
Abstract
Background
Glioblastoma is the most common primary malignant brain tumor. Because of the limited understanding of its pathogenesis, the prognosis of glioblastoma remains poor. This study was conducted to explore potential competing endogenous RNA (ceRNA) network chains and biomarkers in glioblastoma by performing integrated bioinformatics analysis.
Methods
Transcriptome expression data from The Cancer Genome Atlas database and Gene Expression Omnibus were analyzed to identify differentially expressed genes between glioblastoma and normal tissues. Biological pathways potentially associated with the differentially expressed genes were explored by Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway analysis, and a protein-protein interaction network was established using the STRING database and Cytoscape. Survival analysis using Gene Expression Profiling Interactive Analysis was based on the Kaplan–Meier curve method. A ceRNA network chain was established using the intersection method to align data from four databases (miRTarBase, miRcode, TargetScan, and lncBace2.0), and expression differences and correlations were verified by quantitative reverse-transcription polymerase chain reaction analysis and by determining the Pearson correlation coefficient. Additionally, an MTS assay and the wound-healing and transwell assays were performed to evaluate the effects of complement C1s (C1S) on the viability and migration and invasion abilities of glioblastoma cells, respectively.
Results
We detected 2842 differentially expressed (DE) mRNAs, 2577 DE long non-coding RNAs (lncRNAs), and 309 DE microRNAs (miRNAs) that were dysregulated in glioblastoma. The final ceRNA network consisted of six specific lncRNAs, four miRNAs, and four mRNAs. Among them, four DE mRNAs and one DE lncRNA were correlated with overall survival (p < 0.05). C1S was significantly correlated with overall survival (p= 0.015). In functional assays, knockdown of C1S inhibited the proliferation and invasion of glioblastoma cell lines.
Conclusions
We established four ceRNA networks that may influence the occurrence and development of glioblastoma. Among them, the MIR155HG/has-miR-129-5p/C1S axis is a potential marker and therapeutic target for glioblastoma. Knockdown of C1S inhibited the proliferation, migration, and invasion of glioblastoma cells. These findings clarify the role of the ceRNA regulatory network in glioblastoma and provide a foundation for further research.
Similar content being viewed by others


Background
Glioblastoma (GBM) is a common malignant primary brain tumor, accounting for approximately 57 % of all gliomas and 48 % of all primary malignant central nervous system tumors[1]. The three standard modes of treatment (maximal surgical resection, radiotherapy, and chemotherapy) result in an average survival of only 14 months after diagnosis[2, 3]; without treatment, the life expectancy is less than 6 months[4]. The pathogenesis, development, reproduction, and molecular mechanisms of GBM are not well-understood; this hinders the research and development of precise treatments for this condition[5]. Therefore, studies of the molecular mechanisms underlying the action of GBM and new therapeutic strategies are urgently needed.
High-throughput sequencing technologies are powerful tools for identifying potential biomarkers useful for the diagnosis and treatment of cancer. Integrating bioinformatics knowledge with transcriptome sequencing can make data analysis and differential gene identification faster and more accurate[6, 7]. Consequently, studies aimed at applying bioinformatics strategies to analyze molecular markers and diagnostic indicators of GBM are urgently needed.
The competing endogenous RNA (ceRNA) hypothesis proposed in 2011 described an intricate post-transcriptional regulatory network that mainly includes long non-coding RNAs (lncRNAs), microRNAs (miRNAs), mRNAs, circular RNAs (circRNAs), and other types of RNAs[8]. Recent studies have focused on roles of ceRNAs in the tumorigenesis and development of various cancers[9, 10], including lung cancer[10], gallbladder cancer[10], glioblastoma[11], gastric cancer[12], pancreatic cancer[13], and colorectal cancer[14]. The molecular mechanisms underlying the role of the ceRNA regulatory network in the oncogenesis and development of various cancers have also been examined[15, 16]. However, to date, few ceRNA networks for GBM have been constructed using bioinformatics analysis.
To improve the understanding of ceRNA-based regulatory mechanisms in GBM, we constructed an aberrant lncRNA-miRNA-mRNA network using a comprehensive bioinformatics approach. The expression of crucial RNAs in clinical specimens was analyzed by real-time quantitative reverse-transcription polymerase chain reaction (qRT-PCR). Additionally, the biological functions of crucial RNAs were evaluated in vitro. Our study may facilitate comprehensive analysis of essential molecules, aid in the identification of novel mechanisms underlying glioblastoma pathogenesis, and reveal potential therapeutic targets for treating GBM.
Methods
Microarray data
Three expression profile datasets [GSE90604 (GPL17692 platform), GSE65626 (GPL17586 platform), and GSE116520 (GPL10558 platform)] and two microRNA expression profile datasets [GSE65626 (GPL19117 platform) and GSE90604 (GPL21572 platform)] were downloaded from the National Center of Biotechnology Information Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/) database. The lncRNA transcriptome data for GBM were downloaded from The Cancer Genome Atlas (TCGA; https://cancergenome.nih.gov/) database; these data included 5 cases of para-cancerous tissues and 156 cases of tumor tissues. The GSE90604 dataset included 16 GBM tissue samples and 7 healthy brain tissue samples; GSE65626 included 6 GBM and 6 healthy brain tissue samples; and GSE116520 included 17 GBM tumor core tissue samples, 17 GBM peritumoral brain zone tissue samples, and 17 healthy brain tissue samples. Two GEO datasets (GSE65626, GSE90604) with miRNA and mRNA simultaneously detected by Affymetrix microarray were included to reduce variability between samples. Data were normalized using limma software and the edgeR package[17].
Identification of differentially expressed genes (DEGs)
The three expression profiling datasets used for analysis included CEL format files. These datasets of differential RNA expression in GBM were compared with data from normal brain tissue using the limma software package in R. To establish statistical significance, the GSE90604 (miRNA and mRNA) and TCGA datasets were filtered using false discovery rate (FDR) < 0.01 and |fold-change (FC)| > 1 as cutoffs. GSE65626 (miRNA and mRNA) was filtered using cutoffs of p < 0.05 and |FC| > 1. GSE116520 (mRNA) was filtered using cutoffs of FDR < 0.01 and |FC| > 1.5. A Venn diagram was constructed using Venny 2.1.
Gene ontology (GO) and pathway enrichment analysis
GO is widely used in bioinformatics to mainly study three aspects of biology: biological processes (BP), molecular functions (MF), and cellular components (CC). The Kyoto Encyclopedia of Genes and Genomes (KEGG) reflects biological molecular interactions and chemical reactions. We used the Database for Annotation, Visualization, and Integrated Discovery (DAVID) to analyze the DEGs. Values of p< 0.05 and gene counts ≥ 8 were considered as statistically significant.
Protein‐protein interaction (PPI) network and gene module analysis
First, the Search Tool for the Retrieval of Interacting Genes (STRING) database was used to obtain DEG-encoded protein and PPI information. Second, PPI pairs with a combined score > 0.4 were downloaded and analyzed. The PPI networks were constructed using Cytoscape software. The plug-in Molecular Complex Detection (MCODE) was used to screen modules or clusters in large PPI networks. The following parameters were used for DEG clustering and scoring: MCODE score > 5, degree cutoff = 2, node score cutoff = 0.2, max depth = 100, and k-score = 2.
Survival analysis of DEGs
Gene Expression Profiling Interactive Analysis (GEPIA), a web server for cancer and normal gene expression profiling and interactive analysis, was used to perform survival analysis of the DEGs, confirm their expression, and identify the median expression among tumor and normal samples in BodyMap[18].
Prediction of target lncRNA-miRNA genes and initial lncRNA-miRNA-mRNA network construction
The initial lncRNA-miRNA-mRNA network was constructed according to the ceRNA hypothesis. miRcode, the miRTarBase database, and TargetScan were used to predict target differentially expressed (DE) mRNAs, and only miRNA-mRNAs that were aligned across the three databases were incorporated into the ceRNA network. Next, DE lncRNA-DE miRNA interactions were predicted based on the miRcode database. Cytoscape (Version 3.7.1) was used to construct the preliminary ceRNA network.
Gene expression and final ceRNA network construction
One-way analysis of variance was used to analyze the DEGs and DE lncRNAs. The cutoffs for the gene expression boxplot were set to |Log2FC| = 1 and p = 0.01. lncBace2.0 and GEPIA (http://gepia.cancer-pku.cn/index.html) were used to validate the DE lncRNAs and screen for genes showing significant expression, respectively. A Ji mulberry was used to construct the final lncRNA-miRNA-mRNA network. The flowchart for the simplified procedure is shown in Fig. 1.

Flow chart of ceRNA network and control axis construction in glioblastoma
Patient samples
The 37 primary glioma samples were obtained from patients who underwent surgical treatment at the Xiangya Hospital of Central South University from September 2016 to May 2017 but were not administered chemotherapy or radiotherapy. Six normal brain tissues from patients with cerebral trauma surgery were collected as controls. All samples were snap frozen in liquid nitrogen until total RNA was extracted. This study was approved by the Central South University Xiangya Hospital Medical Ethics Committee (No. 201,803,806).
Cell culture
The U87 and T98G cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA). The cells were cultured in Dulbecco’s modified Eagle’s medium (HyClone, Logan, UT, USA) growth medium supplemented with 10 % fetal bovine serum (Gibco, Grand Island, NY, USA) at 37 °C in a humidified 5 % CO2 incubator.
Cell transfection
The U87 and T98G cells were seeded into six-well plates and cultured until they reached 50–60 % confluence. They were then transiently transfected with small-interfering RNA (siRNA-C1S) and negative control (NC) siRNA, which were designed by RiboBio (Guangzhou, China), using Lipofectamine® RNAiMAX Transfection Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. This transfection reagent and siRNA were diluted in Opti-MEM (Invitrogen) without antibiotics and used for experiments at 48 h after transfection.
RNA isolation and qRT-PCR
Total RNA was extracted from tissues using TRIzol reagent (TaKaRa, Shiga, Japan) according to the manufacturer’s protocol. cDNA was transcribed using the PrimeScript RT Reagent Kit (TaKaRa, RR047A). A SYBR Green PCR Kit (Takara, RR820A) was used to measure the quantity of isolated RNA according to the manufacturer’s instructions. qRT-PCR was performed using the CFX Connect Real-Time System (Bio-Rad, Hercules, CA, USA) as follows: 95 °C for 3 min, and 40 cycles of 95 °C for 10 s and 60 °C for 30 s. GAPDH was used as an internal control. The 2−ΔΔCt method was used to calculate the relative expression levels of the RNAs. The qRT-PCR primers included the bulge-loop RT primer and qPCR primers specific for has-miR-129-5p that were designed and synthesized by RiboBio. The primers for lncRNA MIR155HG were F: 5′-CCACCCAATGGAGATGGCTCT A-3′; and R: 5′- GCAAAAACCCCTATCACGATTA-3′. The primers for mRNA C1S were F: 5′- TCCAAGTCCCATACAACAAACTC-3′, and R: 5′-CAAACCCCGTAA AACGCTCT-3′. GAPDH: F: 5′-CCCATCACCATCTTCCAGGAG-3′, R: 5′-GTTGTCATGGATGACCTTGGC-3′.
Cell proliferation assay
Cell viability was measured in an MTS assay (Cell Titer 96 aqueous one solution reagent, Promega, Madison, WI, USA) according to the manufacturer’s instructions. Briefly, at 24 h before the experiment, cells (2 × 103 cells) were seeded into 96-well plates, divided into two groups (NC, si-C1S-3), and cultured for 0, 24, 48, 72, 96 h. At the specified time, 10 µL of CellTiter 96 Aqueous One Solution Reagent was added to each well and incubated at 37 °C for 30 min. The quantity of formazan product was determined by measuring the absorbance at 490 nm with a microplate reader (Bio-Rad). All experiments were repeated three times.
Wound healing assay
The migration abilities of U87 and T98G cells were assessed via a wound-healing assay. When cells grown on the plate reached a density of approximately 90 %, an artificial wound was created by scraping the confluent monolayer with a 200-µL plastic pipette tip. Cell migration was monitored under a microscope at 0 and 24 h, and images were taken immediately. The average wound closure rate was measured in triplicate and the results were evaluated with ImageJ software (NIH, Bethesda, MD, USA).
Cell migration and invasion assays
Cell migration and invasion were evaluated using 24-well transwell chambers (Corning, Inc., Corning, NY, USA) coated with Matrigel or not. Briefly, transfected U87 and T98G cells (2 × 104) were seeded into the upper chambers containing serum-free medium, whereas medium containing 20 % fetal bovine serum was added to the lower chambers. After incubation for 24 h (migration assays) and 48 h (invasion assays) at 37 °C, the cells were fixed with 4 % formalin and stained with 0.1 % crystal violet. The cell invasion assay was performed in the same manner, but the upper chamber was covered with Matrigel. Cells were counted under a light microscope in randomly selected fields and analyzed using ImageJ software.
Statistical analyses
Statistical analyses were conducted using R (version 3.6.1, The R Foundation, Vienna, Austria) and SPSS (version 24.0, SPSS, Inc., Chicago, IL, USA). Statistical analysis involved Student’s t -test, Mann-Whitney U test, analysis of variance, or two-way repeated measures analysis of variance with post-hoc Student’s t-test or paired Student’s t-test. Differences with p values < 0.05 were considered as statistically significant.
Results
Identification of DEGs and differentially expressed mRNAs (DEMs)
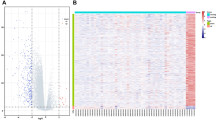
We evaluated data from 35 patients with GBM and 18 healthy controls. GSE90604, GSE65626, and GSE116520 were analyzed using RStudio software. We identified 163 DEGs; 82 were upregulated and 81 were downregulated (Table 1; Fig. 2a–d). Among the DEMs, we identified 49 microRNAs, including 29 and 20 up- and downregulated miRNAs, respectively (Table 2; Fig. 2e). The top 10 upregulated genes were TM1P1, COL4A1, TNC, CA12, PLAU, CKS2, TMEM45A, PLOD2, NCAPG, and SERPINH1. The top 10 downregulated genes were CAMK2A, OPALIN, AK5, GRIN1, SLC17A7, SH3GL3, MOBP, VSNL1, UNC13C, and SYN1.

Expression profiles of DEGs and DEMs. a–c Heat map and volcano map of DEG expression levels in the a GSE65626 dataset, b GSE116520 dataset and c GSE116520 dataset. d Venn diagram showing DEGs in the three datasets. e Venn diagram showing DEMs in the two microRNA datasets. The red nodes represent upregulated DEGs with FDR < 0.01 and logFC > 1 or > 1.5; the green nodes represent downregulated DEGs with FDR < 0.01 and logFC < − 1.0 or < − 1.5. DEGs differentially expressed genes. DEMs differentially expressed microRNAs
Functional and signal pathway enrichment analysis
We uploaded the selected DEGs to the online website DAVID to identify the enriched GO terms and KEGG pathways. The DEGs were classified as BP, CC, or MF (Fig. 3a). The first two significant functions were selected for analysis (Fig. 3b). As shown in Table 3, GO analysis showed that the DEGs were most significantly enriched in the transmission of nerve impulses. Moreover, upregulated DEGs were significantly enriched in the extracellular matrix (ECM) receptor interaction pathway and basement membrane, and particularly in protein binding (Fig. 3c). Downregulated DEGs were enriched in functions such as transmission of nerve impulse, synapse, and synapse part (Fig. 3d). The results of KEGG pathway analysis indicated enrichment in adrenergic signaling in cardiomyocytes (Fig. 3e).

Gene Ontology and Kyoto Encyclopedia of Genes and Genomes enrichment analysis of DEGs. GO enrichment analysis of DEGs. a DEGs were classified as BP, CC, or MF. b, c Ranking of significantly enriched GO terms for upregulated DEGs. d, e Ranking of significantly enriched GO terms for downregulated DEGs. GO Gene Ontology, BP biological process, CC cellular component, MF molecular function, DEGs differentially expressed genes
PPI analysis and gene module analysis
A total of 132 DEGs was screened using the STRING online database and Cytoscape software. Sixty-seven upregulated genes and sixty-five downregulated genes were included in the DEG PPI network (Fig. 4a); 30 genes were excluded from the PPI network (132 nodes and 484 edges). After filtering with a cutoff of node degree ≥ 6, the top 10 hub genes were SYN1, SYT1, SNAP25, SYN2, SLC17A7, GRIN1, ATP2B2, DLG2, CAMK2A, and SNAP91. Using Cytoscape’s MCODE plug-in, module 1 (score = 9.071), consisting of 29 nodes and 127 edges (Fig. 4b), was selected as an important module in the PPI network. In the functional module, the functions of important genes were annotated and analyzed. Functional enrichment analysis showed that module genes were mainly involved in the synaptic vesicle cycle (Fig. 4c).

PPI network of DEGs. Node color: red indicates upregulated genes, sky-blue indicates downregulated genes. a PPI network based on the STRING online database contained 132 nodes and 484 edges. b Most significant module identified by MCODE (score = 9.071). c GO enrichment analysis of modules. PPI protein-protein interaction, DEGs differentially expressed genes, GO Gene Ontology
Prognostic significance of DEGs
The GEPIA database was used to analyze the overall survival (OS) rates of patients with GBM at 20, 40, 60, and 80 months, as well as the correlation between the DEGs and survival prognosis of patients with GBM. The results showed that 12 DEGs (C1S, CFI, DCBLD2, FAM20C, FNDC3B, IFI30, KDELR2, RCAN2, PLP2, SERPINH1, STEAP3, and TNFRSF1A) were significantly correlated with the OS rate of patients with GBM (p < 0.05) (Fig. 5).

Kaplan–Meier curve of 12 DEGs significantly correlated with overall survival. DEGs differentially expressed genes
Construction of ceRNA network
We identified 2577 DE lncRNAs in TCGA dataset using the edgeR package (Fig. 6a). miRcode, miRTarBase, and TargetScan were used to predict the miRNA-mRNA pairs. Only miRNA-mRNA pairs aligned across the three databases were incorporated into the ceRNA network. Four DEMs met the requirements of these databases (Fig. 6b). Thirty-five overlapping lncRNAs were screened out from the 2577 DE lncRNAs in TCGA and 338 LncRNAs that could bind to four DEMs and were predicted by miRcode. (Fig. 6c). According to the ceRNA hypothesis, lncRNA can act as a ceRNA to enhance the expression of target genes by sponging miRNAs. Finally, Cytoscape (Version 3.7.1) was used to verify the initial ceRNA network (Fig. 6d and Table 3).

LncRNA-miRNA-mRNA ceRNA network analysis. a DElncRNAs from TCGA. DE lncRNAs with FDR-adjusted p < 0.01 and logFC > 1. b Four DEMs conformed to the miRcode, miRTarBase, and TargetScan databases according to the Venn diagram. c Venn diagrams showing 35 overlapping lncRNAs were screened out between the 2577 DE lncRNAs in TCGA and 338 lncRNAs predicted by miRcode, which could bind to four DEMs. d Red color represents miRNA downregulation, fluorescent green polygons represent mRNA upregulation, and light-purple diamonds represent lncRNA upregulation. DElncRNAs differentially expressed lncRNAs, TCGA The Cancer Genome Atlas, DEMs differentially expressed mRNAs
To evaluate the DE lncRNAs in the ceRNA network, we screened highly expressed DE lncRNAs from the boxplots in the GEPIA module. We next validated the potential DE lncRNAs targeted by four DEMs using the lncBace2.0 database. Third, we verified the expression of DLEU2, H19, HOTAIRM1, LINC00152, LINC00461, MIR155HG, C1S, DCBLD2, and SERPINH1 in GBM tumor tissues and normal brain tissues using TCGA database. DLEU2, H19, HOTAIRM1, LINC00152, LINC00461, and MIR155HG were identified as DE lncRNAs (p < 0.05), whereas C1S, DCBLD2, and SERPINH1 were identified as DE mRNAs (p < 0.05) (Fig. 7a). The final ceRNA network was used to construct regulatory axes and included 6 lncRNAs, 4 miRNAs, and 3 mRNAs (Fig. 7b and Table 4).

Expression of genes and construction of the GBM lncRNA-miRNA-mRNA network. a Expression of nine key genes, including 6 lncRNAs and 3 mRNAs, in GBM and normal tissue samples from the GEPIA databases (*p < 0.05). b Ji mulberry figure revealing four pairs of ceRNA networks: H19/miR-29b-3p/SERPINH1, H19/miR-29c-3p/SERPINH1, LINC00152 LINC00461/miR-139-5p/DCBLD2, and MIR155HG HOTAIRM1 DLEU2/ miR-129-5p/C1S. GBM glioblastoma, GEPIA Gene Expression Profiling Interactive Analysis
Validation of potential ceRNA axis and correlation analysis of triads
MIR155HG/miR-129-5p/C1S was identified as a potential regulatory axis from the ceRNA network for the following reasons: the results of qRT-PCR analysis indicated that MIR155HG (Fig. 8a) and C1S (Fig. 8b) were significantly upregulated in GBM (n = 37), whereas miR-129-5p (Fig. 8c) was significantly downregulated compared to in normal brain tissues. The results of OS analysis of MIR155HG indicated that MIR155HG had prognostic value in GBM (Fig. 8d). The glioma RNA sequencing data were downloaded from TCGA and converted into transcripts per million, which was verified by the results of qRT-PCR and Pearson coefficient correlation. The expression of MIR155HG and C1S were positively correlated (Fig. 8e, h). In contrast, the expression of MiR-129-5p and MIR155HG (Fig. 8f, i), as well as miR-129-5p and C1S (Fig. 8 g, j) were negatively correlated. Therefore, MIR155HG/miR-129-5p/C1S may be an important ceRNA axis.

Expression and correlation of the MIR155HG/ miR-129-5p/C1S axis in GBM. a Expression of MIR155HG was significantly increased in gliomas compared to in normal brain tissues according to qRT-PCR analysis. b Expression of C1S in GBM (n = 37) and normal brain tissue samples collected at our institution. c Expression of miR-129-5p was significantly reduced in gliomas compared to in normal brain tissues according to qRT-PCR analysis. d High level of MIR155HG expression was significantly associated with the overall survival of patients with glioma in TCGA dataset. e, g Correlation between MIR155HG/C1S mRNA, miR-129-5p/MIR155HG, and miR-129-5p/C1S mRNA expression in TCGA dataset was analyzed by Pearson test. h, j Correlation between MIR155HG/C1S mRNA, miR-129-5p/MIR155HG, and miR-129-5p/C1S mRNA expression was analyzed by Pearson test and qRT-PCR. GBM glioblastoma, qRT-PCR real-time quantitative reverse-transcription polymerase chain reaction, TCGA The Cancer Genome Atlas. ****p < 0.0001
Knockdown of C1S inhibited the proliferation, migration, and invasion abilities of GBM cells
To investigate the function of C1S in GBM, we evaluated the impact of C1S on GBM cell proliferation, migration, and invasion. First, C1S was detected in four GBM cell lines (HS683, U251, U87, T98G), with U87 and T98G cells showing high C1S expression (Fig. 9a). Next, three specific siRNAs were used to silence C1S expression in the U87 and T98G cell lines. After transfection and incubation for 48 h, evaluation of the interference efficiency of the siRNAs by RT-qPCR in U87 and T98G cells revealed that siRNA-C1S-3 showed the highest silencing efficiency (Fig. 9b, c). Compared with the NC group, the MTS assay showed that C1S knockdown significantly reduced the viability of U87 and T98G cells (Fig. 9d). To further explore the functions of C1S in the migration and invasion of GBM cells, a wound-healing assay was performed; C1S knockdown in U87 and T98G cells significantly inhibited the migration abilities of GBM cells at 24 h of incubation (Fig. 9e). Additionally, transwell assays showed that C1S knockdown decreased the migration and invasion abilities of U87 and T98G cells (Fig. 9f).

Depletion of C1S suppressed the proliferation, migration, and invasion abilities of glioblastoma cell lines. a Expression of C1S expression in glioblastoma cell lines. b, c Expression of C1S was downregulated in the siRNA group in U87 and T98G cells. d Suppression of the proliferation of U87 and T98G cells transfected with si-C1S was detected by MTS assay. e, f Wound-healing assay and transwell assay revealed that downregulation of C1S significantly inhibited the invasion and migration of U87 and T98G cell lines compared with the NC groups. *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001
Discussion
Despite the significant progress made in comprehensive therapy, the five-year prognosis of patients with glioma remains poor[19, 20]. In recent years, the ceRNA hypothesis has gained attention, with studies focusing on how imbalanced expression of ceRNA affects the pathogenicity and progression of cancer[21,22,23]. In the present study, we used expression spectrum data from three mRNA expression profiling datasets to identify intersecting DEGs in the datasets to ensure the accuracy of our results. Twelve DEGs associated with survival prognosis were selected to construct a ceRNA network, which was validated by qRT-PCR. This network provides insights into the involvement of ceRNA in the diagnosis and mechanism of glioma, as well as demonstrates the utility of using the ceRNA axis as a GBM biomarker.
A Venn diagram was constructed to further filter and refine the identified DEMs, DE miRNAs, and DE lncRNAs. Through GO and PPI network analysis, we found that the DE mRNAs were mainly enriched in cell adhesion and ECM-receptor interaction. For example, the interaction between integrins and the ECM promotes chemoresistance by protecting cells from drug-induced apoptosis in several cancer types, including small lung cancer, myeloma, and esophageal squamous-cell carcinoma[24, 25]. The migration and invasion of glioma cells involve a complex combination of multiple molecular processes, including those associated with the ECM, protease secretion, and the actin cytoskeleton, which alter the modification of tumor cell adhesion[25].
In general, lncRNAs can act as miRNA sponges to regulate the expression of downstream coding genes. MiRNAs often adsorb adjacent lncRNAs and mRNAs. First, three databases (miRcode, miRTarBase, and TargetScan) were used to analyze whether the DEGs and DEMs contained binding sites. Second, four selected DEMs were used to reverse-predict the potential binding sites of the lncRNAs using two databases (miRcode and lncBace2.0). Initially, we aimed to build a ceRNA network with 163 DEGs screened from the GEO mRNA microarray datasets. However, when we intersected the DEMs-DEGs, those with too many and non-critical genes were predominant. Unexpectedly, among the 163 DEGs, 12 were related to prognosis and survival. Therefore, we isolated and analyzed these DEGs. Finally, a ceRNA network map was constructed.
The 12 DEGs associated with OS were C1S, CFI, DCBLD2, FAM20C, FNDC3B, IFI30, KDELR2, RCAN2, PLP2, SERPINH1, STEAP3, and TNFRSF1A. Among them, DCBLD2[26], FNDC3B[27], PLP2[28], SERPINH1[29, 30], STEAP1[31, 32], and TNFRSF1A[33] were found to be abnormally expressed in a series of human tumors. IFI30 is the most stable prognostic gene among interferon-stimulated genes in glioma[34]. CFI is a potential therapeutic target for nonmelanoma skin cancer[35]. C1S, FAM20C, KDELR2, and RCAN2 have rarely been reported in cancer. After screening three databases, miRcode, miRTarBase and TargetScan, we finally identified three mRNAs for constructing ceRNA networks, namely, C1S, SERPINH1, and DCBLD2.
The Ji mulberry figure illustrated the four established ceRNA axes: H19/has-miR-29b-3p/ SERPINH1, DLEU2/miR-29c-3p/SERPINH1, LINC00152 LINC00461/miR-139-5p/DCBLD2, and MIR155HG HOTAIRM1 H19/miR-129-5p/C1S. MIR155HG is a promising biomarker for tracking tumor progression and predicting metastasis[36] and should be further investigated in longitudinal studies of patients with early-stage tumors. Additionally, MIR155HG, which can also competitively bind to miR-129-5p, is an important regulatory factor for hematopoietic, inflammatory, immune, and tumor development processes, as well as other physiological and pathological processes[37]. Phosphorylated DCBLD2 can recruit TRAF6 and stimulate the AKT pathway, promoting the migration and invasion abilities of glioma cells[26]. SERPINH1 expression is related to the malignancy of glioma and promotes angiogenesis in glioma through autocrine and paracrine mechanisms[38]. C1S, as a protease involved in the classical complement pathway[39], was recently reported to be associated with tumor immunity and the promotion of tumor growth[40]; however, it has not been studied as an oncogene in gliomas. We identified C1S as a new potential biomarker in glioma tissue based on its prognostic and OS-predictive value and predicted that C1S plays a crucial role in the ceRNA network. Analysis of the function of C1S revealed that downregulation of C1S contributed to the inhibition of proliferation, migration, and invasion of glioblastoma cell lines. Our research improves the understanding of the biological function of C1S in tumors and provides a basis for the further exploration of these aspects.
Although we established three network relationship chains of ceRNAs and verified a new ceRNA axis, providing insights into the role of ceRNA in glioma, there were a few limitations to this study. First, verification of the ceRNA axis based only on the correlation of the differential expression of genes is insufficient; further cell function experiments are needed to confirm our results. Second, three network chains of ceRNAs and only one ceRNA axis, which have not been reported previously, were selected for verification. Third, we only performed in vitro cell experiments to analyze the functions of C1S; further functional experiments should be performed in vivo to validate the in vitro findings.
In conclusion, C1S was found to be associated with a worse prognosis of GBM. Additionally, C1S knockdown inhibited the proliferation, invasion, and migration of GBM cell lines. Thus, C1S may serve as a new biomarker for GBM. Our findings provide insight into the roles of ceRNAs in GBM.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request. The TCGA and GEO datasets could be obtained from the online website.
Change history
29 August 2023
A Correction to this paper has been published: https://doi.org/10.1186/s12935-023-03035-0
Abbreviations
- TCGA:
-
The Cancer Genome Atlas
- DEMs:
-
Differentially expressed mRNAs
- GBM:
-
Glioblastoma
- qRT-PCR:
-
Real-time quantitative reverse-transcription polymerase chain reaction
- GEPIA:
-
Gene Expression Profiling Interactive Analysis
References
Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro Oncol. 2018;20:1–86.
Sun Y, Wang Z, Zhou D. Long non-coding RNAs as potential biomarkers and therapeutic targets for gliomas. Med Hypotheses. 2013;81(2):319–21.
Li W, Graeber MB. The molecular profile of microglia under the influence of glioma. Neuro Oncol. 2012;14(8):958–78.
Lin T-K, Chang C-N, Tsai C-S, Huang Y-C, Lu Y-J, Chen W-J, Lin Y-H, Chung IH, Lin K-H. The long non-coding RNA LOC441204 enhances cell growth in human glioma. Sci Rep. 2017. https://doi.org/10.1038/s41598-017-05688-0.
Wang L, Wei B, Hu G, Wang L, Bi M, Sun Z, Jin Y. Screening of differentially expressed genes associated with human glioblastoma and functional analysis using a DNA microarray. Mol Med Rep. 2015;12(2):1991–6.
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–58.
Kirby J, Heath PR, Shaw PJ, Hamdy FC. Gene expression assays. Adv Clin Chem. 2007;44:247–92.
Dhamija S, Menon MB. Non-coding transcript variants of protein-coding genes - what are they good for? RNA Biol. 2018;15(8):1025–31.
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA Hypothesis: the Rosetta Stone of a Hidden RNA Language? Cell. 2011;146(3):353–8.
Rapicavoli NA, Qu K, Zhang J, Mikhail M, Laberge RM, Chang HY. A mammalian pseudogene lncRNA at the interface of inflammation and anti-inflammatory therapeutics. Elife. 2013;2:e00762.
Chiu Y-C, Hsiao T-H, Chen Y, Chuang EY. Parameter optimization for constructing competing endogenous RNA regulatory network in glioblastoma multiforme and other cancers. BMC Genomics. 2015. https://doi.org/10.1186/1471-2164-16-S4-S1.
Wu Q, Xiang S, Ma J, Hui P, Wang T, Meng W, Shi M, Wang Y. Long non-coding RNA CASC15 regulates gastric cancer cell proliferation, migration and epithelial mesenchymal transition by targeting CDKN1A and ZEB1. Mol Oncol. 2018;12(6):799–813.
Yao K, Wang Q, Jia J, Zhao H. A competing endogenous RNA network identifies novel mRNA, miRNA and lncRNA markers for the prognosis of diabetic pancreatic cancer. Tumor Biol. 2017. https://doi.org/10.1177/1010428317707882.
Shan Y, Ma J, Pan Y, Hu J, Liu B, Jia L. LncRNA SNHG7 sponges miR-216b to promote proliferation and liver metastasis of colorectal cancer through upregulating GALNT1. Cell Death Dis. 2018. https://doi.org/10.1038/s41419-018-0759-7.
Sanchez-Mejias A, Tay Y. Competing endogenous RNA networks: tying the essential knots for cancer biology and therapeutics. J Hematol Oncol. 2015. https://doi.org/10.1186/s13045-015-0129-1.
Qi X, Zhang D-H, Wu N, Xiao J-H, Wang X, Ma W. ceRNA in cancer: possible functions and clinical implications. J Med Genet. 2015;52(10):710–8.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015. https://doi.org/10.1093/nar/gkv007.
Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–102.
Lopes MBS. The 2017 World Health Organization classification of tumors of the pituitary gland: a summary. Acta Neuropathol. 2017;134(4):521–35.
Lapointe S, Perry A, Butowski NA. Primary brain tumours in adults. Lancet. 2018;392(10145):432–46.
Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465(7301):1033–90.
Karreth FA, Tay Y, Perna D, Ala U, Tan SM, Rust AG, DeNicola G, Webster KA, Weiss D, Perez-Mancera PA, et al. In vivo identification of tumor-suppressive PTEN ceRNAs in an oncogenic BRAF-induced mouse model of melanoma. Cell. 2011;147(2):382–95.
Poliseno L, Pandolfi PP. PTEN ceRNA networks in human cancer. Methods. 2015;77–78:41–50.
Sethi T, Rintoul RC, Moore SM, MacKinnon AC, Salter D, Choo C, Chilvers ER, Dransfield I, Donnelly SC, Strieter R, et al. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: A mechanism for small cell lung cancer growth and drug resistance in vivo. Nat Med. 1999;5(6):662–8.
Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS. Cell adhesion mediated drug resistance (CAM-DR): Role of integrins and resistance to apoptosis in human myeloma cell lines. Blood. 1999;93(5):1658–67.
Feng H, Lopez GY, Kim CK, Alvarez A, Duncan CG, Nishikawa R, Nagane M, Su A-JA, Auron PE, Hedberg ML, et al. EGFR phosphorylation of DCBLD2 recruits TRAF6 and stimulates AKT-promoted tumorigenesis. J Clin Invest. 2014;124(9):3741–56.
Zhong Z, Zhang H, Hong M, Sun C, Xu Y, Chen X, Gao C, He M, Liu W, Liang J. FNDC3B promotes epithelial-mesenchymal transition in tongue squamous cell carcinoma cells in a hypoxic microenvironment. Oncol Rep. 2018;39(4):1853–9.
Zimmerman JW, Pennison MJ, Brezovich I, Yi N, Yang CT, Ramaker R, Absher D, Myers RM, Kuster N, Costa FP, et al. Cancer cell proliferation is inhibited by specific modulation frequencies. Br J Cancer. 2012;106(2):307–13.
Yamamoto N, Kinoshita T, Nohata N, Yoshino H, Itesako T, Fujimura L, Mitsuhashi A, Usui H, Enokida H, Nakagawa M, et al. Tumor-suppressive microRNA-29a inhibits cancer cell migration and invasion via targeting HSP47 in cervical squamous cell carcinoma. Int J Oncol. 2013;43(6):1855–63.
Zhu J, Xiong G, Fu H, Evers BM, Zhou BP, Xu R. Chaperone Hsp47 drives malignant growth and invasion by modulating an ECM gene network. Can Res. 2015;75(8):1580–91.
Lee C-H, Chen S-L, Sung W-W, Lai H-W, Hsieh M-J, Yen H-H, Su T-C, Chiou Y-H, Chen C-Y, Lin C-Y, et al. The prognostic role of STEAP1 expression determined via immunohistochemistry staining in predicting prognosis of primary colorectal cancer: a survival analysis. Int J Mol Sci. 2016. https://doi.org/10.3390/ijms17040592.
Rodeberg DA, Nuss RA, Elsawa SF, Celis E. Recognition of six-transmembrane epithelial antigen of the prostate-expressing tumor cells by peptide antigen-induced cytotoxic T lymphocytes. Clin Cancer Res. 2005;11(12):4545–52.
Ullio C, Brunk UT, Urani C, Melchioretto P, Bonelli G, Baccino FM, Autelli R. Autophagy of metallothioneins prevents TNF-induced oxidative stress and toxicity in hepatoma cells. Autophagy. 2015;11(12):2184–98.
Zhu C, Chen X, Guan G, Zou C, Guo Q, Cheng P, Cheng W, Wu A. IFI30 is a novel immune-related target with predicting value of prognosis and treatment response in glioblastoma. Oncotargets Ther. 2020;13:1129–43.
Riihila P, Nissinen L, Farshchian M, Kivisaari A, Ala-aho R, Kallajoki M, Grenman R, Meri S, Peltonen S, Peltonen J, et al. Complement factor I promotes progression of cutaneous squamous cell carcinoma. J Invest Dermatol. 2015;135(2):579–88.
Thiele J-A, Hosek P, Kralovcova E, Ostasov P, Liska V, Bruha J, Vycital O, Rosendorf J, Opattova A, Horak J, et al. lncRNAs in non-malignant tissue have prognostic value in colorectal cancer. Int J Mol Sci. 2018. https://doi.org/10.3390/ijms19092672.
Vargova K, Curik N, Burda P, Basova P, Kulvait V, Pospisil V, Savvulidi F, Kokavec J, Necas E, Berkova A, et al. MYB transcriptionally regulates the miR-155 host gene in chronic lymphocytic leukemia. Blood. 2011;117(14):3816–25.
Wu ZB, Cai L, Lin SJ, Leng ZG, Guo YH, Yang WL, Chu YW, Yang SH, Zhao WG. Heat shock protein 47 promotes glioma angiogenesis. Brain Pathol. 2016;26(1):31–42.
Kerr FK, O’Brien G, Quinsey NS, Whisstock JC, Boyd S, de la Banda MG, Kaiserman D, Matthews AY, Bird PI, Pike RN. Elucidation of the substrate specificity of the C1s protease of the classical complement pathway. J Biol Chem. 2005;280(47):39510–4.
Roumenina LT, Daugan MV, Noé R, Petitprez F, Vano YA, Sanchez-Salas R, Becht E, Meilleroux J, Clec’h BL, Giraldo NA, et al. Tumor cells hijack macrophage-produced complement C1q to promote tumor growth. Cancer Immunol Res. 2019;7(7):1091–105.
Acknowledgements
Not applicable.
Funding
This study was supported in part by grants from The National Natural Science Foundation of China (81960458, 81860664, 81703780), Youth Science Foundation of Science and Technology Department of Jiangxi Provincial (20192BAB215063, 20202BABL216080), Project in the Second Affiliated Hospital of Nanchang University (2014YNLC12009), and Science and Technology Research Project of Education Department of Jiangxi Province (180077).
Author information
Authors and Affiliations
Contributions
SXL planned and supervised the study. YK contributed to conception and design, data acquisition and manuscript drafting. YH, LQL, WLC, JYL, LQX, CJJ, TZL, ZZ, HW, CZ collected glioma tissues and clinical information. All authors drafted the final manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Medical Ethics Committee approved the Xiangya Hospital of Central South University (No. 201803806). In this study, all procedures were performed following the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
In vitro experiment
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yu, K., Yang, H., Lv, Ql. et al. Construction of a competitive endogenous RNA network and analysis of potential regulatory axis targets in glioblastoma. Cancer Cell Int 21, 102 (2021). https://doi.org/10.1186/s12935-021-01789-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-021-01789-z