
Abstract
Signal transducer and activator of transcription 3 (STAT3) induces breast cancer malignancy. Recent clinical and preclinical studies have demonstrated an association between overexpressed and activated STAT3 and breast cancer progression, proliferation, metastasis, and chemoresistance. Resveratrol (RES), a naturally occurring phytoalexin, has demonstrated anti-cancer activity in several disease models. Furthermore, RES has also been shown to regulate the STAT3 signaling cascade via its anti-oxidant and anti-inflammatory effects. In the present review, we describe the STAT3 cascade signaling pathway and address the therapeutic targeting of STAT3 by RES as a tool to mitigate breast cancer.
Similar content being viewed by others

Introduction
Resveratrol (RES), also known as trans-3,4′,5-trihydroxystilbene, was first extracted from the white hellebore roots in 1939 by Michio Takaoka [1]. It is ubiquitous in various plants, including grape skin and by inference in wine, berries, peanuts, polygonum cuspidatum, and Rheum officinale Baill, which are traditional Chinese medicines [2, 3]. RES has been used to mitigate inflammation, reduce oxidative stress, suppress growth, and increase apoptosis [4]. These properties have led to its use as a therapeutic agent in multiple disorders such as cancer, cardiovascular, inflammatory, and neurodegenerative diseases [5]. RES exerts its efficacy through different molecules and pathways [6]. One of the target factors of RES is signal transducers and activators of transcription 3 (STAT3), an important transcription factor in several cancers, including breast cancer [7].
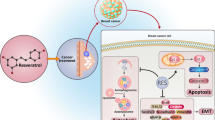
Among cancer-related deaths globally, breast cancer is the second most common in women [8]. Only few treatment approaches are available for it, given chemoresistance to most treatment modalities [9]. Various transcription factors (TFs) are directly associated with breast cancer progression and development [10, 11]. The STAT family comprises seven TFs, STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6, among the most critical TFs in breast cancer. These TFs are structurally similar and highly conserved [11,12,13]. Overall, six functional domains have been recognized, comprised of an N-terminal (NH2) domain, also known as STAT_int, DNA-binding (DBD) domain, SRC homology 2 (SH2) domain, coiled-coil (CDC) domain, transactivation (TAD) domain, and linker domain [14]. The involvement of STAT3 in the development, metastasis, multidrug resistance, and proliferation of cancer has been subject to extensive research [15, 16]. Classical STAT3 signaling cascade has been shown in several investigations [7, 17, 18]. Figure 1 provides an overview of STAT3 signaling cascades. As noted, several cytokines, such as interleukin 6 (IL-6) and interleukin 10 (IL-10), and growth factors comprising epidermal growth factor (EGF), fibroblast growth factor (FGF) and insulin-like growth factor (IGF), initiate the activation of STAT3 [19, 20]. Binding of these elements to their relevant receptors, leads to the activation of Janus kinases (JAKs) [21]. Cognate receptor’s cytoplasmic tail is phosphorylated by JAKs, followed by binding the SH2 domain of STAT3 to phosphorylated tyrosine residues. The phosphorylation of STAT3 enables the translocation of signals from the cytoplasm to the nucleus by forming homodimers. Once translocated, pSTAT3 binds to the target genes’ promoter site, by shaping a complex with several coactivators, such as p68, and leading to their transcription [22]. In the present review, we summarize the role of RES, via its effects on STAT3, in the development of breast cancer, focusing on the most recent and pertinent studies.

The classical STAT3 Signaling Pathway in Cancer Cells. Binding of IL-6 to its membrane-bound IL-6 receptor α (IL-6R) and IL-6 receptor β (also known as gp130) results in formation of IL-6/IL-6R/gp130 complex. The IL-6/IL-6R/gp130 complex activates the JAKs by phosphorylation which results in STAT3 phosphorylation and activation. STAT3 activation can be achieved by phosphorylation via other factors such as growth factors (i.e. FGF, IGF and EGF). These growth factors bind to their cognate membrane receptors. Upon phosphorylation, STAT3 forms a homodimer which translocates into the nucleus to bind to the promotor region of target genes and activates target gene transcription
The role of STAT3 in breast cancer progression, apoptosis, metastasis, proliferation, and chemoresistance will be discussed first. Once the interplay between STAT3 and breast cancer is delineated, next, the effect of RES on STAT3, and finally, the effects of RES on breast cancer through STAT3 will be addressed (Fig. 2).

Effects of Resveratrol on STAT3 Signaling Pathway in Breast Cancer Cells. Resveratrol can directly affect the STAT3 and its downstream molecular targets in breast cancer cells. Also, resveratrol can affect upstream regulators of STAT3 which results in changes in downstream molecular targets affecting the growth, progression and metastasis of breast cancer cells. Bcl-xL: B-cell lymphoma-extra large; MCL-1: Myeloid cell leukemia-1; TSG: tumor suppressor genes; COX-2: Cyclooxygenase-2; Akt: Protein kinase B (PKB) aka Akt
STAT3 and breast cancer progression
Oncostatin M (OSM), a cytokine and a member of the IL-6 family, can upregulate and phosphorylate IL-6 and STAT3, respectively, advancing breast cancer progression [23]. OSM can also activate STAT3 and hypoxia-inducible factor-1 alpha (HIF-1α) in estrogen receptor (E.R.)- breast cancer cells or in ER + breast cancer cells in concert with IL-6 [24]. Moreover, other factors enhance breast cancer progression, including several interleukins, such as IL-35 and IL-8. IL35 suppresses conventional T cells (T-conv). It induces of breast cancer progression by activating STAT1 and STAT3 [25]. In contrast, IL-8 and growth-regulated oncogene (GRO) chemokines induce the activation of STAT3 and inflammatory breast cancer progression [25]. Activation of STAT3 has been shown to prevent breast cancer progression by diminishing the expression of IL-17 [26].
There are several other regulators for STAT3, comprised of suppressors, such as microRNA and protein tyrosine phosphatase 2 (PTPN2), as well as activators, such as prostaglandin E2, cyclooxygenase-2 (COX2), SET and MYND (myeloid-Nervy-DEAF-1) domain-containing protein (SMYD2). In recent studies, epigenetics mediators have also been investigated and recognized as STAT3 mediators in the genesis of breast cancer. A class II histone deacetylase mediates upregulated activation of STAT3 in breast cancer, histone deacetylase 6 (HDAC6), as well as prostaglandin E2 and COX2 [27]. Activated STAT3 binds to the COX2/ prostaglandin (PGE)2 gene promoter and leads to the activation of COX-2 and PGE2 expression.
Further investigations have shown that STAT3 methylation or phosphorylation is regulated by lysine methyltransferase SMYD2, resulting in breast cancer progression [28]. Cyclin-dependent kinases 4/6 (CDK4/6) interacts with SMYD2, in turn, leading to the phosphorylation and activation of SMYD2, and resulting in SMYD2 mediating STAT3 methylation.
microRNAs (miR) have also been of great interest in the study of cancer progression and development. Nuclear enriched abundant transcript 1 (NEAT1) has been shown to cause breast cancer progression by a feedback loop with STAT3, and miR-124 has been shown to inhibit NEAT1 [29]. Hosea et al. posited that glucosamine leads to the prevention of STAT3 activation followed by a reduction in breast cancer stemness and progression [30]. Moreover, EGF-modulated STAT3 activation has been noted upon PTPN2 knockdown.
Chronic inflammation is involved in breast cancer progression, and it can be suppressed by STAT3 inhibition [31]. However, stat3 signaling cascades are also regulated by several other modulators. For example, inflammation and progression of breast cancer and the elevation of forming breast cancer stem cells are mediated through the IL-6/STAT3/ROS pathway [32]. Additionally, the FAM3 cytokine family includes an oncogenic member, TGFβ-modulated FAM3C/Interleukin-like EMT Inducer (ILEI), which can lead to the formation of breast cancer stem cells, accelerating the progression of breast cancer [8, 9]. Furthermore, STAT3 has been shown to regulate TNFRSF1A, a gene encoding one of the transmembrane receptors for TNF-α, resulting in the activation of NF-κB signaling in breast cancer [33].
It has also been shown that the progression and the proliferation of breast cancer are affected by several STAT3 co-factors. For example, progranulin (PGRN) has been shown to impart chemoresistance and worsen breast cancer prognosis [34, 35]. In agreement, progranulin antisense oligonucleotide has been shown to suppress STAT3 oncogenesis in CRC cells [36]. The cyclin-dependent kinase 5 (CDK5) regulatory subunit-related protein 3 (CDK5RAP3, also known as C53/LZAP), has been considered as a p53 co-activator [37]. It has been recently shown that CDK5RAP3 is associated with the proliferation and progression of breast cancer, ameliorating STAT3-dependent genes expression [38]. Therefore, targeting the STAT3 co-factors might represent a novel therapeutic modality in the regulation of breast cancer.
STAT3 and metastasis
One of the critical factors in breast cancer metastasis is matrix metallopeptidases (MMPs). One of the most investigated STAT3-modulated mechanisms is via the upregulation of MMP2, MMP9, Twist, Snail, Slug, and vimentin [39,40,41]. It has been demonstrated that vasodilator-stimulated phosphoprotein (VASP), MMP2, and MMP9 expression in breast cancer is diminished upon repression of STAT3 phosphorylation [42]. The binding of cytokines and growth factors with their cognate receptors on the plasma membrane induces the activation of STAT3 signaling. The inhibition of receptor binding by Wwox suppresses metastasis in breast cancer [43]. Additionally, induction of JAK/STAT3 signaling cascade activation, mesoderm-specific transcript (MEST) stimulates Twist expression [44], while inhibition of the JAK/STAT3 and protein kinase B (Akt) cascade, GRAM domain-containing protein 1B (GRAMD1B) prevents breast cancer cell migration [45].
Further, it has been shown that OSM/SMAD3 induces the activation of STAT3 and modulates the expression of Snail. It also induces epithelial-mesenchymal transition (EMT) in breast cancer, instead of erstwhile binding of ligand/receptor in the plasma membrane for the activation of STAT3, alluding to a novel mechanism for the activation of STAT3 via cytoplasmic molecules and endogenous signaling [46]. Other molecules regulate STAT3-modulated breast cancer metastasis, such as proto-oncogene serine/threonine-protein kinase (PIM1), miRNA, Mucin-1-C (MUC1-C), natriuretic peptide receptor A (NPRA), and RhoU. Han et al. have demonstrated that modulating the exogenous signaling cascade, known as Krüppel-like factor 11 (KLF-11), induces the activation of STAT3 by binding to its transmembrane receptor KLF-11R, while miR-30d regulates breast cancer cell migration and invasion [47]. Furthermore, the JAK/STAT3 cascade in breast cancer bone metastasis is modulated by IL-11 [48]. The IL-6/STAT3 signaling cascade modulates PIM1, a proto-oncogene that induces cell invasion and upregulates the expression of EMT in breast cancer [49]. An oncogenic protein, MUC1-C, has also been shown to activate STAT3 and stimulate Twist transactivation, leading to the induction of EMT [50]. Additionally, a natriuretic peptide receptor, NPRA, increases STAT3 and MMP9 expression, resulting in the induction of breast cancer cells migration and invasion [51]. STAT3 also stimulates the expression of high Ras Homolog Family Member U (RhoU) by collaborating with Specificity Protein 1 (SP1), leading to breast cancer cell migration [52].
Moreover, upon STAT3 posttranscriptional changes, several enzymes are effective in breast cancer metastasis. Dai et al. have demonstrated that the induction of STAT3 phosphorylation and the elevation of MMP2 and MMP9 expression in breast cancer cells are stimulated by Rac-specific Rho GTPase-activating protein (Rho GAP), also known as ARHGAP24 [53]. Zhao et al. have shown that the upregulation of p-STAT3, p-AKT, MMP9, and E2F1 expression by a histone acetyltransferase, called GCN5, leading to breast cancer migration and invasion [54].
Hypoxia is a state of stress that is widely investigated in cancer. Hypoxia can stimulate STAT3 activation, causing the promotion of breast cancer stemness and metastasis [55]. These findings afford new approaches for research on STAT3 therapy targets in breast cancer. In addition, it has been recently shown that estrogen-associated receptor alpha may induce triple-negative breast cancer metastasis as a STAT3 target gene [56].
STAT3, proliferation, and apoptosis
Apoptosis has a central role in the pathogenesis of several cancers; therefore, it is necessary to identify the underlying mechanism of apoptosis in cancer [57]. Active STAT3 in tumor cells contributes to the preservation of tumor cells by inhibiting apoptosis [58]. Considerable evidence corroborates the presence of active STAT3 in greater than 95% of cancers, leading to diminished apoptosis [59].
Stat3 protein activation by Src has been shown to be required for cell transformation and promotes cell proliferation.
There is a significant association between high anti-apoptotic proteins such as BCL-2 and Bcl-xL and cancer [60]. IL- 6 mediated Stat3 signaling to induce Bcl-xL gene expression [61]. In addition, over-expression of Bcl-xL protects cells from the anti-apoptotic effect of Stat3 signaling.
Recently, a clinical study indicated that constitutive STAT3 activation caused the expression of Mcl-1 and Bcl-xL, resulted in cancer cell progression and survival [62]. Evidence indicated that Stat3 regulates several genes related to the promotion of cell cycle progression and inhibition of the apoptotic pathway, leading to tumorigenesis [63]. In breast cancer cells, activation of STAT3 via the IL-6/JAK2 cascade causes suppression of Bax/Bcl-2-associated caspase-dependent apoptosis [64]. STAT3 has been shown to upregulate cyclin D-1, c-myc, and bcl-2, leading to the prevention of breast cancer cells apoptosis, suggesting an association between STAT3 in cell cycle and survival [65]. Down-regulation or up-regulation of several mi-RNAs can promote the activation of STAT3 signaling pathways in breast cancer cells. In this regards, it was found that down-regulation of DPF3 [64], miR-125a and let-7e [66], miR-124 [67], miR-9 [68] can activate STAT3 signaling pathway. In contrast, up-regulation of several miRNAs, including miR-93-5p and miR-25-3p, have been shown to induce the proliferation of breast cancer by activating STAT3 [69].
STAT3 and chemoresistance
Studies in triple-negative breast cancer cells have shown that Src/STAT3 signaling cascade is associated with multidrug resistance (MDR) [70]. Castellaro et al. have demonstrated that by activating the NF-κB/STAT3/ERK cascade, an association between breast cancer cells and macrophages stimulates tamoxifen and ICI 182,780 resistance [71]. There are several identified STAT3-modulated chemoresistance downstream targets, such as carnitine palmitoyltransferase 1B (CPT1B), fatty acid beta-oxidation (FAO), mitogen-activated protein kinase (MAPK)/AKT, HIF-1, and octamer-binding transcription factor-4 (Oct-4). Wang et al. have shown increased CPT1B and FAO levels, secondary to activation of the JAK2/STAT3 signaling cascade, leading to breast cancer chemoresistance [72]. It has also been noted that activation of the JAK-STAT3/MAPKs/AKT cascade, IL-22 stimulates the migration and paclitaxel resistance in breast cancer [73]. Furthermore, via the STAT3/hypoxia-inducible factor 1 (HIF-1) cascade target, miR-124 causes the reversion of breast cancer cells doxorubicin (DOX) resistance [74]. Cheng et al. investigated the formation of a signal circuit via Oct-4 and c-myc concerning increased Adriamycin resistance in breast cancer [75]. On the other hand, IL-24 generation in breast cancer cells via STAT3 and NF-B-activation, confers radiation resistance by Oct-4 [76]. Moreover, phosphorylation of STAT3 has been shown to regulate survivin, in turn conferring resistance to paclitaxel, which is extensively used in breast cancer treatment [77].
Several upstream modulators of STAT3-regulated chemoresistance have been recognized. Upon STAT3 activation, the COOH-terminal proline-rich area of 78-kDa glucose-modulated protein (GRP78), has been shown to play a vital role in the progression of tamoxifen-resistant breast cancer cells [78]. Moreover, induction of STAT3 activation caused by leukemia inhibitory factor receptor (LIFR) leads to resistance to Trastuzumab-emtansine (T-DM1) in breast cancer [79]. Feng et al. also showed that miR-4532 promotes resistance to Adriamycin in breast cancer by inhibiting hypermethylated in cancer-1 (HIC-1) and IL-6/STAT3 [80].
On the other hand, several other small molecules mediate chemoresistance regulated by STAT3. For example, Chen et al. have found that via JAK/STAT3 cascade, a combination of Piperlongumine with DOX stimulates apoptosis and suppresses breast cancer cells’ DOX resistance [81]. Furthermore, utilizing the combination of STAT3 suppressor with a poly ADP-ribose polymerase (PARP) suppressor, IL6/STAT3 activity could be targeted, resulting in the successful treatment of palbociclib resistance in breast cancer cells [82].
RES phenotypically influences breast cancer
As we previously mentioned, RES, a natural polyphenolic phytoalexin, exerts anti-inflammatory and anti-oxidative activity. RES administration is associated with anti-breast cancer efficacy. RES has a leading role against breast cancer cell growth at all stages, including initiation, promotion, and progression [83]. In addition, its anti-oxidant and anti-inflammatory activities enable RES to inhibit the development of breast cancer by inducing apoptosis and cell cycle arrest. RES’s estrogenic activity also contributes to its protective effects against breast cancer cell growth modulating estrogen receptor activity [84].
RES was effective against different molecular subtypes of breast cancer, including luminal A [ER+, PR+/-, Her-2-, low Ki 67]; luminal B [ER+, PR±, Her-2+, high Ki 67], E.R. -, Her-2 + and basal [E.R. -, P.R. -, Her-2-, EGFR + and cytokeratin 5/6+, high Ki 67] [85].
RES effects on the upstream and the downstream of STAT3 pathways
In vivo investigations have indicated that RES suppresses tumor growth and stimulates apoptosis by inhibiting STAT3 [86]. In this context, Kotha et al. have shown that RES suppresses the activation of Src tyrosine kinase. In turn, it causes the inhibition of constitutive activation of Stat3 protein in tumorigenesis cells [40]. Further, they reported that malignant cells with constitutively active STAT3, treated with RES, showed irreparable cell cycle arrest at the G0-G1 phase or the S phase of several cancer cell lines, including human breast (MDA-MB-231) cell line, as well as apoptosis, leading to loss of viability [40]. On the other hand, they observed that RES administered to cells that did not include abnormal STAT3 activity, led to reversible growth arrest and a loss of viability. Additionally, RES administration to malignant cells with constitutively induced active STAT3 caused inhibition of STAT3-modulated cyclin D1, and Bcl-xL, and Mcl-1 genes, suggesting that the anti-cancer activity of RES is mediated by blocking STAT3-modulated dysregulation of growth and survival pathways [40].
Yu et al. reported the potential effect of RES in STAT3 signaling cascade in two cancer cell lines [87]. The expression and phosphorylation of STAT3 were observed in regular cultured cell lines, with RES reducing both parameters. Furthermore, the expression of downstream genes of STAT3, including survivin, cyclin D1, Cox-2, and c-Myc, was prevented. concomitantly, Bcl-2 was elevated secondary to RES administration [87]. Moreover, the production and secretion of a STAT3 activator, leukemia suppressive factor, was activated in RES-treated cells. Overall, these findings suggest STAT3 signaling cascade might be a significant target for RES [87] Li et al. have shown that RES can suppress cell proliferation through G1 phase cell cycle arrest and induce cell apoptosis in cancer cells, while not affecting normal cells, indicating that the AKT/STAT3 pathway may represent a novel target for RES [88].
It was also found that RES inhibited proliferation, migration, and invasion of human breast cancer cells exposed to cancer-associated fibroblast-conditioned media (CAF-CM) by suppressing the CAF-CM-induced expression of Cyclin D1, c-Myc, MMP-2 and MMP-9, Sox2 expression and also the activation of Akt and STAT3. Additionally, RES decreased the expression of self-renewal signaling molecules in stem-like breast cancer cells [89].
The endothelial to mesenchymal transition (EndMT) is responsible for cancer metastasis via modulating the complexity of the tumor microenvironment (TME). RES, an inhibitor of STAT3 acetylation, reduced the expression of Ac-STAT3, p-STAT3, and EndMT markers in human umbilical vein endothelial cells (HUVECs) exposed to 27-hydroxycholesterol (27HC). In addition, inhibition of STAT3 signaling by RES prevents Cross-talk between 27HC-induced EndMT in the TME [90].
RES blocks Stat3 activation by inhibiting the Src tyrosine kinase activity in human breast cancer cells, leading to dysregulation of growth and survival pathways [91].
In tumor cells, an increase in the lysine acetylation of the STAT3 induces cell growth. It was indicated that genetically modifying STAT3 at Lys685 decreased tumor growth due to demethylation and reactivation of several tumor-suppressor genes. Treatment with RES decreased acetylated STAT3 in triple-negative breast cancer cells resulted in demethylation and activation of the estrogen receptor-α gene, sensitizing the breast cancer cells to antiestrogens [92]. RES reduced STAT3 acetylation and inhibited tumor suppressor genes (TSG) expression, leading to apoptosis in breast cancer cells. STAT3 bound the Sin3A transcriptional repressor. This complex bound the promoter region of silenced TSG following Res administration into the breast cancer cells [93]. 6-methyl-2-propylimino-6, 7-dihydro-5 H-benzo [1, 3]-oxathiol- 4-one (LYR71), a derivative of trimeric resveratrol, was also effective against breast cancer growth via inhibiting the STAT3-mediated MMP-9 expression [94].
Conclusions
This novel review highlights STAT3 as a critical transcriptional activator in breast cancer, which can mediate breast cancer progression, metastasis, chemoresistance, apoptosis, and proliferation. Several of its upstream regulators and downstream targets are inhibitable by naturally occurring substrates, such as RES. Specifically, we highlighted studies that show the propensity of RES to suppress breast cancer cells proliferation and induce apoptosis via the STAT3 signaling pathway. Taken together, modulation of STAT3 levels should be further explored as a potential novel pharmaceutical modality for the treatment of breast cancer as well as other cancer types.
Availability of data and materials
The authors confirm that the data supporting the findings of this study are available within the article.
References
Takaoka M. Resveratrol, a new phenolic compound, from Veratrum grandiflorum. Nippon Kagaku Kaishi. 1939;60:1090–100.
Borriello A, Bencivenga D, Caldarelli I, Tramontano A, Borgia A, Pirozzi AV, et al. Resveratrol and cancer treatment: is hormesis a yet unsolved matter. Curr Pharm Des. 2013;19(30):5384–93.
Xu Q, Zong L, Chen X, Jiang Z, Nan L, Li J, et al. Resveratrol in the treatment of pancreatic cancer. Ann N Y Acad Sci. 2015;1348(1):10–9.
Singh CK, Ndiaye MA, Ahmad N. Resveratrol and cancer: challenges for clinical translation. Biochimica et Biophysica Acta (BBA) Mol Basis Dis. 2015;1852(6):1178–85.
Ko J-H, Sethi G, Um J-Y, Shanmugam MK, Arfuso F, Kumar AP, et al. The role of resveratrol in cancer therapy. Int J Mol Sci. 2017;18(12):2589.
Rouse M, Egan JM. Resveratrol in aging and age-related diseases. Conn’s Handbook of Models for Human Aging. Amsterdam: Elsevier; 2018. pp. 1133–42.
Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell. 1999;98(3):295–303.
Qiu C, Zhang T, Zhu X, Qiu J, Jiang K, Zhao G, et al. Methylseleninic acid suppresses breast cancer growth via the JAK2/STAT3 pathway. Reprod Sci. 2019;26(6):829–38.
Woosley AN, Dalton AC, Hussey GS, Howley BV, Mohanty BK, Grelet S, et al. TGFβ promotes breast cancer stem cell self-renewal through an ILEI/LIFR signaling axis. Oncogene. 2019;38(20):3794–811.
Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, et al. The human transcription factors. Cell. 2018;172(4):650–65.
Adams CC, Workman JL. Binding of disparate transcriptional activators to nucleosomal DNA is inherently cooperative. Mol Cell Biol. 1995;15(3):1405–21.
Levy DE, Lee C-K. What does Stat3 do? J Clin Investig. 2002;109(9):1143–8.
Furtek SL, Backos DS, Matheson CJ, Reigan P. Strategies and approaches of targeting STAT3 for cancer treatment. ACS Chem Biol. 2016;11(2):308–18.
Mitchell TJ, John S. Signal transducer and activator of transcription (STAT) signalling and T-cell lymphomas. Immunology. 2005;114(3):301–12.
Siveen KS, Sikka S, Surana R, Dai X, Zhang J, Kumar AP, et al. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochimica et Biophysica Acta (BBA) Rev cancer. 2014;1845(2):136–54.
Darnell JE, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264(5164):1415–21.
Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14(11):736–46.
Akira S. Roles of STAT3 defined by tissue-specific gene targeting. Oncogene. 2000;19(21):2607–11.
Boskabady MH, Karimi GR, Samarghandian S, Farkhondeh T. Tracheal responsiveness to methacholine and ovalbumin; and lung inflammation in guinea pigs exposed to inhaled lead after sensitization. Ecotoxicol Environ Saf. 2012;86:233–8.
Farkhondeh T, Samarghandian S. Antidotal effects of curcumin against agents-induced cardiovascular toxicity. Cardiovasc Hematol Disord Drug Targets. 2016;16(1):30–7.
Farkhondeh T, Samarghandian S, Pourbagher-Shahri AM, Sedaghat M. The impact of curcumin and its modified formulations on Alzheimer's disease. J Cell Physiol. 2019;234(10):16953–65
Hashemi V, Masjedi A, Hazhir-karzar B, Tanomand A, Shotorbani SS, Hojjat‐Farsangi M, et al. The role of DEAD‐box RNA helicase p68 (DDX5) in the development and treatment of breast cancer. J Cell Physiol. 2019;234(5):5478–87.
Tawara K, Scott H, Emathinger J, Wolf C, LaJoie D, Hedeen D, et al. HIGH expression of OSM and IL-6 are associated with decreased breast cancer survival: synergistic induction of IL-6 secretion by OSM and IL-1β. Oncotarget. 2019;10(21):2068.
Tawara K, Scott H, Emathinger J, Ide A, Fox R, Greiner D, et al. Co-expression of VEGF and IL-6 family cytokines is associated with decreased survival in HER2 negative breast cancer patients: subtype-specific IL-6 family cytokine-mediated VEGF secretion. Transl Oncol. 2019;12(2):245–55.
Valeta-Magara A, Gadi A, Volta V, Walters B, Arju R, Giashuddin S, et al. Inflammatory breast cancer promotes development of M2 tumor-associated macrophages and cancer mesenchymal cells through a complex chemokine network. Cancer Res. 2019;79(13):3360–71.
Samarghandian S, Azimi-Nezhad M, Farkhondeh T. Thymoquinone-induced antitumor and apoptosis in human lung adenocarcinoma cells. J Cell Physiol. 2019;234(7):10421–31.
Li A, Chen P, Leng Y, Kang J. Histone deacetylase 6 regulates the immunosuppressive properties of cancer-associated fibroblasts in breast cancer through the STAT3–COX2-dependent pathway. Oncogene. 2018;37(45):5952–66.
Li LX, Zhou JX, Calvet JP, Godwin AK, Jensen RA, Li X. Lysine methyltransferase SMYD2 promotes triple negative breast cancer progression. Cell Death Dis. 2018;9(3):1–17.
Pang Y, Wu J, Li X, Wang C, Wang M, Liu J, et al. NEAT1/miR–124/STAT3 feedback loop promotes breast cancer progression. Int J Oncol. 2019;55(3):745–54.
Hosea R, Hardiany NS, Ohneda O, Wanandi SI. Glucosamine decreases the stemness of human ALDH + breast cancer stem cells by inactivating STAT3. Oncol Lett. 2018;16(4):4737–44.
Sun Y, Li X, Zhang L, Liu X, Jiang B, Long Z, et al. Cell permeable NBD peptide-modified liposomes by hyaluronic acid coating for the synergistic targeted therapy of metastatic inflammatory breast cancer. Mol Pharm. 2019;16(3):1140–55.
Kim S-L, Choi HS, Kim J-H, Jeong DK, Kim K-S, Lee D-S. Dihydrotanshinone-induced NOX5 activation inhibits breast cancer stem cell through the ROS/Stat3 signaling pathway. Oxid Med Cell Longev. 2019;2019:9296439.
Egusquiaguirre SP, Yeh JE, Walker SR, Liu S, Frank DA. The STAT3 target gene TNFRSF1A modulates the NF-κB pathway in breast cancer cells. Neoplasia. 2018;20(5):489–98.
Abrhale T, Brodie A, Sabnis G, Macedo L, Tian C, Yue B, et al. GP88 (PC-cell derived growth factor, progranulin) stimulates proliferation and confers letrozole resistance to aromatase overexpressing breast cancer cells. BMC Cancer. 2011;11(1):1–10.
Wang W, Hayashi J, Serrero G. PC cell–derived growth factor confers resistance to dexamethasone and promotes tumorigenesis in human multiple myeloma. Clin Cancer Res. 2006;12(1):49–56.
Laudisi F, Cherubini F, Di Grazia A, Dinallo V, Di Fusco D, Franzè E, et al. Progranulin sustains STAT 3 hyper-activation and oncogenic function in colorectal cancer cells. Mol Oncol. 2019;13(10):2142–59.
Wang J-b, Wang Z-w, Li Y, Huang C-q, Zheng C-h, Li P, et al. CDK5RAP3 acts as a tumor suppressor in gastric cancer through inhibition of β-catenin signaling. Cancer Lett. 2017;385:188–97.
Egusquiaguirre SP, Liu S, Tošić I, Jiang K, Walker SR, Nicolais M, et al. CDK5RAP3 is a co-factor for the oncogenic transcription factor STAT3. Neoplasia. 2020;22(1):47–59.
Zhang F, Yin G, Han X, Jiang X, Bao Z. Chlorogenic acid inhibits osteosarcoma carcinogenesis via suppressing the STAT3/Snail pathway. J Cell Biochem. 2019;120(6):10342–50.
Kamran MZ, Patil P, Gude RP. Role of STAT3 in cancer metastasis and translational advances. BioMed Res Int. 2013;2013:421821.
Li Z, Chen Y, An T, Liu P, Zhu J, Yang H, et al. Nuciferine inhibits the progression of glioblastoma by suppressing the SOX2-AKT/STAT3-slug signaling pathway. J Exp Clin Cancer Re. 2019;38(1):1–15.
Ma Q, Gao FF, He X, Li K, Gao Y, Xu XL, et al. Antitumor effects of saikosaponin b2 on breast cancer cell proliferation and migration. Mol Med Rep. 2019;20(2):1943–51.
Chang R, Song L, Xu Y, Wu Y, Dai C, Wang X, et al. Loss of Wwox drives metastasis in triple-negative breast cancer by JAK2/STAT3 axis. Nat Commun. 2018;9(1):1–12.
Kim MS, Lee HS, Kim YJ, Lee DY, Kang SG, Jin W. MEST induces Twist-1-mediated EMT through STAT3 activation in breast cancers. Cell Death Differ. 2019;26(12):2594–606.
Khanna P, Lee JS, Sereemaspun A, Lee H, Baeg GH. GRAMD1B regulates cell migration in breast cancer cells through JAK/STAT and Akt signaling. Sci Rep. 2018;8(1):1–10.
Doherty MR, Parvani JG, Tamagno I, Junk DJ, Bryson BL, Cheon HJ, et al. The opposing effects of interferon-beta and oncostatin-M as regulators of cancer stem cell plasticity in triple-negative breast cancer. Breast Cancer Res. 2019;21(1):1–12.
Han M, Wang Y, Guo G, Li L, Dou D, Ge X, et al. MicroRNA-30d mediated breast cancer invasion, migration, and EMT by targeting KLF11 and activating STAT3 pathway. J Cell Biochem. 2018;119(10):8138–45.
Liang M, Ma Q, Ding N, Luo F, Bai Y, Kang F, et al. IL-11 is essential in promoting osteolysis in breast cancer bone metastasis via RANKL-independent activation of osteoclastogenesis. Cell Death Dis. 2019;10(5):1–12.
Gao X, Liu X, Lu Y, Wang Y, Cao W, Liu X, et al. PIM1 is responsible for IL-6-induced breast cancer cell EMT and stemness via c-myc activation. Breast Cancer. 2019;26(5):663–71.
Hata T, Rajabi H, Yamamoto M, Jin C, Ahmad R, Zhang Y, et al. Targeting MUC1-C inhibits TWIST1 signaling in triple-negative breast cancer. Mol Cancer Ther. 2019;18(10):1744–54.
Qu J, Zhao X, Liu X, Sun Y, Wang J, Liu L, et al. Natriuretic peptide receptor a promotes breast cancer development by upregulating MMP9. Am J Cancer Res. 2019;9(7):1415.
Monteleone E, Orecchia V, Corrieri P, Schiavone D, Avalle L, Moiso E, et al. SP1 and STAT3 functionally synergize to induce the RhoU small GTPase and a subclass of non-canonical WNT responsive genes correlating with poor prognosis in breast cancer. Cancers. 2019;11(1):101.
Dai X, Geng F, Dai J, Li M, Liu M. Rho GTPase activating protein 24 (ARHGAP24) regulates the anti-cancer activity of sorafenib against breast cancer MDA-MB-231 cells via the Signal Transducer and Activator of Transcription 3 (STAT3) signaling pathway. Med Sci Monit. 2018;24:8669.
Zhao L, Pang A, Li Y. Function of GCN5 in the TGF–β1–induced epithelial–to–mesenchymal transition in breast cancer. Oncol Lett. 2018;16(3):3955–63.
Abyaneh HS, Gupta N, Alshareef A, Gopal K, Lavasanifar A, Lai R. Hypoxia induces the acquisition of cancer stem-like phenotype via upregulation and activation of signal transducer and activator of transcription-3 (STAT3) in MDA-MB-231, a triple negative breast cancer cell line. Cancer Microenviron. 2018;11(2):141–52.
Ma J-H, Qi J, Lin S-Q, Zhang C-Y, Liu F-y, Xie W-D, et al. STAT3 targets ERR-α to promote epithelial–mesenchymal transition, migration, and invasion in triple-negative breast cancer cells. Mol Cancer Res. 2019;17(11):2184–95.
Bold RJ, Termuhlen PM, McConkey DJ. Apoptosis, cancer and cancer therapy. Surg Oncol. 1997;6(3):133–42.
Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19(21):2474–88.
Pencik J, Pham HTT, Schmoellerl J, Javaheri T, Schlederer M, Culig Z, et al. JAK-STAT signaling in cancer: from cytokines to non-coding genome. Cytokine. 2016;87:26–36.
Hata AN, Engelman JA, Faber AC. The BCL2 family: key mediators of the apoptotic response to targeted anticancer therapeutics. Cancer Discov. 2015;5(5):475–87.
Catlett-Falcone R, Dalton WS, Jove R. STAT proteins as novel targets for cancer therapy. Curr Opin Oncol. 1999;11(6):490.
Xie T-x, Wei D, Liu M, Gao AC, Ali-Osman F, Sawaya R, et al. Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene. 2004;23(20):3550–60.
Fathi N, Rashidi G, Khodadadi A, Shahi S, Sharifi S. STAT3 and apoptosis challenges in cancer. Int J Biol Macromol. 2018;117:993–1001.
Xie Q, Yang Z, Huang X, Zhang Z, Li J, Ju J, et al. Ilamycin C induces apoptosis and inhibits migration and invasion in triple-negative breast cancer by suppressing IL-6/STAT3 pathway. J Hematol Oncol. 2019;12(1):1–14.
Chun J, Song K, Kim YS. Sesquiterpene lactones-enriched fraction of Inula helenium L. induces apoptosis through inhibition of signal transducers and activators of transcription 3 signaling pathway in MDA‐MB‐231 breast cancer cells. Phytother Res. 2018;32(12):2501–9.
Park Y, Kim J. Regulation of IL-6 signaling by miR-125a and let-7e in endothelial cells controls vasculogenic mimicry formation of breast cancer cells. BMB Rep. 2019;52(3):214.
Shi P, Chen C, Li X, Wei Z, Liu Z, Liu Y. MicroRNA–124 suppresses cell proliferation and invasion of triple negative breast cancer cells by targeting STAT3. Mol Med Rep. 2019;19(5):3667–75.
Zhang Y, Dai G. Overexpression of MicroRNA-9 inhibits proliferation of human breast cancer cells by targeting STAT3. Trop J Pharm Res. 2018;17(9):1753–8.
Li JP, Xiang Y, Fan LJ, Yao A, Li H, Liao XH. Long noncoding RNA H19 competitively binds miR-93-5p to regulate STAT3 expression in breast cancer. J Cell Biochem. 2019;120(3):3137–48.
Tzeng Y-DT, Liu P-F, Li J-Y, Liu L-F, Kuo S-Y, Hsieh C-W, et al. Kinome-wide siRNA screening identifies Src-enhanced resistance of chemotherapeutic drugs in triple-negative breast cancer cells. Front Pharmacol. 2018;9:1285.
Castellaro AM, Rodriguez-Baili MC, Di Tada CE, Gil GA. Tumor-associated macrophages induce endocrine therapy resistance in ER + breast cancer cells. Cancers. 2019;11(2):189.
Wang T, Fahrmann JF, Lee H, Li Y-J, Tripathi SC, Yue C, et al. JAK/STAT3-regulated fatty acid β-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metabol. 2018;27(1):136–50.
Wang S, Yao Y, Yao M, Fu P, Wang W. Interleukin-22 promotes triple negative breast cancer cells migration and paclitaxel resistance through JAK-STAT3/MAPKs/AKT signaling pathways. Biochem Biophys Res Commun. 2018;503(3):1605–9.
Liu C, Xing H, Guo C, Yang Z, Wang Y, Wang Y. MiR-124 reversed the doxorubicin resistance of breast cancer stem cells through STAT3/HIF-1 signaling pathways. Cell Cycle. 2019;18(18):2215–27.
Cheng C-C, Shi L-H, Wang X-J, Wang S-X, Wan X-Q, Liu S-R, et al. Stat3/Oct-4/c-Myc signal circuit for regulating stemness-mediated doxorubicin resistance of triple-negative breast cancer cells and inhibitory effects of WP1066. Int J Oncol. 2018;53(1):339–48.
Kim J-Y, Kim J-C, Lee J-Y, Park M-J. Oct4 suppresses IR–induced premature senescence in breast cancer cells through STAT3-and NF–κB-mediated IL–24 production. Int J Oncol. 2018;53(1):47–58.
Xiang S, Dauchy RT, Hoffman AE, Pointer D, Frasch T, Blask DE, et al. Epigenetic inhibition of the tumor suppressor ARHI by light at night-induced circadian melatonin disruption mediates STAT3‐driven paclitaxel resistance in breast cancer. J Pineal Res. 2019;67(2):e12586.
Tseng C-C, Zhang P, Lee AS. The COOH-terminal proline-rich region of GRP78 is a key regulator of its cell surface expression and viability of tamoxifen-resistant breast cancer cells. Neoplasia. 2019;21(8):837–48.
Wang L, Wang Q, Gao M, Fu L, Li Y, Quan H, et al. STAT 3 activation confers trastuzumab-emtansine (T‐DM 1) resistance in HER 2‐positive breast cancer. Cancer Sci. 2018;109(10):3305–15.
Feng F, Zhu X, Wang C, Chen L, Cao W, Liu Y, et al. Downregulation of hypermethylated in cancer-1 by miR-4532 promotes adriamycin resistance in breast cancer cells. Cancer Cell Int. 2018;18(1):1–12.
Chen D, Ma Y, Li P, Liu M, Fang Y, Zhang J, et al. Piperlongumine induces apoptosis and synergizes with doxorubicin by inhibiting the JAK2-STAT3 pathway in triple-negative breast cancer. Molecules. 2019;24(12):2338.
Kettner NM, Vijayaraghavan S, Durak MG, Bui T, Kohansal M, Ha MJ, et al. Combined inhibition of STAT3 and DNA repair in palbociclib-resistant ER-positive breast cancer. Clin Cancer Res. 2019;25(13):3996–4013.
Le Corre L, Chalabi N, Delort L, Bignon YJ, Bernard-Gallon DJ. Resveratrol and breast cancer chemoprevention: molecular mechanisms. Mol Nutr Food Res. 2005;49(5):462–71.
Basly J-P, Marre-Fournier F, Le Bail J-C, Habrioux G, Chulia AJ. Estrogenic/antiestrogenic and scavenging properties of (E)-and (Z)-resveratrol. Life Sci. 2000;66(9):769–77.
Sinha D, Sarkar N, Biswas J, Bishayee A, editors Resveratrol for breast cancer prevention and therapy: preclinical evidence and molecular mechanisms; Amsterdam: Elsevier; 2016.
Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CWW, et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science. 1997;275(5297):218–20.
Yu L-J, Wu M-L, Li H, Chen X-Y, Wang Q, Sun Y, et al. Inhibition of STAT3 expression and signaling in resveratrol-differentiated medulloblastoma cells. Neoplasia. 2008;10(7):736–44.
Li D, Wang G, Jin G, Yao K, Zhao Z, Bie L, et al. Resveratrol suppresses colon cancer growth by targeting the AKT/STAT3 signaling pathway. Int J Mol Med. 2019;43(1):630–40.
Suh J, Kim D-H, Surh Y-J. Resveratrol suppresses migration, invasion and stemness of human breast cancer cells by interfering with tumor-stromal cross-talk. Arch Biochem Biophys. 2018;643:62–71.
Jiao K, Zhen J, Wu M, Teng M, Yang K, Zhou Q, et al. 27-Hydroxycholesterol-induced EndMT acts via STAT3 signaling to promote breast cancer cell migration by altering the tumor microenvironment. Cancer Biol Med. 2020;17(1):88.
Kotha A, Sekharam M, Cilenti L, Siddiquee K, Khaled A, Zervos AS, et al. Resveratrol inhibits Src and Stat3 signaling and induces the apoptosis of malignant cells containing activated Stat3 protein. Mol Cancer Ther. 2006;5(3):621–9.
Lee H, Zhang P, Herrmann A, Yang C, Xin H, Wang Z, et al. Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc Natl Acad Sci. 2012;109(20):7765–9.
Gambi G, Di Simone E, Basso V, Ricci L, Wang R, Verma A, et al. The transcriptional regulator Sin3A contributes to the oncogenic potential of STAT3. Cancer Res. 2019;79(12):3076–87.
Kim JE, Kim HS, Shin Y-J, Lee CS, Won C, Lee S-A, et al. LYR71, a derivative of trimeric resveratrol, inhibits tumorigenesis by blocking STAT3-mediated matrix metalloproteinase 9 expression. Exp Mol Med. 2008;40(5):514–22.
Acknowledgements
None.
Funding
This research did not receive funding.
Author information
Authors and Affiliations
Contributions
ZK and SS were involved in the conceptualization, validation of resources, and data extraction. ZK, TF, AMPS, and SS wrote the manuscript, SS and MA conceptualized, reviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kohandel, Z., Farkhondeh, T., Aschner, M. et al. STAT3 pathway as a molecular target for resveratrol in breast cancer treatment. Cancer Cell Int 21, 468 (2021). https://doi.org/10.1186/s12935-021-02179-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-021-02179-1