
Abstract
Recent advances in omics technology have prompted extraordinary attempts to define the molecular changes underlying the onset and progression of a variety of complex human diseases, including cancer. Since the advent of sequencing technology, cancer biology has become increasingly reliant on the generation and integration of data generated at these levels. The availability of multi-omic data has transformed medicine and biology by enabling integrated systems-level approaches. Multivariate signatures are expected to play a role in cancer detection, screening, patient classification, assessment of treatment response, and biomarker identification. This review reports current findings and highlights a number of studies that are both novel and groundbreaking in their application of multi Omics to prostate cancer.
Similar content being viewed by others

Introduction
Recent advances in omics technology have prompted unexpected attempts to define the molecular changes underlying the onset and progression of a variety of complex human diseases, including cancer [1]. Identifying accurate, early signs of disease is a primary goal of biomedical research, which has entered an unprecedented era due to technological advances. New tools now allow characterization of large biological systems in great detail and with unprecedented resolution, rather than focusing research efforts on individual chemicals, metabolic pathways, or cells of interest [2]. The goal of omics technology is to study changes in DNA, RNA, proteins, and other biological molecules of various types in different species and individuals [3]. Continued developments in sequencing technology and the incorporation of high-throughput Omics data provided invaluable data to untangle the complexity of biological systems at several dimensions. To name a few, this comprises genomics, transcriptomics, proteomics, epigenomics, and metabolomics. Omics and functional genomics investigations have begun to discover and reveal characteristic features of tumor growth, such as main drivers of oncogenic signaling, and therapy response mechanisms [4]. Cancer biology has become increasingly reliant on the generation and integration of data generated at these levels.
Prostate cancer (PCa) is the second most common malignancy and the fifth leading cause of cancer-related mortality [5]. Up to 40% of men with PCa have no clinical signs because it is generally a slow-growing tumor. However, if PCa is detected at an early stage, patients have a survival rate of more than 99%; if it is detected at an advanced and highly metastatic stage, the survival rate is only 30% [6]. Clinicopathologic indices such as tumor stage, Gleason score, and (prostate specific antigen) PSA level are currently used to assigned patients into clinical risk groups (low, intermediate, or high-risk) [7]. In the early stages of the disease, current clinicopathologic indices do not discriminate well across patients with varied survival prospects [8]. Early PCa detection is primarily based on PSA testing, which has significant limitations. This test is far from ideal, however, because even slightly elevated PSA levels can be associated with confounding factors such as benign prostatic hyperplasia (BPH) or prostatitis [9]. In addition, more than 25% of men with PCa are found to have normal PSA levels [10, 11].
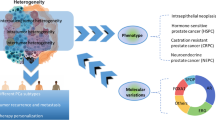
All these facts highlight the urgent need for innovative biomarkers to improve clinical outcomes and treatment of PCa. Rapid improvements in molecular technology have enabled the identification of a number of putative PCa biomarkers. Thus, the aim of this review is to present the main findings on cancer from recently published Omics, single-cell Omics, and multi-Omic studies, as well as current computational algorithms (CAs) and cancer databases for integrating and deciphering the increasing data. The necessity of combining multi-omic data over single-omics analysis is emphasized in this review. We believe that the information provided will help readers gain a more systematic and thorough understanding of the application of joint analysis of multiple Omics data in PCa. The general picture of single Omics application in PCa is presented first, followed by a summary of research advances in the joint analysis of Omics data (Fig. 1).

A summary of the applications of several omics technologies, as well as studies on PCa
Single omics in PCa
Genomics
A vast number of susceptibility loci have been discovered as a result of genome-wide association studies, which have highlighted the significance of genetic variations in the etiology of PCa [12]. The characterization of PCa gene drivers is the base for defining disease subgroups and developing therapeutic options of precision medicine strategies. Signature somatic gene alterations in AR, PI3K-PTEN, WNT, DNA repair, and cell cycle signaling pathways are found in almost all metastatic and most primary PCa patients [13]. In multiple large genomic cohort studies of primary PCa and metastatic castration-resistant prostate cancer (mCRPC), a number of DNA copy number variations (CNV), gene mutations, gene rearrangements, and gene fusions have been associated to disease recurrence [13, 14]. Furthermore, familial mutations in transcription factors such as HOXB13 [15], tumor suppressor genes such as TP53 and APC [16], and DNA repair genes such as POLD1, BRCA1 and BRCA2 [17, 18], have also been discovered in PCa genomic studies.
In addition to single nucleotide variants, other classes of driver aberrations such as gene fusions in 60%, driver CNV in 54%, and homozygous driver deletions in 50% were also very common [19]. While TP53, PTEN and RB1 showed deletion peaks, AR and CCND1 showed predicted recurrent CNV peaks [20]. Another study by Seifert and colleagues demonstrated that radio-resistance of the DU145 and LNCaP PCa cell lines had a direct association with CNVs using a network-based approach. Remarkably, based on driver oncogenes, it was possible to distinguish between early and late relapse groups in irradiated PCa patients [21]. Translocations of androgen-regulated promoter regions and transcription factors of the ETS family, such as ERG and ETV, are the most common genomic alterations in PCa [22, 23]. Chinnaiyan’s group analyzed a recurrent fusion of the 5′-UTR of TMPRSS2 and ERG (TMPRSS2: ERG) as the first translocation discovered in PCa. Localized PCa has a 50% chance of TMPRSS2: ERG fusion [24, 25]. In nearly one-third of fatal mCRPC, the ETS2 gene is deleted, which is usually achieved by fusion of TMPRSS2 and ERG. Moreover, mouse intraepithelial prostatic neoplasia, a precursor lesion of prostate cancer, develops in transgenic mice producing the TMPRSS2: ERG fusion [26]. Recent genomic investigations have revealed that mCRPC with neuroendocrine symptoms frequently has RB1 and TP53 gene deletions, as well as a lower AR signal [27].
Interestingly, the incidence and prognosis of PCa vary by ethnic group, with African men having the greatest rates of incidence and mortality [28]. Later research discovered that some of the distinctions between races were attributable to genetics [29]. In a study conducted in a Chinese cohort, FOXA1 mutations were detected in 41% compared with 4% in the Western population based on the TCGA database [30]. Moreover, the mutational spectrum of FOXA1 in the Chinese cohort was centered on the fork-head domain, whereas in tissue samples from the Western cohort, mutations covered the entire coding sequence. Moreover, FOXA1 mutations correlated with higher FOXA1 expression in tumors and were associated with poor prognosis. Similarly, ZMYM3, SPOP, and KDM6A were also significantly mutated in the Chinese cohort compared with the Western population [30].
Transcriptomics
Transcriptomics is the study of an organism's total number of RNA transcripts. At least 11 RNAs have been found, with mRNA, which is formed by DNA transcription and eventually translated into proteins, currently being the most interesting RNA in cancer [31]. The transcriptomics is often created as a measure of gene expression to capture the subtypes of specific tumors, as many genes are expressed similarly and are closely associated to each other [32].
Marzec et al. used a holistic method to reconstruct the molecular profile of PCa and trace the changes in mRNA levels from normal prostate to high-grade prostatic intraepithelial neoplasia and metastatic disease, providing the first full insight into its progression. They reported nine previously undiscovered stage-specific candidate genes with prognostic value. GSTP1 and MYC, as well as TP63, EZH2, CENPA, and PIK3CB, were linked to tumor initiation and progression, respectively [33]. Similarly, Alkhateeb and colleagues combining several PCa gene expression datasets, have found that DDC, HEATR5B, and GABPB1-AS1 genes show differential expression in malignant samples, suggesting that they could be used as biomarkers for PCa [34]. A valuable source of noninvasive PCa biomarkers is the urinary transcriptome. In a study by Goicoechea et al., the total urine of low-stage PCa patients (LS), high-stage (HS) PCa patients, and benign prostatic hyperplasia (BPH) patients was analyzed. UACA content could be distinguished between BPH and PCa patients and healthy controls. In addition, OSBP, BRPF1, and PHC3 discriminate between LS and HS PCa patients and could be used as biomarkers to identify the early stages of the disease to guide the application of local therapy [35].
Non-coding RNAs (ncRNAs) are emerging as key regulators in the development of a variety of disorders, including cancer. With a 200-nucleotide threshold, there are two types of ncRNAs: Long ncRNAs (lncRNAs) are divided into subgroups based on their genomic localization and evolutionary decent, including sense intron RNA, antisense RNA, long intergenic RNA (lincRNA), enhancer RNA, and pseudogenes; Short ncRNAs include miRNAs and siRNAs [36]. The lncRNAs LINC00261 and LINC00665 were elevated after radiotherapy and were found to be a possible negative prognostic sign for overall survival in prostate cancer patients. In long-term survivors, silencing of LINC00665 and LINC00261 reduced survival after re-irradiation and impaired repair of DNA double-strand breaks [37]. Recently, Tang et al., have shown that hypoxia-inhibited mir-133a-3p promotes PCa bone metastasis PI3K/AKT signaling and its low expression was significantly correlated with advanced clinicopathological characteristics [38]. Lekchnov and colleagues examined 84 miRNAs in urine samples. They reported that in the supernatant urine fraction, the most diagnostically significant miRNA pairs were miR-26b.5p, miR-107, and miR-375.3p. In addition, miR-31.5p, miR-200b, miR-16.5p, and miR-660.5p existed in the fraction of extracellular vesicles, which differed between healthy men and BPH patients [39].
Circular RNAs (circRNAs) and lncRNAs have been proposed as a new form of non-coding RNA with regulatory potential in recent years [40]. To investigate circular transcripts found in prostate tumors, ultra-deep RNA-Seq was performed without poly-A selection. In several patient cohorts, circRNAs production was associated with disease progression and an average of 7,232 circRNAs were expressed in each sample. In a loss-of-function screening, it was discovered that 11.3% of circRNAs, which are extremely abundant, are necessary for cell proliferation. For example, circCSNK1G3 stimulates cell growth by interacting with miR-181 [41]. Similarly, Yan et al. discovered 827 upregulated and 1279 downregulated circRNAs by high-throughput sequencing and reported that has-circ-0001165, has-circ-0001085, and has-circ-0004916 were differentially expressed in IFN-γ treated cells by qPCR confirmation [42].
Proteomics
According to recent cancer studies, transcriptome changes account for only 10 to 20% of proteome changes [43]. Proteomics deals with the identification of proteins and the evaluation of their quantitative properties. Proteomics has been employed in various studies seeking for PCa biomarkers since it can directly reflect cell activity and detect deregulations in the most treatable cellular components [44]. Cell cycle control, DNA repair, proteasomal degradation, and metabolic activity have all been associated with proteomic changes. A study by Shina et al. compared different Omics approaches and evaluated the accuracy of each biomarker. They found that proteomic features were significantly more informative than genomic, epigenomic, or transcriptomic features for predicting biochemical relapse. They also discovered that proteomic subsets of prostate tumors were only weakly linked to their genetic counterparts [45].
Tissue obtained after a biopsy or radical prostatectomy can be utilized for biomarker discovery to and to identify therapy targets based on knowledge of the drug's genome and proteome. Researchers can also explore the molecular basis of cancer growth and progression using in situ histopathology. PPP1CB, UBE2N, and PSMB6 were found to be protein indicators for PCa diagnosis in a study using two-dimensional differential gel electrophoresis-mass spectrometry and Western blot [46]. According to Launonen and colleagues, deletion of the chromatin remodeler SMARCA4 affects chromatin accessibility and expression of a limited selection of AR target genes, as well as proliferation and metastasis of CRPC cells [47].
The proteome of PCa tumor tissue and surrounding tissue is also used for comparative studies to determine the process of carcinogenesis. To rule out interindividual variations, researchers must analyze the same prostate tissue with different histological patterns. Proteins related to smooth muscle contraction, calcium binding, and intercellular interstitial interaction were identified to be increased in the tumor stroma of PCa patients when compared to the neighboring normal stroma [48]. Aiello and coworkers used PCa tissue with a Gleason grade of 6 to look at 132 differentially expressed proteins. Oncogene products, nuclear receptor-supported activators, cytoskeletal proteins, G-protein-coupled receptors, and antiproliferative proteins were all shown to be significantly overexpressed in PCa [49]. In addition, combining proteomics with post-translational adaptations yields more detailed data. In a study by Maria et al. in four prostate cell lines (PC3, DU145, LNCaP, and PNT1A), protein candidates associated with PCa development were identified [50]. The importance of proteomics in the diagnosis and targeted therapy of PCa was reviewed in detail by Tonry et al. [51].
Epigenomics
Abnormal gene expression is one of the changes that lead to tumor formation. In addition to changes in DNA nucleotide sequence, epigenetic mechanisms can also result in abnormal gene expression. Modification of DNA after transcription and modification of proteins after translation are responsible for epigenetic alteration of DNA structure. Epigenetic changes, unlike gene mutations, are reversible and dynamic. Aberrant DNA methylation, histone modification, chromatin remodeling, and noncoding RNA-mediated abnormal gene expression are all epigenetic processes that have been implicated in the development of PCa [52].
Primary PCa can be distinguished from mCRPC based on the binding of transcription factors and metastasis-specific histone acetylation patterns (H3K27ac) to regulatory elements of the genes AR, HOXB13, and FOXA1, according to a study that integrated the epigenome with genomic and transcriptomic data. Interestingly, during metastatic disease progression, reprogrammed regulatory elements access sites related to prostate organogenesis and hijack latent developmental programs [53]. In addition, there was also a strong association between the inheritance of PCa risk and the density of somatic mutations and prostate-specific regulatory elements. Similarly, in a genome-wide sequencing analysis of histone acetylation, Baca et al. showed that the FOXA1 cistrome was reprogrammed in the development of neuroendocrine prostate carcinoma (NEPC) after endocrine treatment. NKX2-1 and ASCL1 are sufficient to induce de novo H3K27ac at NEPC-enriched FOXA1 binding sites, leading to NEPC gene expression. Although NEPC is not dependent on AR, it maintains expression of FOXA1 and requires it for gene expression and proliferation that determine neuroendocrine lineage [54].
DNA methylation is a critical component of the epigenetic system. DNA methylation regulates the transcription of the genome, therefore aberrant methylation can cause a variety of diseases, including cancer [55, 56]. Methylation suppresses transcription activity in the promoter region (CpG repeats) by blocking methyl DNA transferases from entering the cell [57]. In over 55% of cases, CpG repeats form clusters whose methylation or demethylation state impedes or activates the transcription process [58]. When malignant tumors, such as PCa, form, these areas are often methylated. In PCa, both hypermethylation and hypomethylation occur, and both methylation status changes contribute to the course of the disease [59, 60]. Hypermethylated genes include CDH1 and CD44 are involved in cell adhesion, PYCARD is involved in apoptosis regulation, CDKN2A is involved in cell cycle regulation, and GSTP1 and MGMT are involved in DNA repair [61]. Anticancer genes such as RAR, RARRES1, RASSF1, and APC are also hypermethylated in PCa [62]. Xu and colleagues discovered a modest increase in overall methylation levels with increasing Gleason score after genome-wide DNA methylation profiling in leukocyte DNA from 280 African American PCa patients. They also discovered 77 differentially methylated regions/genes, including 10 homeobox genes and six zinc finger protein genes, which may be valuable biomarkers for aggressive PCa [63].
Interestingly, studies have shown that the decrease in methylation content in the genome occurs very late in PCa progression and contributes to the heterogeneity of the metastatic tumor. Furthermore, testis antigen genes including MAGEA1, CTAG1B undergoes CpG island hypomethylation and only expressed in those cell lines that had significant hypomethylation [64]. Similarly, after analysis of CpG methylation signatures in bone metastases and primary PCa by Ylitalo and colleagues, hypomethylation was found to be more common in metastases. In addition, they proposed a methylation classification signature for androgen receptor activity (MCA) that divided metastases into two groups. The MCA-positive metastases had low methylation levels in genes associated with higher AR activity and had a more favorable prognosis after androgen deprivation therapy [65]. However, primary PCa exhibit more pronounced hypomethylation of the LINE1 (Long Interspersed Nuclear Element-1 s) promoter, which can be used for cancer screening, risk assessment, tumor staging, and prognosis [66].
Metabolomics
In recent years, many metabolomics studies have been performed on PCa samples to characterize the particular metabolic profile associated with PCa progression and to detect metabolic abnormalities that could be used as clinical biomarkers. Mondul et al. studied sera from 74 men with PCa and 74 healthy cases 20 years after blood collection using gas chromatography/mass spectrometry (GC–MS) and liquid chromatography/mass spectrometry (LC–MS). There was a significant negative association between 1stearoylglycerol and PCa risk [67]. They were unable to duplicate the association of 1-stereoylglycerol and thyroxine with higher PCa risk when they performed metabolomics analysis with an additional sample of 200 confirmed PCa cases and 200 controls. Nonetheless, alpha-ketoglutarate and citrate have been associated to an increased risk of aggressive PCa [68]. Positive lipid correlations with overall PCa risk as well as risk for aggressive PCa were also detected in the PLCO cohort study performed on 380 sera from PCa cases and 380 healthy individuals. In contrast to the above studies [67, 68], an association between thyroxine and aggressive PCa was not confirmed. However, the correlation of 2′-deoxyuridine with overall risk of PCa as well as bile acid tauro-beta-muricholate could be repeated [69].
To predict treatment response, Huang and colleagues used LC–MS, to screen 36 PCa patients receiving androgen deprivation therapy (ADT), 18 untreated diagnosed PCa, and 18 healthy controls. Arachidonic acid, pyridinoline, deoxycholic acid, tryptophan, and the nucleotide deoxycytidine triphosphate were discovered as possible biomarkers for predicting ADT response. All of these markers were altered in PCa patients compared with healthy controls. In contrast, serum levels returned to those of the control group in subjects who responded to ADT. This suggests that serum levels of these metabolites could be used to predict response to endocrine therapy [70]. Using high-performance liquid chromatography and mass spectrometry (HPLC–MS), Andras and colleagues analyzed the sera of 90 individuals with PCa and benign prostatic hyperplasia (BPH). From the data set, a discovery set (n = 59) and a validation set (n = 31) were created. In the training set, the resulting score, which comprised markers including homocysteine inosine, methyladenosine, lipoic acid, hydroxymelatonin, and decanoilcarnitine, had a sensitivity of 74% and a specificity of 76%. In the validation set, the score distinguished PCa from BPH with a sensitivity of 88% and a specificity of 60% [71].
Many noninvasive assessments have been used to predict PCa risk. Urinary changes linked to PCa risk were described by Kosti et al. [72]. They employed LC–MS to evaluate urine concentrations of 15 estrogen metabolites in 77 incident PCa cases, 77 healthy controls, and 37 subjects lacking PCa evidence based on prostate biopsy. When compared to healthy controls, PCa patients had considerably lower levels of 16-ketoestradiol (16-KE2) and 17-epiestriol (17-epiE3). Roberts et al. used nuclear magnetic resonance spectroscopy to analyze seminal plasma prior to or at least one month after prostate biopsy. The presence of a primary Gleason pattern 4 (present versus absent) in these samples was associated with higher levels of lipids/lipoproteins, lactate, and pyruvate and lower levels of citrate, spermine, and myoinositol [73]. For a comprehensive analysis of studies performed on metabolomics and PCa, see review by Kdadra et al. [74]. Cerrato and colleagues discovered that amino acids and carnitine derivatives were associated with PCa by comparing urine samples from BPH and PCa. Although previous research points to their importance in cancer biomarker discovery, these families of chemicals are often overlooked in conventional metabolomics testing [75].
Single-cell omics
Advances in DNA sequencing technology have enabled more thorough investigation of genomic features in treatment-resistant tumors, although the associated analytical tools are only now beginning to demonstrate their value. For most omics, single-cell (SC) omics is theoretically possible. In many different cell types, SC analysis reveals DNA mutations and altered gene expression and can identify resistant cells before and after treatment. It can also quantify heterogeneity within and between tumors, characterize mutation rates, and identify unusual cell types, ultimately helping to develop diagnostic and treatment guidelines [76]. Researchers could also use SC omics to analyze tumor microenvironment (TME) function in cancers that are resistant to immune treatment, such as PCa, which has a high degree of clinical heterogeneity and clonal genetic diversity [77]. The first level of gene control is DNA accessibility, and transcriptome changes are increasingly used to find molecular predictors of response to cancer treatment.
At PCa, non-genetic alterations in the transcriptome, chromatin structure, and DNA accessibility of transcription factor binding motifs are increasingly common but less well understood. Taavitsainen et al. have taken a molecular look at the emergence of resistance to AR -targeted treatments. They show that chromatin remodeling and gene expression changes in single cell populations are accompanied by several PCa-associated transcription factors such as MYC, HOXB13, and GATA2, which are outnumbered in several cell clusters. Moreover, patient responses to treatment can be stratified using transcriptional patterns specific to persisted cells [78]. More recently, Anti-programmed cell death protein 1 (PD-1) and YY001 (a novel EP4 antagonist) have been proposed to have anti-cancer activity in vitro and in vivo studies on clinical samples. EP4 regulates the TME of PCa patients by expression in epithelial cells and various immune cells. The development, maturation, and immunosuppressive function of myeloid-derived suppressor cells (MDSCs) were hindered by YY001, while the proliferation of T cells and their ability to fight cancer were increased. When combined with anti PD-1 antibodies, YY001 transforms non-responsive PCa into responsive tumors, resulting in significant tumor shrinkage, long-term survival and enhanced immunological memory [79].
To decipher the cellular architecture of neuroendocrine differentiation in human PCa, the transcriptome of 21,292 cells from 6 CRPC was sequenced. It became clear that neuroendocrine tumor cells have a luminal-like epithelial phenotype and are derived from luminal-like malignant cells rather than the basal compartment. These results were validated by microarray analyzes, and gene signatures associated with neuroendocrine differentiation were also clarified [80]. Song et al. identified club cells in radical prostatectomy specimens and localized PCa biopsies by diagnosing highly expressed genes such MMP7, PIGR, LTF, and CP, as well as the significantly reduced expression of LCN2 and SCGB3A1. These club cells are more sensitive to androgens, which may predispose to tumor cell transformation and promote prostate tumorigenesis [81]. Similarly, Chen et al. found that ectopic expression of KLK3 was connected with micrometastases in a transcriptome analysis of 36,424 single cells from 13 prostate carcinomas. In addition, there is close cell–cell communication between cells and activated endothelial cells are enriched in CRPC cells and promote cancer cell invasion [82].
Multi omics
PCa is associated with multisystemic and multilayered pathogenic changes during its development and progression. Omics studies at one level often reach their limits. In contrast, combining multiple Omics data to develop targeted indicators for PCa therapy may be more effective and thorough. Therefore, integrating data generated at each level is essential to understand the complex nature of cancer and get a holistic overview of genomic instability events, which otherwise is not possible by single Omics data analysis [83]. In the recent decade, several clinical and preclinical studies showed the importance of data integration to get a clear and concise picture of the disease under investigation. The PCa multi-Omic combination study strategy is depicted in Fig. 2.

Multi-omic strategy in Prostate cancer study. Omics investigations discover molecular characteristics of the PCa. Integration of multiple omics provide a more comprehensive view of the PCa, leading to cancer detection, screening, patient classification, and assessment of treatment response. SNV single-nucleotide variation, CNV copy number variation
PCa is correlated with molecular abnormalities, as shown by integration of metabolomics and transcriptomics. According to this study, the metabolite sphingosine has high specificity and sensitivity for distinguishing prostate cancer from benign prostatic hyperplasia. Loss of the tumor suppressor gene sphingosine-1-phosphate receptor 2, which is downstream of sphingosine, suggests a potentially important oncogenic pathway that can be therapeutically targeted [20]. Ren et al., by a genome-wide sequencing and transcriptomic study on samples from 65 untreated PCa patients, found that the high frequency of CHD1 deletion in Chinese patients was associated with a higher proportion of mutations in the androgen receptor upstream activator gene and a low TMPRSS2-ERG fusion rate. They also identified PCDH9 as a critical tumor suppressor gene because it is missing in 23% of cancers. Besides, PLXNA1 gene was amplified in approximately 17 percent of tumors in a group of PCa patients from multiple institutions, which was confirmed by functional and clinical analyses. PLXNA1 overexpression was found to accelerate prostate tumor development and to predict metastasis, biochemical recurrence, and poor survival rates independently [84].
Several research papers have recently demonstrated that merging omics datasets leads to a better knowledge and picture of the system under study. Kwon et al. discovered 70 mutant peptides in PCa cell lines. After parallel reaction screening, they found that the levels of seven mutant peptides changed in PCa, with CAPN2 D22E being the most dramatically increased. They concluded that modified mutant peptides in PCa could be exploited to generate novel biomarkers for advanced PCa [85]. Semen is an ideal sample for biomarker discovery because it is close to the prostate and can be analyzed noninvasively. Using data from histology, transcriptomics, seminal fluid proteomics, tissue specificity, androgen regulation, and cell line secretion omics, Drabovich et al. found 147 potential markers. Prostate-specific and androgen-regulating protein TGM4 has been shown to be more effective than age and blood PSA levels in predicting PCa by biopsy. TGM4 was increased 3.7-fold and the AUC was 0.66 in the independent validation data set for PCa patients with a serum PSA level of 4 ng/ml and age of 50 years [86].
Due to the vast range of molecular characteristics of PCa, identification of metabolic alteration is an important step in obtaining a more accurate diagnosis and prognosis. Gao et al. discovered significant changes in cell phenotype between the LASCPC-01 and LNCAP cell lines based on the transcriptomic and metabolomic integration. Upregulation of 62 genes in LASCPC-01 and 112 genes in LNCAP was discovered through enrichment analysis of transcriptome data. Chemical enrichment analysis of metabolome and liposome Omics revealed 25 significantly distinct metabolite groups. LNCAP had increased one-carbon metabolism as a glycolytic intermediate pathway and reduced levels of the mitochondrial lipid transporter carnitine, whereas LASCPC-01 had more glycolytic activity and lower triglyceride levels [87].
Multiple cancer-related pathways can be inhibited simultaneously with drugs, increasing treatment options and decreasing drug resistance. A team of researchers processed DNA sequencing, gene copy number, DNA methylation, and RNA-Seq data from cancer patients to create a network of driver signaling pathways using a combined analysis algorithm [88]. Drake et al. used phosphoproteomics, transcriptomics, and genomics to identify the biological mechanisms that impede response to antiandrogenic therapy in patients with mCRPC. According to their analysis, a number of signaling proteins found in these pathways, including PTK2, PRKAA2, RPS6KA4, PRKDC and members of the CDK family, may serve as novel therapeutic targets or biomarkers for prostate cancer [89]. In addition, the combination of multi-Omic and network pharmacology is becoming increasingly common [90]. A network of putative PCa targets and active chemicals extracted from Hedyotis diffusa Willd was constructed. Quercetin and Ursolic acid were found to be the major components involved in the treatment of PCa [88] (Table 1).
Computational algorithms for data integration
Since the introduction of high-throughput technologies such as next-generation sequencing (NGS) and microarrays the amount of biological data available has increased dramatically, throwing biology into the era of Big Data [91]. Conventional data analysis tools have struggled to extract biological insights about various features of genes, proteins, or biomolecules from these high-dimensional datasets in a timely and cost-effective manner [92]. The physiological and computational complexity of multi-omic datasets, as well as noise ratios and lack of statistical power, have further complicated the interpretation of data in cancer research [93]. To gain new insights and expand our understanding of cancer, improve diagnostics, and develop individualized treatments, biomedicine requires advanced informatics tools. There are numerous attempts in computational biology to develop new algorithm to integrate datasets generated by omics techniques.
Drake et al. combined several datasets of mutations, transcriptional alterations, and phosphoproteome activities using the TieDIE algorithm, which provide an integrative, pathway-based reference for drug prioritization in individual patients by shedding light on the diversity of activated signaling pathways in metastatic CRPC [89]. Sinha et al. conducted one of the intriguing studies combining omic methods on 76 PCa specimens [94]. They employed supervised machine learning to train and evaluate CNV, methylation, RNA, and protein biomarkers after profiling the genome, transcriptome, proteome, and epigenome of intermediate-risk PCa patients. They discovered biomarkers using matched biomolecule combinations and found that predictive biomarkers that combined genomic or epigenomic with proteomic features significantly outperformed biomarkers based on a single data type.
These studies have demonstrated the value of integrating multi-omic data compared to single-omics analyzes. The use of a multi-omic approach has led to the development of a variety of tools, methods, and platforms for the analysis, visualization, and interpretation of multi-omic data. The Integrated Heterogeneous Multi-Omic Data Analysis (iODA) tool is a system-level graphical user interface that uses common pathways from single Omics datasets such as mRNA, miRNA, and protein-DNA interaction (ChIP-Seq) data. The pathways shared by multiple Omics data have been evaluated as a key component of iODA's systematic description of disease progression, which is critical for the in-depth study of complicated pathogenic mechanisms [95]. Recently, Saghaleyni and colleagues developed a module with eight machine learning algorithms for analyzing secreted protein profiles using gene expression data from thousands of normal human tissue and tumor samples. KIF20A and KIF23, members of the kinesin family, were consistently among the top genes linked to malignant transformation [96]. Multi-Omic Graph Convolutional NETworks (MOGONET) effectively categorize multi-omic data, by both omics-specific learning and cross-omics correlation learning. Using mRNA expression data, microRNA expression data, and DNA methylation data, it identifies important biomarkers in various cancers [97]. Kaur et al. published a comprehensive review of CAs that can handle multi-Omic data [98].
Large collections of genomic datasets from diverse samples, such as biopsies, cell lines, and images, have been stored in public databases, which has improved our understanding of the genomic heterogeneity of cancer cells and supported the identification of novel patient-specific treatments. The Gene Expression Omnibus (GEO) and ArrayExpress are high-throughput gene expression databases that also includes hybridization arrays, and microarrays in various diseases [93]. Similarly, The Cancer Genome Atlas (TCGA) is a groundbreaking cancer genome project that has scrutinized more than 20,000 samples of primary and benign cancers from 33 cancer types for molecular signatures [99]. These data have increased our ability to identify, treat, and prevent cancer, and they will be available to the scientific community. Using TGCA, transcriptomics and survival data from 498 patients with PCa were analyzed to find potential biomarkers for prognosis. TMED10, TMED2, and SEC31A were found to be positive prognostic biomarkers for PCa because their high expression was associated with better overall survival in PCa patients [100]. Gao et al. discovered MiR-452-5p is downregulated in prostate cancer by combining data from GEO, ArrayExpress, and the TCGA database on 1007 prostate cancer samples and 387 noncancerous samples [101]. Following bioinformatic analysis revealed that the genes targeted by MiR-452-5p are thought to be involved in biological processes such as Ras signaling, signaling pathways regulating stem cell pluripotency, and transforming growth factor beta signaling. Table 2 contains a list of valuable cancer-related databases.
Challenges
Although it is the most reliable method, assessment of tumor stage and biology using biopsies remains challenging and tedious and carries significant potential side effects and complications. In addition, the inherent risk of missing more advanced/aggressive tumor areas and the high variability between observers lead to misclassification of samples [102, 103]. More importantly, these classifications for clinical decision making do not take into account the different tumor phenotypes and therefore do not reliably predict individual patient risk [104]. Furthermore, clinicians and patients often opt for intensive therapy based on findings and psychological distress without clear evidence of aggressive disease, which contributes significantly to extensive overtreatment [105, 106]. Current estimates suggest that up to 50% of PCa cases eventually receive intensive treatment, while only 20% have aggressive cancer. After intensive therapy, patients who undergo surgery or radiation therapy experience severe side effects, with up to 50% of patients relapsing.
Although significant progress has been made in recent years in identifying potential biomarkers, there are still gaps between discovery research and clinical application [107]. Biomarkers for PCa have been proposed, including branched-chain amino acids and citrate [108,109,110], which have been reported for other diseases [111], implying that the biomarkers found are likely not specific to prostate cancer. Another crucial concern is whether the alterations in plasma metabolites are reflective of tumor cell changes or not. To improve the predictive power of potential diagnostic biomarkers, experiments must be carefully designed. For example, sample size should be determined according to the type of sample, technical characteristics of omics, and statistical analysis methods. In addition, standardization of sample collection and storage may help reduce biological variability between studies [112, 113]. Follow-up studies are also needed to track survival rates and determine whether certain screening methods can reduce mortality [114]. Finally, more thorough validation is needed before results can be used in routine clinical care. Fortunately, the less invasive and rapid liquid biopsy procedure meets the need for large screens in healthy individuals. However, current liquid biopsy tests lack accuracy and consistency [115]. Standardization of liquid biopsy testing procedures and analysis platforms will be necessary in the future so that results from different studies can be compared and combined [116].
Integrating biomarkers across multiple omics systems dramatically improves predictive accuracy. Omics enables researchers to organize, monitor, compare, and assess patterns of molecular changes such as DNA mutations and CNV, protein modifications, mRNAs and miRNAs expression patterns, and epigenetic changes in individual patients to uncover the molecular signatures underlying complex cellular phenotypes [117]. The introduction of technologies to capture and integrate information from these large, multidimensional data sets will improve the translation of omics data into clinical practice. Luckily, CAs for the analysis of multi-omic data, such as machine learning techniques, have been proposed that enable the identification of multi-omic signatures associated with disease phenotypes [118]. These many molecular profiles will provide a comprehensive picture of PCa screening and diagnosis and accelerate the search for candidate biomarkers. There are two approaches to data integration. The first strategy relies primarily on prior knowledge of known cancer pathways and processes. However, linking separate molecular data sets between databases is challenging. For example, regulatory data from ENCODE are not necessarily linked to specific genes that can subsequently be mapped to KEGG pathways. Similarly, metabolite data are weakly linked in current versions of gene-centric pathway networks. The second approach neglected existing knowledge of metabolic pathways and network interactions in cells and tissues and prioritized finding queries that change in a coordinated manner [119].
While the use of omics data, particularly genomics, has led to insights that have been used in clinical oncology to support treatment decisions, SC analysis, which has recently emerged as a promising approach for capturing invaluable cellular-level data, such as TME and metastasis status [75, 120]. However, it is limited by artifacts caused by evolutionary dynamics during the growth of a laboratory clone and by the fact that it can only examine a small number of founder cells [121]. Similarly, since proteins, lipids, glycans, and metabolites cannot be replicated like nucleic acids, single cell omics would not be able to effectively analyze proteomics, lipidomics, glycomics, or metabolomics.
Because most RNA sequencing data are from the bulk of malignancies, they cannot account for PC heterogeneity. This is because the transcriptome is the consequence of numerous biological processes that contribute to differential gene regulation, and these activities are not always synchronized in the tumor bulk [122, 123]. Cancer stem cells and circulating tumor cells are among the rare tumor cells that can be effectively detected by the SC method. It is interesting that topographic SC sequencing can detect tumor cell invasion and metastasis [124]. In addition, SC multiple sequencing examines DNA methylation and the state of chromatin in the SC to define intratumoural heterogeneity, which in turn leads to tailored therapeutic approaches [125]. Moreover, coupling SC sequencing with other technologies, such as CRISPR screening, enables the functional study of heterogeneous cell populations and facilitates the study of the interplay between genes and regulatory elements. A significant cost reduction is also foreseen [126]. Recently, the development of novel Nano-microarrays has made it possible to process thousands of single cells in parallel, which, combined with dynamic secondary ion mass spectrometry that has three dimensional scanning capacity and higher resolution, could greatly improve the sensitivity of single-molecule quantification for all classes of biomolecules [127].
Conclusion
Overall, this work highlights the need for the use of multi omic approach to achieve better outcomes in the treatment of PCa patients. We are just beginning to collect complete, unbiased multi-omic data to develop the appropriate statistical and annotation tools to help us understand these complicated data sets and extract biologically and clinically relevant information. Because multi-omic data are expensive and time-consuming, access to appropriate tissue samples and biopsy material is essential for generating multi-omic data. As described in this review, various molecular characteristics of tumor cells revealed by the multi-omic approach lead to effective screening methods for early cancer detection, screening, patient selection strategies, or assessment of response to treatment. In the future, more advanced and innovative approaches to the integration and interpretation of multiple omics data should be developed.
Availability of data and materials
All the data generated and analyzed during this study are included in the manuscript and the additional materials.
Abbreviations
- APC:
-
APC regulator of WNT signaling pathway
- AR:
-
Androgen receptor
- CAPN2:
-
Calpain 2
- CDH1:
-
Cadherin 1
- CDKN2A:
-
Cyclin dependent kinase inhibitor 2A
- CENPA:
-
Centromere protein A
- CHD1:
-
Chromodomain helicase DNA binding protein 1
- CP:
-
Ceruloplasmin
- DDC:
-
Dopa decarboxylase
- ERG:
-
ETS transcription factor ERG
- EZH2:
-
Enhancer of zeste 2 polycomb repressive complex 2 subunit
- FOXA1:
-
Forkhead box A1
- GABPB1:
-
GA binding protein transcription factor subunit beta 1
- GATA2:
-
GATA binding protein 2
- GSTP1:
-
Glutathione S-transferase pi 1
- HEATR5B:
-
HEAT repeat containing 5B
- HOXB13:
-
Homeobox B13
- JMJD6:
-
Jumonji domain containing 6, arginine demethylase and lysine hydroxylase
- KIF20A:
-
Kinesin family member 20A
- KIF23:
-
Kinesin family member 23
- KLK3:
-
Kallikrein related peptidase 3
- LCN2:
-
Lipocalin 2
- LTF:
-
Lactotransferrin
- MED4:
-
Mediator complex subunit 4
- MGMT:
-
O-6-methylguanine-DNA methyltransferase
- MMP7:
-
Matrix metallopeptidase 7
- MYC:
-
MYC proto-oncogene, bHLH transcription factor
- PCDH9:
-
Protocadherin 9
- PIGR:
-
Polymeric immunoglobulin receptor
- PIK3CB:
-
Phosphatidylinositol-4, 5-bisphosphate 3-kinase catalytic subunit beta
- PLXNA1:
-
Plexin A1
- PPP1CB:
-
Protein phosphatase 1 catalytic subunit beta
- PSMB6:
-
Proteasome 20S subunit beta 6
- PYCARD:
-
PYD and CARD domain containing
- RARRES1:
-
Retinoic acid receptor responder 1
- RASSF1:
-
Ras association domain family member 1
- RB1:
-
RB transcriptional corepressor 1
- RFX6:
-
Regulatory factor X6
- SCGB3A1:
-
Secretoglobin family 3A member 1
- SEC31A:
-
SEC31 homolog A, COPII coat complex component
- SMARCA4:
-
SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a, member 4
- TELO2:
-
Telomere maintenance 2
- TGM4:
-
Transglutaminase 4
- TMED2:
-
Transmembrane p24 trafficking protein 2
- TMED10:
-
Transmembrane p24 trafficking protein 10
- TMPRSS2:
-
Transmembrane serine protease 2
- TNC:
-
Tenascin C
- TP53:
-
Tumor protein p53
- TP63:
-
Tumor protein p63
- UBE2N:
-
Ubiquitin conjugating enzyme E2 N
- ZMYND19:
-
Zinc finger MYND-type containing 19
- ZNF292:
-
Zinc finger protein 292
- SPOP:
-
Speckle-type POZ protein
- MATH:
-
Meprin and TRAF homology
- CDK9:
-
Cyclin-dependent kinase 9
- AMPK:
-
AMP-activated protein kinase
- HO-1:
-
Heme oxygenase 1
- 1-MA:
-
1-Methyladenosine phosphatidic acid
- Apo-AIV:
-
Apolipoprotein A1V
- PDK4:
-
Pyruvate dehydrogenase kinase 4
References
Karczewski KJ, Snyder MP. Integrative omics for health and disease. Nat Rev Genet. 2018;19(5):299–310.
Suravajhala P, Kogelman LJ, Kadarmideen HN. Multi-omic data integration and analysis using systems genomics approaches: methods and applications in animal production, health and welfare. Genet Sel Evol. 2016;48(1):1–14.
Karahalil B. Overview of systems biology and omics technologies. Curr Med Chem. 2016;23(37):4221–30.
Vucic EA, Thu KL, Robison K, Rybaczyk LA, Chari R, Alvarez CE, et al. Translating cancer ‘omics’ to improved outcomes. Genome Res. 2012;22(2):188–95.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34.
Wang G, Zhao D, Spring DJ, DePinho RA. Genetics and biology of prostate cancer. Genes Dev. 2018;32(17–18):1105–40.
Parry M, Cowling T, Sujenthiran A, Nossiter J, Berry B, Cathcart P, et al. Risk stratification for prostate cancer management: value of the Cambridge Prognostic Group classification for assessing treatment allocation. BMC Med. 2020;18(1):1–9.
Eggener SE, Cifu AS, Nabhan C. Prostate cancer screening. JAMA. 2015;314(8):825–6.
Thompson IM, Pauler DK, Goodman PJ, Tangen CM, Lucia MS, Parnes HL, et al. Prevalence of prostate cancer among men with a prostate-specific antigen level ≤4.0 ng per milliliter. N Engl J Med. 2004;350(22):2239–46.
Merriel SW, Pocock L, Gilbert E, Creavin S, Walter FM, Spencer A, et al. Systematic review and meta-analysis of the diagnostic accuracy of prostate-specific antigen (PSA) for the detection of prostate cancer in symptomatic patients. BMC Med. 2022;20(1):1–11.
Heidenreich A, Pfister D, Merseburger A, Bartsch G. Castration-resistant prostate cancer: where we stand in 2013 and what urologists should know. Eur Urol. 2013;64(2):260–5.
Taşan M, Musso G, Hao T, Vidal M, MacRae CA, Roth FP. Selecting causal genes from genome-wide association studies via functionally coherent subnetworks. Nat Methods. 2015;12(2):154–9.
Weischenfeldt J, Simon R, Feuerbach L, Schlangen K, Weichenhan D, Minner S, et al. Integrative genomic analyses reveal an androgen-driven somatic alteration landscape in early-onset prostate cancer. Cancer Cell. 2013;23(2):159–70.
Grasso CS, Wu Y-M, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487(7406):239–43.
Dupont WD, Breyer JP, Johnson SH, Plummer WD, Smith JR. Prostate cancer risk variants of the HOXB genetic locus. Sci Rep. 2021;11(1):11385.
Caspari R, Friedl W, Mandl M, Möslein G, Kadmon M, Knapp M, et al. Familial adenomatous polyposis: mutation at codon 1309 and early onset of colon cancer. Lancet (London, England). 1994;343(8898):629–32.
McKinley JM, Weideman PC, Jenkins MA, Friedlander ML, Hopper JL, McLachlan S-A, et al. Prostate screening uptake in Australian BRCA1 and BRCA2 carriers. Hered Cancer Clin Pract. 2007;5(3):161.
Mur P, García-Mulero S, Del Valle J, Magraner-Pardo L, Vidal A, Pineda M, et al. Role of POLE and POLD1 in familial cancer. Genet Med. 2020;22(12):2089–100.
Zhuang Y, Wang H, Jiang D, Li Y, Feng L, Tian C, et al. Multi gene mutation signatures in colorectal cancer patients: predict for the diagnosis, pathological classification, staging and prognosis. BMC Cancer. 2021;21(1):380.
Robinson D, Van Allen EM, Wu Y-M, Schultz N, Lonigro RJ, Mosquera J-M, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215–28.
Seifert M, Peitzsch C, Gorodetska I, Börner C, Klink B, Dubrovska A. Network-based analysis of prostate cancer cell lines reveals novel marker gene candidates associated with radioresistance and patient relapse. PLoS Comput Biol. 2019;15(11): e1007460.
Tomlins SA, Bjartell A, Chinnaiyan AM, Jenster G, Nam RK, Rubin MA, et al. ETS gene fusions in prostate cancer: from discovery to daily clinical practice. Eur Urol. 2009;56(2):275–86.
Klezovitch O, Risk M, Coleman I, Lucas JM, Null M, True LD, et al. A causal role for ERG in neoplastic transformation of prostate epithelium. Proc Natl Acad Sci. 2008;105(6):2105–10.
Henzler C, Li Y, Yang R, McBride T, Ho Y, Sprenger C, et al. Truncation and constitutive activation of the androgen receptor by diverse genomic rearrangements in prostate cancer. Nat Commun. 2016;7(1):1–12.
Chen M, Zhang J, Sampieri K, Clohessy JG, Mendez L, Gonzalez-Billalabeitia E, et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat Genet. 2018;50(2):206–18.
Tomlins SA, Laxman B, Varambally S, Cao X, Yu J, Helgeson BE, et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia (New York, NY). 2008;10(2):177–88.
Tan H-L, Sood A, Rahimi HA, Wang W, Gupta N, Hicks J, et al. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin Cancer Res. 2014;20(4):890–903.
Pritchard CC, Offit K, Nelson PS. DNA-repair gene mutations in metastatic prostate cancer. N Engl J Med. 2016;375(18):1804–5.
Shenoy D, Packianathan S, Chen AM, Vijayakumar S. Do African-American men need separate prostate cancer screening guidelines? BMC Urol. 2016;16(1):1–6.
Li J, Xu C, Lee HJ, Ren S, Zi X, Zhang Z, et al. A genomic and epigenomic atlas of prostate cancer in Asian populations. Nature. 2020;580(7801):93–9.
Brouwer I, Lenstra TL. Visualizing transcription: key to understanding gene expression dynamics. Curr Opin Chem Biol. 2019;51:122–9.
Pope SD, Medzhitov R. Emerging principles of gene expression programs and their regulation. Mol Cell. 2018;71(3):389–97.
Marzec J, Ross-Adams H, Pirrò S, Wang J, Zhu Y, Mao X, et al. The transcriptomic landscape of prostate cancer development and progression: an integrative analysis. Cancers. 2021;13(2):345.
Alkhateeb A, Rezaeian I, Singireddy S, Cavallo-Medved D, Porter LA, Rueda L. Transcriptomics signature from next-generation sequencing data reveals new transcriptomic biomarkers related to prostate cancer. Cancer Inf. 2019;18:1176935119835522.
Solé C, Goicoechea I, Goñi A, Schramm M, Armesto M, Arestin M, et al. The urinary transcriptome as a source of biomarkers for prostate cancer. Cancers (Basel). 2020;12(2):513.
Fabbri M, Girnita L, Varani G, Calin GA. Decrypting noncoding RNA interactions, structures, and functional networks. Genome Res. 2019;29(9):1377–88.
Eke I, Bylicky MA, Sandfort V, Chopra S, Martello S, Graves EE, et al. The lncRNAs LINC00261 and LINC00665 are upregulated in long-term prostate cancer adaptation after radiotherapy. Mol Ther-Nucleic Acids. 2021;24:175–87.
Tang Y, Pan J, Huang S, Peng X, Zou X, Luo Y, et al. Downregulation of miR-133a-3p promotes prostate cancer bone metastasis via activating PI3K/AKT signaling. J Exp Clin Cancer Res. 2018;37(1):1–16.
Lekchnov EA, Amelina EV, Bryzgunova OE, Zaporozhchenko IA, Konoshenko MY, Yarmoschuk SV, et al. Searching for the novel specific predictors of prostate cancer in urine: the analysis of 84 miRNA expression. Int J Mol Sci. 2018;19(12):4088.
Du WW, Zhang C, Yang W, Yong T, Awan FM, Yang BB. Identifying and characterizing circRNA-protein interaction. Theranostics. 2017;7(17):4183.
Chen S, Huang V, Xu X, Livingstone J, Soares F, Jeon J, et al. Widespread and functional RNA circularization in localized prostate cancer. Cell. 2019;176(4):831–43.
Yan Z, Xiao Y, Chen Y, Luo G. Screening and identification of epithelial-to-mesenchymal transition-related circRNA and miRNA in prostate cancer. Pathol Res Pract. 2020;216(2): 152784.
Kumar D, Bansal G, Narang A, Basak T, Abbas T, Dash D. Integrating transcriptome and proteome profiling: strategies and applications. Proteomics. 2016;16(19):2533–44.
Tanase CP, Codrici E, Popescu ID, Mihai S, Enciu A-M, Necula LG, et al. Prostate cancer proteomics: current trends and future perspectives for biomarker discovery. Oncotarget. 2017;8(11):18497.
Valdes-Mora F, Clark S. Prostate cancer epigenetic biomarkers: next-generation technologies. Oncogene. 2015;34(13):1609–18.
Davalieva K, Kostovska IM, Kiprijanovska S, Markoska K, Kubelka-Sabit K, Filipovski V, et al. Proteomics analysis of malignant and benign prostate tissue by 2D DIGE/MS reveals new insights into proteins involved in prostate cancer. Prostate. 2015;75(14):1586–600.
Launonen K-M, Paakinaho V, Sigismondo G, Malinen M, Sironen R, Hartikainen JM, et al. Chromatin-directed proteomics-identified network of endogenous androgen receptor in prostate cancer cells. Oncogene. 2021;40(27):4567–79.
Iglesias-Gato D, Thysell E, Tyanova S, Crnalic S, Santos A, Lima TS, et al. The proteome of prostate cancer bone metastasis reveals heterogeneity with prognostic implications. Clin Cancer Res. 2018;24(21):5433–44.
Aiello D, Casadonte F, Terracciano R, Damiano R, Savino R, Sindona G, et al. Targeted proteomic approach in prostatic tissue: a panel of potential biomarkers for cancer detection. Oncoscience. 2016;3(7–8):220.
Katsogiannou M, Boyer J-B, Valdeolivas A, Remy E, Calzone L, Audebert S, et al. Integrative proteomic and phosphoproteomic profiling of prostate cell lines. PLoS ONE. 2019;14(11): e0224148.
Tonry C, Finn S, Armstrong J, Pennington SR. Clinical proteomics for prostate cancer: understanding prostate cancer pathology and protein biomarkers for improved disease management. Clin Proteomics. 2020;17(1):41.
Widschwendter M, Jones A, Evans I, Reisel D, Dillner J, Sundström K, et al. Epigenome-based cancer risk prediction: rationale, opportunities and challenges. Nat Rev Clin Oncol. 2018;15(5):292–309.
Pomerantz MM, Qiu X, Zhu Y, Takeda DY, Pan W, Baca SC, et al. Prostate cancer reactivates developmental epigenomic programs during metastatic progression. Nat Genet. 2020;52(8):790–9.
Baca SC, Takeda DY, Seo J-H, Hwang J, Ku SY, Arafeh R, et al. Reprogramming of the FOXA1 cistrome in treatment-emergent neuroendocrine prostate cancer. Nat Commun. 2021;12(1):1–12.
Donkena KV, Young CY, Tindall DJ. Oxidative stress and DNA methylation in prostate cancer. Obstet Gynecol Int. 2010;2010.
Wu A, Cremaschi P, Wetterskog D, Conteduca V, Franceschini GM, Kleftogiannis D, et al. Genome-wide plasma DNA methylation features of metastatic prostate cancer. J Clin Investig. 2020;130(4):1991–2000.
Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38(1):23–38.
Kobayashi Y, Absher DM, Gulzar ZG, Young SR, McKenney JK, Peehl DM, et al. DNA methylation profiling reveals novel biomarkers and important roles for DNA methyltransferases in prostate cancer. Genome Res. 2011;21(7):1017–27.
Willard SS, Koochekpour S. Regulators of gene expression as biomarkers for prostate cancer. Am J Cancer Res. 2012;2(6):620.
López JI, Angulo JC, Martín A, Sánchez-Chapado M, González-Corpas A, Colás B, et al. A DNA hypermethylation profile reveals new potential biomarkers for the evaluation of prognosis in urothelial bladder cancer. APMIS. 2017;125(9):787–96.
Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. 2011;17(2):211–5.
Boldrini L, Bartoletti R, Giordano M, Manassero F, Selli C, Panichi M, et al. C-MYC, HIF-1α, ERG, TKT, and GSTP1: an axis in prostate cancer? Pathol Oncol Res. 2019;25(4):1423–9.
Xu Y, Tsai C-W, Chang W-S, Han Y, Huang M, Pettaway CA, et al. Epigenome-wide association study of prostate cancer in African Americans identifies DNA methylation biomarkers for aggressive disease. Biomolecules. 2021;11(12):1826.
Yegnasubramanian S, Haffner MC, Zhang Y, Gurel B, Cornish TC, Wu Z, et al. DNA hypomethylation arises later in prostate cancer progression than CpG island hypermethylation and contributes to metastatic tumor heterogeneity. Cancer Res. 2008;68(21):8954–67.
Ylitalo EB, Thysell E, Landfors M, Brattsand M, Jernberg E, Crnalic S, et al. A novel DNA methylation signature is associated with androgen receptor activity and patient prognosis in bone metastatic prostate cancer. Clin Epigenetics. 2021;13(1):133.
Kitkumthorn N, Mutirangura A. Long interspersed nuclear element-1 hypomethylation in cancer: biology and clinical applications. Clin Epigenetics. 2011;2(2):315–30.
Mondul AM, Moore SC, Weinstein SJ, Männistö S, Sampson JN, Albanes D. 1-Stearoylglycerol is associated with risk of prostate cancer: results from a serum metabolomic profiling analysis. Metabolomics. 2014;10(5):1036–41.
Mondul AM, Moore SC, Weinstein SJ, Karoly ED, Sampson JN, Albanes D. Metabolomic analysis of prostate cancer risk in a prospective cohort: the alpha-tocopherol, beta-carotene cancer prevention (ATBC) study. Int J Cancer. 2015;137(9):2124–32.
Zhang X, Xia B, Zheng H, Ning J, Zhu Y, Shao X, et al. Identification of characteristic metabolic panels for different stages of prostate cancer by 1H NMR-based metabolomics analysis. J Transl Med. 2022;20(1):275.
Huang G, Liu X, Jiao L, Xu C, Zhang Z, Wang L, et al. Metabolomic evaluation of the response to endocrine therapy in patients with prostate cancer. Eur J Pharmacol. 2014;729:132–7.
Andras I, Crisan N, Vesa S, Rahota R, Romanciuc F, Lazar A, et al. Serum metabolomics can predict the outcome of first systematic transrectal prostate biopsy in patients with PSA< 10 ng/mL. Future Oncol. 2017;13(20):1793–800.
Kosti O, Xu X, Veenstra TD, Hsing AW, Chu LW, Goldman L, et al. Urinary estrogen metabolites and prostate cancer risk: a pilot study. Prostate. 2011;71(5):507–16.
Roberts MJ, Richards RS, Chow CW, Buck M, Yaxley J, Lavin MF, et al. Seminal plasma enables selection and monitoring of active surveillance candidates using nuclear magnetic resonance-based metabolomics: a preliminary investigation. Prostate Int. 2017;5(4):149–57.
Kdadra M, Höckner S, Leung H, Kremer W, Schiffer E. Metabolomics biomarkers of prostate cancer: a systematic review. Diagnostics. 2019;9(1):21.
Cerrato A, Bedia C, Capriotti AL, Cavaliere C, Gentile V, Maggi M, et al. Untargeted metabolomics of prostate cancer zwitterionic and positively charged compounds in urine. Anal Chim Acta. 2021;1158: 338381.
Bocci F, Gearhart-Serna L, Boareto M, Ribeiro M, Ben-Jacob E, Devi GR, et al. Toward understanding cancer stem cell heterogeneity in the tumor microenvironment. Proc Natl Acad Sci. 2019;116(1):148–57.
Boyd LK, Mao X, Lu Y-J. The complexity of prostate cancer: genomic alterations and heterogeneity. Nat Rev Urol. 2012;9(11):652–64.
Taavitsainen S, Engedal N, Cao S, Handle F, Erickson A, Prekovic S, et al. Single-cell ATAC and RNA sequencing reveal pre-existing and persistent cells associated with prostate cancer relapse. Nat Commun. 2021;12(1):1–16.
Peng S, Hu P, Xiao Y-T, Lu W, Guo D, Hu S, et al. Single-cell analysis reveals EP4 as a target for restoring T-cell infiltration and sensitizing prostate cancer to immunotherapy. Clin Cancer Res. 2021;28:552.
Dong B, Miao J, Wang Y, Luo W, Ji Z, Lai H, et al. Single-cell analysis supports a luminal-neuroendocrine transdifferentiation in human prostate cancer. Commun Biol. 2020;3(1):1–15.
Song H, Weinstein HN, Allegakoen P, Wadsworth MH, Xie J, Yang H, et al. Single-cell analysis of human primary prostate cancer reveals the heterogeneity of tumor-associated epithelial cell states. Nat Commun. 2022;13(1):1–20.
Chen S, Zhu G, Yang Y, Wang F, Xiao Y-T, Zhang N, et al. Single-cell analysis reveals transcriptomic remodellings in distinct cell types that contribute to human prostate cancer progression. Nat Cell Biol. 2021;23(1):87–98.
Cantini L, Zakeri P, Hernandez C, Naldi A, Thieffry D, Remy E, et al. Benchmarking joint multi-omics dimensionality reduction approaches for the study of cancer. Nat Commun. 2021;12(1):1–12.
Ren S, Wei G-H, Liu D, Wang L, Hou Y, Zhu S, et al. Whole-genome and transcriptome sequencing of prostate cancer identify new genetic alterations driving disease progression. Eur Urol. 2018;73(3):322–39.
Kwon OK, Ha Y-S, Lee JN, Kim S, Lee H, Chun SY, et al. Comparative proteome profiling and mutant protein identification in metastatic prostate cancer cells by quantitative mass spectrometry-based proteogenomics. Cancer Genomics Proteomics. 2019;16(4):273–86.
Drabovich AP, Saraon P, Drabovich M, Karakosta TD, Dimitromanolakis A, Hyndman ME, et al. Multi-omics biomarker pipeline reveals elevated levels of protein-glutamine gamma-glutamyltransferase 4 in seminal plasma of prostate cancer patients*[S]. Mol Cell Proteomics. 2019;18(9):1807–23.
Gao B, Lue H-W, Podolak J, Fan S, Zhang Y, Serawat A, et al. Multi-omics analyses detail metabolic reprogramming in lipids, carnitines, and use of glycolytic intermediates between prostate small cell neuroendocrine carcinoma and prostate adenocarcinoma. Metabolites. 2019;9(5):82.
Song Y, Wang H, Pan Y, Liu T. Investigating the multi-target pharmacological mechanism of Hedyotis diffusa Willd acting on prostate cancer: a network pharmacology approach. Biomolecules. 2019;9(10):591.
Drake JM, Paull EO, Graham NA, Lee JK, Smith BA, Titz B, et al. Phosphoproteome integration reveals patient-specific networks in prostate cancer. Cell. 2016;166(4):1041–54.
Ru J, Li P, Wang J, Zhou W, Li B, Huang C, et al. TCMSP: a database of systems pharmacology for drug discovery from herbal medicines. J Cheminf. 2014;6(1):1–6.
Cook CE, Bergman MT, Finn RD, Cochrane G, Birney E, Apweiler R. The European Bioinformatics Institute in 2016: data growth and integration. Nucleic Acids Res. 2016;44(D1):D20–6.
Argelaguet R, Cuomo AS, Stegle O, Marioni JC. Computational principles and challenges in single-cell data integration. Nat Biotechnol. 2021;39(10):1202–15.
Chen H, Li J, Wang Y, Ng PK-S, Tsang YH, Shaw KR, et al. Comprehensive assessment of computational algorithms in predicting cancer driver mutations. Genome Biol. 2020;21(1):1–17.
Sinha R, Sharma B, Dangi AK, Shukla P. Recent metabolomics and gene editing approaches for synthesis of microbial secondary metabolites for drug discovery and development. World J Microbiol Biotechnol. 2019;35(11):1–14.
Yu C, Qi X, Lin Y, Li Y, Shen B. iODA: An integrated tool for analysis of cancer pathway consistency from heterogeneous multi-omics data. J Biomed Inform. 2020;112: 103605.
Saghaleyni R, Sheikh Muhammad A, Bangalore P, Nielsen J, Robinson JL. Machine learning-based investigation of the cancer protein secretory pathway. PLoS Comput Biol. 2021;17(4): e1008898.
Wang T, Shao W, Huang Z, Tang H, Zhang J, Ding Z, et al. MOGONET integrates multi-omics data using graph convolutional networks allowing patient classification and biomarker identification. Nat Commun. 2021;12(1):1–13.
Kaur P, Singh A, Chana I. Computational techniques and tools for omics data analysis: state-of-the-art, challenges, and future directions. Arch Comput Methods Eng. 2021;28(7):4595–631.
Tomczak K, Czerwińska P, Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol. 2015;19(1A):A68.
Chen Z, Hu H. Identification of prognosis biomarkers of prostatic cancer in a cohort of 498 patients from TCGA. Curr Probl Cancer. 2019;43(6): 100503.
Gao L, Zhang L-j, Li S-h, Wei L-l, Luo B, He R-q, et al. Role of miR-452-5p in the tumorigenesis of prostate cancer: a study based on the Cancer Genome Atl (TCGA), Gene Expression Omnibus (GEO), and bioinformatics analysis. Pathol Res Pract. 2018;214(5):732–49.
Penney KL, Sinnott JA, Fall K, Pawitan Y, Hoshida Y, Kraft P, et al. mRNA expression signature of Gleason grade predicts lethal prostate cancer. J Clin Oncol. 2011;29(17):2391.
Hou J, Li X, Xie K-P. Coupled liquid biopsy and bioinformatics for pancreatic cancer early detection and precision prognostication. Mol Cancer. 2021;20(1):1–12.
Devos G, Joniau S. PREDICT Prostate, a useful tool in men with low-and intermediate-risk prostate cancer who are hesitant between conservative management and active treatment. BMC Med. 2020;18(1):1–3.
Loeb S, Bjurlin MA, Nicholson J, Tammela TL, Penson DF, Carter HB, et al. Overdiagnosis and overtreatment of prostate cancer. Eur Urol. 2014;65(6):1046–55.
Kim Y, Park YH, Lee JY, Choi IY, Yu H. Discovery of prostate specific antigen pattern to predict castration resistant prostate cancer of androgen deprivation therapy. BMC Med Inform Decis Mak. 2016;16(1):1–9.
Couñago F, López-Campos F, Díaz-Gavela AA, Almagro E, Fenández-Pascual E, Henríquez I, et al. Clinical applications of molecular biomarkers in prostate cancer. Cancers. 2020;12(6):1550.
Olivier M, Asmis R, Hawkins GA, Howard TD, Cox LA. The need for multi-omics biomarker signatures in precision medicine. Int J Mol Sci. 2019;20(19):4781.
Mayers JR, Wu C, Clish CB, Kraft P, Torrence ME, Fiske BP, et al. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat Med. 2014;20(10):1193–8.
Icard P, Fournel L, Coquerel A, Gligorov J, Alifano M, Lincet H. Citrate targets FBPase and constitutes an emerging novel approach for cancer therapy. Cancer Cell Int. 2018;18(1):1–2.
Dougan MM, Li Y, Chu LW, Haile RW, Whittemore AS, Han SS, et al. Metabolomic profiles in breast cancer: a pilot case-control study in the breast cancer family registry. BMC Cancer. 2018;18(1):1–8.
Sawyers CL. The cancer biomarker problem. Nature. 2008;452(7187):548–52.
Bou-Dargham MJ, Sha L, Sang Q-XA, Zhang J. Immune landscape of human prostate cancer: immune evasion mechanisms and biomarkers for personalized immunotherapy. BMC Cancer. 2020;20(1):1–10.
Litwin MS, Tan H-J. The diagnosis and treatment of prostate cancer: a review. JAMA. 2017;317(24):2532–42.
Shen L. Liquid biopsy: a powerful tool to monitor trastuzumab resistance in HER2-positive metastatic gastric cancer. Cancer Commun. 2018;38(1):1–3.
Vaidyanathan R, Soon RH, Zhang P, Jiang K, Lim CT. Cancer diagnosis: from tumor to liquid biopsy and beyond. Lab Chip. 2019;19(1):11–34.
Kiebish MA, Cullen J, Mishra P, Ali A, Milliman E, Rodrigues LO, et al. Multi-omic serum biomarkers for prognosis of disease progression in prostate cancer. J Transl Med. 2020;18(1):1–10.
Tian R, Basu MK, Capriotti E. Computational methods and resources for the interpretation of genomic variants in cancer. BMC Genomics. 2015;16(8):1–19.
Zhang H, Guo Y, Prosperi M, Bian J. An ontology-based documentation of data discovery and integration process in cancer outcomes research. BMC Med Inform Decis Mak. 2020;20(4):1–22.
Yan X, Xie Y, Yang F, Hua Y, Zeng T, Sun C, et al. Comprehensive description of the current breast cancer microenvironment advancements via single-cell analysis. J Exp Clin Cancer Res. 2021;40(1):1–15.
Nagasawa S, Kashima Y, Suzuki A, Suzuki Y. Single-cell and spatial analyses of cancer cells: toward elucidating the molecular mechanisms of clonal evolution and drug resistance acquisition. Inflamm Regen. 2021;41(1):1–15.
Zhang Z, Zhou C, Li X, Barnes SD, Deng S, Hoover E, et al. Loss of CHD1 promotes heterogeneous mechanisms of resistance to AR-targeted therapy via chromatin dysregulation. Cancer Cell. 2020;37(4):584–98.
Su J-H, Zheng P, Kinrot SS, Bintu B, Zhuang X. Genome-scale imaging of the 3D organization and transcriptional activity of chromatin. Cell. 2020;182(6):1641–59.
Guo F, Li L, Li J, Wu X, Hu B, Zhu P, et al. Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells. Cell Res. 2017;27(8):967–88.
Mannarapu M, Dariya B, Bandapalli OR. Application of single-cell sequencing technologies in pancreatic cancer. Mol Cell Biochem. 2021;476(6):2429–37.
Datlinger P, Rendeiro AF, Schmidl C, Krausgruber T, Traxler P, Klughammer J, et al. Pooled CRISPR screening with single-cell transcriptome readout. Nat Methods. 2017;14(3):297–301.
Misevic GN, BenAssayag G, Rasser B, Sales P, Simic-Krstic J, Misevic NJ, et al. Design and construction of wall-less nano-electrophoretic and nano in micro array high throughput devices for single cell ‘omics’ single molecule detection analyses. J Mol Struct. 2014;1073:142–9.
Latonen L, Afyounian E, Jylhä A, Nättinen J, Aapola U, Annala M, et al. Integrative proteomics in prostate cancer uncovers robustness against genomic and transcriptomic aberrations during disease progression. Nat Commun. 2018;9(1):1176.
Yan M, Qi H, Li J, Ye G, Shao Y, Li T, et al. Identification of SPOP related metabolic pathways in prostate cancer. Oncotarget. 2017;8(61):103032–46.
Oberhuber M, Pecoraro M, Rusz M, Oberhuber G, Wieselberg M, Haslinger P, et al. STAT3-dependent analysis reveals PDK4 as independent predictor of recurrence in prostate cancer. Mol Syst Biol. 2020;16(4): e9247.
Murphy K, Murphy BT, Boyce S, Flynn L, Gilgunn S, O’Rourke CJ, et al. Integrating biomarkers across omic platforms: an approach to improve stratification of patients with indolent and aggressive prostate cancer. Mol Oncol. 2018;12(9):1513–25.
Itkonen HM, Poulose N, Walker S, Mills IG. CDK9 inhibition induces a metabolic switch that renders prostate cancer cells dependent on fatty acid oxidation. Neoplasia (New York, NY). 2019;21(7):713–20.
Kamoun A, Cancel-Tassin G, Fromont G, Elarouci N, Armenoult L, Ayadi M, et al. Comprehensive molecular classification of localized prostate adenocarcinoma reveals a tumour subtype predictive of non-aggressive disease. Ann Oncol. 2018;29(8):1814–21.
Gómez-Cebrián N, García-Flores M, Rubio-Briones J, López-Guerrero JA, Pineda-Lucena A, Puchades-Carrasco L. Targeted metabolomics analyses reveal specific metabolic alterations in high-grade prostate cancer patients. J Proteome Res. 2020;19(10):4082–92.
Paez AV, Pallavicini C, Schuster F, Valacco MP, Giudice J, Ortiz EG, et al. Heme oxygenase-1 in the forefront of a multi-molecular network that governs cell-cell contacts and filopodia-induced zippering in prostate cancer. Cell Death Dis. 2016;7(12): e2570.
Sial N, Saeed S, Ahmad M, Hameed Y, Rehman A, Abbas M, et al. Multi-omics analysis identified TMED2 as a shared potential biomarker in six subtypes of human cancer. Int J General Med. 2021;14:7025.
Wang T-H, Lee C-Y, Lee T-Y, Huang H-D, Hsu JB-K, Chang T-H. Biomarker identification through multiomics data analysis of prostate cancer prognostication using a deep learning model and similarity network fusion. Cancers. 2021;13(11):2528.
Kiebish MA, Cullen J, Mishra P, Ali A, Milliman E, Rodrigues LO, et al. Multi-omic serum biomarkers for prognosis of disease progression in prostate cancer. J Transl Med. 2020;18(1):10.
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res. 2012;41(D1):D991–5.
Parkinson H, Kapushesky M, Shojatalab M, Abeygunawardena N, Coulson R, Farne A, et al. ArrayExpress—a public database of microarray experiments and gene expression profiles. Nucleic Acids Res. 2007;35(suppl_1):D747–50.
Zhang J, Baran J, Cros A, Guberman JM, Haider S, Hsu J, et al. International Cancer Genome Consortium Data Portal—a one-stop shop for cancer genomics data. Database. 2011;2011.
Krupke DM, Begley DA, Sundberg JP, Richardson JE, Neuhauser SB, Bult CJ. The mouse tumor biology database: a comprehensive resource for mouse models of human cancer. Can Res. 2017;77(21):e67–70.
Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia (New York, NY). 2004;6(1):1–6.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1.
Yuan H, Yan M, Zhang G, Liu W, Deng C, Liao G, et al. CancerSEA: a cancer single-cell state atlas. Nucleic Acids Res. 2018;47(D1):D900–8.
Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47(D1):D941–7.
Samur MK, Yan Z, Wang X, Cao Q, Munshi NC, Li C, et al. canEvolve: a web portal for integrative oncogenomics. PLoS ONE. 2013;8(2): e56228.
Bhattacharya A, Cui Y, Somami R. 2.0: a database of cancer somatic mutations altering microRNA–ceRNA interactions. Nucleic Acids Res. 2016;44(D1):D1005–10.
Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–7.
He X, Chang S, Zhang J, Zhao Q, Xiang H, Kusonmano K, et al. MethyCancer: the database of human DNA methylation and cancer. Nucleic Acids Res. 2007;36(suppl_1):D836–41.
Zhao Y, Li H, Fang S, Kang Y, Wu W, Hao Y, et al. NONCODE 2016: an informative and valuable data source of long non-coding RNAs. Nucleic Acids Res. 2016;44(D1):D203–8.
Funding
None.
Author information
Authors and Affiliations
Contributions
MN conceptualized the project. All authors were involved in preparing and agreeing the final manuscript. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
None.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gholami, N., Haghparast, A., Alipourfard, I. et al. Prostate cancer in omics era. Cancer Cell Int 22, 274 (2022). https://doi.org/10.1186/s12935-022-02691-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-022-02691-y