts as an immune-checkpoint molecule on macrophages in glioblastoma, restricting T-cell priming and immunotherapy response. Nat Cancer. 2023.
Aggen DH, Ager CR, Obradovic AZ, Chowdhury N, Ghasemzadeh A, Mao W, et al. Blocking IL1 Beta promotes Tumor Regression and Remodeling of the myeloid compartment in a renal cell Carcinoma Model: multidimensional analyses. Clin Cancer Res. 2021;27(2):608–21.
Li R, Ferdinand JR, Loudon KW, Bowyer GS, Laidlaw S, Muyas F, et al. Mapping single-cell transcriptomes in the intra-tumoral and associated territories of kidney cancer. Cancer Cell. 2022;40(12):1583–99. e10.
Garcia Garcia CJ, Huang Y, Fuentes NR, Turner MC, Monberg ME, Lin D, et al. Stromal HIF2 regulates Immune suppression in the Pancreatic Cancer Microenvironment. Gastroenterology. 2022;162(7):2018–31.
Peng YL, Xiong LB, Zhou ZH, Ning K, Li Z, Wu ZS, et al. Single-cell transcriptomics reveals a low CD8 + T cell infiltrating state mediated by fibroblasts in recurrent renal cell carcinoma. J Immunother Cancer. 2022;10(2):e004206.
Zhu GQ, Tang Z, Huang R, Qu WF, Fang Y, Yang R, et al. CD36 + cancer-associated fibroblasts provide immunosuppressive microenvironment for hepatocellular carcinoma via secretion of macrophage migration inhibitory factor. Cell Discov. 2023;9(1):25.
Halaby MJ, Hezaveh K, Lamorte S, Ciudad MT, Kloetgen A, MacLeod BL, et al. GCN2 drives macrophage and MDSC function and immunosuppression in the Tumor microenvironment. Sci Immunol. 2019;4(42):eaax8189.
Croucher DC, Richards LM, Tsofack SP, Waller D, Li Z, Wei EN, et al. Longitudinal single-cell analysis of a Myeloma mouse model identifies subclonal molecular programs associated with progression. Nat Commun. 2021;12(1):6322.
Liu Y, Wang H, Zhao X, Zhang J, Zhao Z, Lian X et al. Targeting the immunoglobulin IGSF9 enhances anti-tumor T cell activity and sensitivity to anti-PD-1 immunotherapy. Cancer Res. 2023.
Luo Q, Zhang H. Emergence of Bias during the synthesis and amplification of cDNA for scRNA-seq. Adv Exp Med Biol. 2018;1068:149–58.
Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. 2017;14(9):865–8.
Lewis SM, Asselin-Labat ML, Nguyen Q, Berthelet J, Tan X, Wimmer VC, et al. Spatial omics and multiplexed imaging to explore cancer biology. Nat Methods. 2021;18(9):997–1012.
Acknowledgements
Not applicable.
Funding
This work was supported by National High Level Hospital Clinical Research Funding (2022-PUMCH-B-128), CAMS Innovation Fund for Medical Sciences (CIFMS) (2022-I2M-C&T-A-003), CAMS Innovation Fund for Medical Sciences (CIFMS) (2021-I2M-1-061, 2021-I2M-1-003), CSCO-hengrui Cancer Research Fund (Y-HR2019-0239, Y-HR2020MS-0415, Y-HR2020QN-0414), CSCO-MSD Cancer Research Fund (Y-MSDZD2021-0213) and National Ten-thousand Talent Program.
Author information
Authors and Affiliations
Contributions
BYS and ZYX wrote the paper; NZ, KL, and XQC reviewed and edited the paper; HTZ supervised and acquired fundings for this study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sun, B., Xun, Z., Zhang, N. et al. Single-cell RNA sequencing in cancer research: discovering novel biomarkers and therapeutic targets for immune checkpoint blockade. Cancer Cell Int 23, 313 (2023). https://doi.org/10.1186/s12935-023-03158-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-023-03158-4