
Abstract
Background
Gastric cancer (GC) is the fifth most frequently diagnosed cancer and the third leading cause of cancer-related mortality. Lack of prognostic indicators for patient survival hinders GC treatment and survival.
Methods and results
Methylation profile data of patients with GC obtained from The Cancer Genome Atlas (TCGA) database were analyzed to identify methylation sites as biomarkers for GC prognosis. The cohort was divided into training and validation sets. Univariate Cox, LASSO regression,and multivariate Cox analyses revealed a close correlation of a four-DNA methylation signature as a risk score model with the overall survival of patients with GC. The survival between high-risk and low-risk score patients with GC was significantly different. Analyses of receiver operating characteristics revealed a high prognostic accuracy of the four-DNA methylation signature in patients with GC. The subgroup analysis indicated that the accuracy included that for anatomical region, histologic grade, TNM stage, pathological stage, and sex. The GC prognosis based on the four-DNA methylation signature was more precise than that based on known biomarkers.
Conclusions
The four-DNA methylation signature could serve as a novel independent prognostic factor that could be an important tool to predict the prognostic outcome of GC patients. This potential must be verified in a large-scale population cohort study and through basic research studies.
Similar content being viewed by others

Background
Gastric cancer (GC) is the fifth most frequently diagnosed cancer and the third leading cause of cancer-related mortality [1]. According to the 2018 global cancer report, more than one million new cases of GC were reported, and 783,000 deaths were attributed to GC [1]. While the prevalence of stomach cancer has decreased in most Western countries, it remains one of the most common causes of cancer-related deaths in Asia [1,2,3]. Patients are typically asymptomatic during early stages, which can delay diagnosis. Diagnosis at an advanced stage of the disease is associated with poor overall survival (OS). Current treatments for GC include surgical resection, chemotherapy, targeted therapy, and radiotherapy [3, 4]. The tumor, node, metastasis (TNM) stage is vital for defining patient prognosis and the choice of therapeutic strategies [5,6,7]. Accurate staging and treatment following the guidelines could differentiate OS for the patients with the same TNM stage [8, 9]. A precise predictive tool for the prognosis of patients with GC and the choice of individual treatment are hot topics.
An increasing number of studies on the pathogenesis of tumors are directed towards epigenetics. The spatial conformation of DNA and its transcriptional activity are affected by epigenetic changes, which are inherited and reversible. Epigenetics includes abnormal DNA methylation, editing of coding RNA and non-coding RNA, and histone modification [10, 11]. DNA methylation annotates genomic regions, which determines when and how information is read, as well as transcription control [12]. The DNA in tumor cells is more readily methylation compared to that in normal cells [13]. Abnormal DNA methylation is not only a late-stage feature of tumors, but also a drives the early development of tumors [14]. Aging, diet, obesity, H. pylori and Epstein Barr virus infection are all associated with gene methylation in GC [15]. Studies confirmed the involvement of DNA methylation in the transformation of the normal gastric mucosa to GC [16]. The tumor-related genes are hypermethylated in precancerous lesions and intestinal metaplasia [17, 18], and inhibiting DNA methylation reduces the incidence of GC in animal models [19, 20], DNA methylation could be used as a biomarker for diagnosis, treatment and prognosis of GC [15, 21]. DNA methylation research has opened a new field in genomics biology by identifying novel biomarkers for several cancers, such as gastrointestinal tumors, breast and lung cancer, malignant melanoma, and other tumors [22,23,24,25,26,27]. In one study, hypermethylation of the potassium calcium-activated channel subfamily M alpha 1 (KCNMA1) promoter was detected in 68.7% of GC tissues examined and related to shorter survival time. KCNMA1 hypermethylation was implicated as an independent prognosis factor in GC [28]. By analyzing the genome-wide methylation status, Simon et al. [29] showed that a multiple-DNA methylation signature could predict the therapeutic outcome of bevacizumab during metastatic breast cancer treatment, and was the first DNA methylation study that precited the efficacy of bevacizumab as a cancer treatment. Emerging evidence indicates that DNA methylation is closely correlated with the development, invasion, and metastasis of cancer, and could hence, be a valuable prognostic biomarker [14, 24].
We analyzed the genome-wide methylation map of GC tissues in The Cancer Genome Atlas (TCGA) database to explore the use of DNA methylation as a prognostic biomarker. The Kaplan–Meier survival curve and receiver operating characteristic (ROC) analyses were used to determine the specificity and sensitivity of a four-DNA methylation signature as a prognostic biomarker. The independence and repeatability of this signature as a prognostic factor were verified in validation datasets.
Methods
GC DNA methylation data from the TCGA dataset
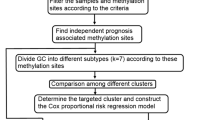
Level three DNA methylation data of patients with GC obtained using the Infinium HumanMethylation450 BeadChip (Illumina Inc., CA, USA) were retrieved from the TCGA database. The CpG sites in the genome were defined following the guideline of the Genome Reference Consortium Human Build 38 (GRCh38). The DNA methylation expression were assessed by β-values and calculated as the ratio of M and M + U, where M represents the signal from methylated beads targeting CpG site, while U represents the signal from unmethylated beads. Data for a total of 368 GC patients comprising 485,578 DNA methylation sites were included after excluding data for patients with a short survival time (< 30 days) and cases for whom clinical survival information was lacking. The relationship between DNA methylation levels and the corresponding survival time of patients with GC was analyzed. The 368 patients with GC were divided evenly into the training (n = 184) and validation set (n = 184) to cross-validate the prognostic indicators. The training set was used to construct the prognostic model, and the validation set was used to evaluate the prognostic accuracy of the model for the survival status of patients with GC. There were no significant differences in the sets concerning clinicopathological data.
Statistical analyses
Univariate Cox regression analysis was conducted in the training set based on the OS time and the CpG expression in patients with GC. A total of 1274 methylation sites that were significantly related to GC prognosis (P < 0.005) were screened for the prognostic methylation markers. To avoid overfitting of the prognostic model, LASSO regression analysis (Glmnet R package) and cross-validation were first performed; the 1274 methylation sites were screened more than 1000 times. From the screened methylation sites, if specific sited were detected more than 800 times, they were regarded as candidate biomarkers and were analyzed further by multivariate Cox regression analysis designed to identify the independent prognostic biomarkers by controlling confounding factors or covariates. Risk score models were constructed based on the risk coefficient and expression of methylation sites. The median risk score was regarded as the cutoff point. Patients with GC were categorized into “high-risk” or “low-risk” groups according to a high and low score, respectively. Log-rank testing of the Kaplan–Meier curve was performed to calculate the difference in OS of the two groups. We conducted ROC analysis for patients with OS < 3 years in terms of the methylation biomarkers. The area under the ROC curve (AUC) was calculated with 95% confidence intervals (CIs) to assess the predictive accuracy of the biomarkers. The Z-test further compared the AUCs of these diverse biomarkers. All statistical analyses were performed using R software (version 3.4.4).
Results
Clinical characteristics of patients
The 368 patients with GC from the STAD cohort of the TCGA database who had been diagnosed clinically and pathologically were analyzed. The median age was 67 years (range, 58–73 years) and 66.6% of the patients were male (Table 1). The median OS was 489 days (range, 31–3720 days) encompassing the different pathological stages of GC. The 3-year OS rate of all patients was 13.3%. Based on the pathological characteristics of TNM staging of GC, histological grade, histological type, and anatomical subdivision following the World Health Organization criteria, the majority of the STAD cohort of GC were diagnosed initially with invasive disease (T3–4, 74.7%), lymphatic metastasis (69.3%), and extensive malignancy (G3–4, 62.2%). The occurrence rate of the intestinal type and unspecific type of GC was 46.5% and 32.3%, respectively, and were higher than the rate of the diffuse type. Anatomic subdivisions of GC were obtained from different regions, such as gastroesophageal junction (GEJ), cardia fundus, and antrum. Among them, stomach antrum (36.4%) and fundus (36.4%) were the predominant sites for GC (Table 1).
Methylation markers associated with prognosis of GC patients in training set
From the DNA methylation expression data in patients with GC, the DNA methylation markers related to the OS of patients with GC in the training set were screened by univariate Cox proportional hazard regression analysis (Fig. 1a–c). A total of 1274 DNA methylation sites correlated significantly with the prognosis of patients with GC (P < 0.005). Of these, four methylation sites including cg05413957 (GRID2IP), cg07020967 (TMEM117), cg10674684 (intergenic region), and cg20100408 (HLA-DP) were selected as the optimal model sites for the prognostic assessment of patient with GC by LASSO regression and multivariate Cox regression analysis. The risk scoring formula was calculated as follows: Risk score = − 0.33126 × β value of cg05413957 + 0.53604 × β value of cg07020967 − 0.38673 × β value of cg10674684 − 0.21447 × β value of cg20100408. The scores revealed strong associations of poor prognosis with hypermethylation of cg07020967 and the hypomethylation of cg05413957, cg10674684, and cg20100408 sites.

Development of the prognostic index based on prognosis‐related DNA methylation sites. a Rank of prognostic index and distribution of groups. b Survival status of patients in different groups. c Heatmap of expression profiles of included DNA methylation sites
Association between the four-DNA methylation biomarker and prognosis
Hazard ratios (HRs) obtained from the Cox regression analysis showed a relation between the four-DNA methylation signature and the OS (P < 0.0001, HR 2.72, 95% CI 2.127–3.474). The prognostic value of the four-DNA methylation signature was explored by Kaplan–Meier survival analyses of the training and validation datasets. The median risk scores as cutoff value were selected to define the high-risk and low-risk for GC prognosis. As shown in Fig. 2a–c, survival was significantly longer in the low-risk group compared to the high-risk group in the training dataset (P < 0.001) and the validation dataset (P < 0.001). A similar result was found in the all-cohort dataset (P < 0.001). The findings indicated the utility of the four biomarkers as prognostic indicators in patients with GC. The differential expressions of the four methylation biomarkers were analyzed individually, and distinct, aberrant regulation was observed (Fig. 2d). Three methylation sites (cg05413957, cg10674684, and cg20100408) were obviously downregulated in the high-risk group compared to the low-risk group, whereas the expression of cg07020967 was higher in the high-risk group compared to the low-risk group in the training and validation datasets. The predictive performance of the four-DNA methylation signature was assessed ROC analysis. The AUC of the four-DNA methylation signature was 0.72 (95% CI 0.67–0.78) in the total cohort, 0.79 (95% CI 0.72–0.86) in the training dataset and 0.66 (95% CI 0.58–0.74) in the validation dataset (Fig. 3a–c), implying the high predictive accuracy of four-DNA methylation signature for GC prognosis.

The overall survival (OS) and methylation levels of gastric cancer patients, The Kaplan–Meier curve of the OS for high-risk and low-risk scores ranking by the four-DNA methylation indicator in the training dataset (N = 184) (a), the validation dataset (N = 184) (b) and all cohort dataset (c). The log-rank test illustrated the higher risk scores are significantly associated with worse OS (P < 0.001). d The differential expression level of methylation sites in training dataset of GC cohort. Mann–Whitney U test was used to evaluate the differences between the high-risk score and low-risk score

ROC analysis of the four-DNA methylation biomarker for predicting the OS of GC patients in total dataset (a), the training dataset (b) and the validation dataset (c)
Predictive performance of the four-DNA methylation biomarker in different patient subgroups
Many confounding clinicopathological factors could influence the predictive performance of the four-DNA methylation biomarker. Subgroup analyses were conducted on the TNM stage, histologic type, histologic grade, and anatomic subdivisions to gauge the clinical applicability and predictive accuracy of the four-DNA methylation biomarker for prognosis of GC with regard to different clinicopathological characteristics (Table 2). The biomarker in the high-risk group of males and females associated significantly with poor prognosis, compared to the case for the low-risk group (both P < 0.001). The AUC value of prognostic accuracy for sex was 0.69 and 0.68, respectively (Additional file 1: Figure S1). The risk scoring system could be applied to the extent of infiltration, with discrimination of extensive infiltration (T3–4) from scant infiltration (T1–2), to lymphatic infiltration, with discrimination of lymphatic metastasis (N1–3) from non-lymphatic metastasis (N0), with an AUC value of 0.67 and 0.72, respectively. Risk score was also valuable in discriminating non-distant organ metastasis (M0) from extensive metastasis (M1–3). However, the risk score could not discriminate the survival time of M1–3 Patients with GC (Additional file 1: Figure S2). Although the risk scoring system could be applied to the entire pathologic stage and histologic grade for OS prediction, the predictive accuracy was lower than that for the pathologic stage III-IV (AUC: 0.60 vs. 0.72) and histologic grade G3–4 (AUC: 0.64 vs. 0.70) (Additional file 1: Figure S3–4). Concerning anatomic subdivisions, in addition to the non-specific classification, the risk scoring system was valuable for prediction of OS for cancers from the antrum, fundus, cardia, and GEJ of the stomach. Survival was worse for high-risk score patients than for the low-risk score patients in the antrum group (P = 0.003), cardia group (P = 0.005), fundus group (P = 3.79E−08), and GEJ group (P < 0.001). The respective AUC values of 0.59, 0.71, 0.75, and 0.82 indicated the predictive accuracy of the four-DNA methylation signature (Additional file 1: Figure S5). The collective results support the application of the signature as an independent prognostic predictor of survival in patients with GC.
Comparison of the four-DNA methylation signature with other known prognostic biomarkers
Prior studies identified some independent prognostic biomarkers for GC. For example, an analysis of microRNA (miRNA) expression profile and risk score identified a seven-miRNA biomarker (miR-10b, miR-223, miR-30a-5p, miR-126 miR-21, miR-338, and let-7a) for OS prediction, and was validated using an independent dataset [30]. An examination of the Oncomine GC database and 32 paired fresh GC tissues revealed upregulated expression of podocalyxin-like protein 1 (PODXL). PODXL downregulation could inhibit tumor development, as shown by the inhibition of epithelial mesenchymal transformation, invasion, and metastasis in vitro and reduction of tumorigenesis in vivo. The Cox proportional hazards model showed PODXL as an independent prognostic biomarker for OS of patients with GC [31]. To verify the predictive performance of the four-DNA methylation indicator, we evaluated the sensitivity and specificity of the seven-miRNA signature and PODXL from other studies in our dataset. Comparison of AUCs revealed that the four-DNA methylation indicator was superior to other known prognostic biomarkers, including mRNA, miRNA, multi-/single-molecule models (Fig. 4). A statistical comparison using the Z-test indicated that the four-DNA methylation indicator had significantly higher (P < 0.05) predictive performance compared to other known biomarkers (Additional file 1: Table 1S). Concerning the prediction of the OS of patients with GC, all the results evidenced that the four-DNA methylation indicator showed a better predictive ability.

ROC curves show the AUC of the four-DNA methylation biomarker (0.71, 95% CI 0.66–0.77) and other known biomarker such as miRNAs (0.52, 95% CI 0.45–0.58) and PODXL (0.55, 95% CI 0.49–0.61) in predicting the OS of GC patients, meth_RS: methylation ROC analysis, miRNA_RS: miRNA ROC analysis
Discussion
GC is a highly heterogeneous disease involving a complex interplay between host and environmental risk factors including Helicobacter pylori, alcohol consumption, tobacco smoking, dietary habits, and others [1]. GC tumorigenesis and progression involve complex regulatory networks [4]. Consequently, multi-molecule models are superior to single-molecule models for the diagnosis and prognosis of GC. DNA methylation is a reliable molecular biomarker that can easily be evaluated by PCR. It is an appropriate biomarker for non-invasive clinical measurements of blood and other body fluids and feces [21, 26, 32, 33]. For instance, methylation of CDH1 could be detected in tissue samples, preoperative peritoneal washes, and serum of patients with GC [32,33,34]. The aberrant methylation of Vimentin in feces is the basis of a commercial test for colorectal cancer [35]. Here, we analyzed the whole genome-wide methylation map in patients with GC. DNA methylation sites that correlated with OS in GC patients were screened, and the candidate biomarkers were used to construct a model for predicting survival time in patients with GC using the high-risk and low-risk scores. Cross-validation of the training and validation datasets tested the stability of the model for prediction of GC prognosis. ROC analysis showed that the four-DNA methylation signature had a superior predictive ability compared to the other tested biomarkers.
Currently, several biomarkers for GC prognosis are available and include mRNAs, non-coding RNAs, and proteins [36,37,38,39]. Long Intergenic Non-Protein Coding RNA 1133 (LINC01133) was identified as a potential prognostic biomarker for patients with GC. LINC01133 inhibits GC progression and metastasis by sponging miR-106a-3p to regulate APC expression and the Wnt/β-catenin pathway [38]. Many studies have established Her-2 as a prognostic biomarker for GC, which informed the development of targeted drugs, such as trastuzumab to treat Her-2 positive GC [40, 41]. Negatively regulated by KLF4, PODXL is a novel prognostic biomarker of GC [31]. The seven-miRNA signature was shown to be an independent biomarker of relapse-free survival and OS in GC [30]. However, some studies lacked large-scale cohort validation, and it was unclear whether the miRNA signature was an independent prognostic indicator with high predictive ability. In this study, using the methylation data from the STAD database, the four-DNA methylation sites related to OS of patients with GC were analyzed via the construction of a regression model. The predictive accuracy of this biomarker was assessed in training and validation datasets by ROC analysis. The interaction of variables was controlled by subgroup analyses, including sex, TNM stages, histologic grade, and anatomic subdivision. The four-DNA methylation biomarker could differentiate early- and advanced-GC and was a novel and independent biomarker with good predictive ability. Additionally, comparisons with the seven-miRNA signature and PODXL demonstrated the superior efficiency of the four-DNA methylation biomarker with regard to the predictive performance.
Three candidate sites of the four-DNA methylation signature were from corresponding genes of GRID2IP (cg05413957), TMEM117 (cg07020967) and HLA-DP (cg20100408). However, the methylation site of cg10674684 did not match to the definite gene due to the intergenic region. GRID2IP is expressed predominantly at the post-synapse regions of parallel fiber-Purkinje cells in the brain and is crucial for synaptogenesis and synaptic plasticity. The formation of excitatory synapses between neurons and tumor cells promotes cancer growth. The molecular chemical signals released from these pseudo-synapses are a likely cause of breast cancer brain metastasis [42,43,44,45]. An analysis of the TCGA database has also implicated GRID2IP as a novel prognostic marker in patients with cholangiocarcinoma [46]. TMEM117 is a six transmembrane protein with a conserved sequence; it is present in pro- and anti-apoptotic BCL-2 family of proteins. Bioinformatic analysis revealed an association between TMEM117 downregulation and pancreatic cancer tumorigenesis and metastasis [47], trans-differentiation to mesenchymal cells in breast cancer [48], phenotypic change of normal cells of gliomas [49], and development of malignant lymphoblastic leukemia [50]. However, experiment data are insufficient to support a role of TMEM117 in cancers, further research is required to identify the tumorigenic role of TMEM117 protein levels in cancer. The HLA-DP protein/peptide-antigen receptor and graft-versus-host disease antigen is an HLA class II beta chain paralog. It is a heterodimer comprised of an alpha chain (DPA) and beta chain (DPB) that are both anchored to the membrane as HLA-DPB1. The biological involvement of HLA-DPB is mainly in the immune response. HLA-DP is participates in the origin and development of cervical cancer, and different HLA-DP polymorphisms correspond to different cervical cancer risks in the Chinese and Swedish populations [51]. Genetic polymorphism of HLA-DP is also associated with the prognosis of Hodgkin’s lymphoma [52]. Although the three genes (GRID2IP, TMEM117, HLA-DP) are involved in the progression of cancers, their role in GC is unclear. Because of the limitations about bioinformatics tools when analyzed large amounts of data, and disadvantages of Infinium HumanMethylation450 BeadChip (Illumina Inc., CA, USA) [53,54,55], we needed validate bioinformatics result by experiment. In the future, We will verify the expression level and predictive efficacy of the four methylation sites in GC cells, mouse models and tissues to explore the genetic mechanism underlying the development of GC.
Conclusion
We established a GC prognostic classifier model consisting of four CpG sites and validated the model in training and validation datasets. The four-DNA methylation signature serves as an biomarker to evaluate the prognosis of patients with GC. The signature remains to be validated in a large-scale cohort and through basic research studies (Additional file 1: Supplemental Figure S1–5 and Supplemental Table S1).
Availability of data and materials
Additional data not presented in the manuscript can be obtained by contacting the authors.
Abbreviations
- GC:
-
Gastric cancer
- TCGA:
-
The Cancer Genome Atlas
- ROC:
-
Receiver operating curve
- OS:
-
Overall survival
- CI:
-
Confidence interval
- AUC:
-
Area under the curve
- GEJ:
-
Gastroesophageal junction
- HR:
-
Hazard ratios
- LASSO:
-
The least absolute shrinkage and selection operator
- TNM:
-
Tumor node metastasis
- STAD:
-
Stomach adenocarcinoma
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. https://doi.org/10.3322/caac.21492.
Allemani C, Weir HK, Carreira H, Harewood R, Spika D, Wang X-S, Bannon F, Ahn JV, Johnson CJ, Bonaventure A, et al. Global surveillance of cancer survival 1995–2009: analysis of individual data for 25,676,887 patients from 279 population-based registries in 67 countries (CONCORD-2). The Lancet. 2015;385(9972):977–1010. https://doi.org/10.1016/s0140-6736(14)62038-9.
Sasako M, Inoue M, Lin JT, Khor C, Yang HK, Ohtsu A. Gastric Cancer Working Group report. Jpn J Clin Oncol. 2010;40(Suppl 1):i28–37. https://doi.org/10.1093/jjco/hyq124.
Van Cutsem E, Sagaert X, Topal B, Haustermans K, Prenen H. Gastric cancer. Lancet. 2016;388(10060):2654–64. https://doi.org/10.1016/s0140-6736(16)30354-3.
National Comprehensive Cancer Network. Gastric cancer (Version 4. 2019). https://www.nccn.org/store/login/login.aspx?ReturnURL=https://www.nccn.org/professionals/physician_gls/pdf/gastric.pdf.
Chinese Society of Clinical Oncology. Gastric cancer (Version 1.2018). http://www.csco.org.cn/cat/147/show/8627.html.
Edge S, Compton C. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol. 2010;17(6):1471–4.
Noh SH, Park SR, Yang H-K, Chung HC, Chung I-J, Kim S-W, Kim H-H, Choi J-H, Kim H-K, Yu W, et al. Adjuvant capecitabine plus oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): 5-year follow-up of an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15(12):1389–96. https://doi.org/10.1016/S1470-2045(14)70473-5.
Jiang Y, Xie J, Han Z, Liu W, Xi S, Huang L, Huang W, Lin T, Zhao L-Y, Hu Y-F, et al. Immunomarker support vector machine classifier for prediction of gastric cancer survival and adjuvant chemotherapeutic benefit. Clin Cancer Res. 2018. https://doi.org/10.1158/1078-0432.ccr-18-0848.
Padmanabhan N, Ushijima T, Tan P. How to stomach an epigenetic insult: the gastric cancer epigenome. Nat Rev Gastroenterol Hepatol. 2017;14(8):467–78. https://doi.org/10.1038/nrgastro.2017.53.
Tahara T, Arisawa T. DNA methylation as a molecular biomarker in gastric cancer. Epigenomics. 2015;7(3):475–86. https://doi.org/10.2217/epi.15.4.
Dor Y, Cedar H. Principles of DNA methylation and their implications for biology and medicine. Lancet (London, England). 2018;392(10149):777–86. https://doi.org/10.1016/s0140-6736(18)31268-6.
Aran D, Hellman A. DNA methylation of transcriptional enhancers and cancer predisposition. Cell. 2013;154(1):11–3. https://doi.org/10.1016/j.cell.2013.06.018.
Klutstein M, Nejman D, Greenfield R, Cedar H. DNA methylation in cancer and aging. Cancer Res. 2016;76(12):3446–50. https://doi.org/10.1158/0008-5472.CAN-15-3278.
Li Y, Liang J, Hou P. Hypermethylation in gastric cancer. Clin Chim Acta. 2015;448:124–32. https://doi.org/10.1016/j.cca.2015.07.001.
Anderson BW, Suh YS, Choi B, Lee HJ, Yab TC, Taylor WR, Dukek BA, Berger CK, Cao X, Foote PH, et al. Detection of gastric cancer with novel methylated DNA markers: discovery, tissue validation, and pilot testing in plasma. Clin Cancer Res. 2018;24(22):5724–34. https://doi.org/10.1158/1078-0432.CCR-17-3364.
Schneider BG, Mera R, Piazuelo MB, Bravo JC, Zabaleta J, Delgado AG, Bravo LE, Wilson KT, El-Rifai W, Peek RM Jr, et al. DNA methylation predicts progression of human gastric lesions. Cancer Epidemiol Biomarkers Prev. 2015;24(10):1607–13. https://doi.org/10.1158/1055-9965.EPI-15-0388.
Lee JH, Park SJ, Abraham SC, Seo JS, Nam JH, Choi C, Juhng SW, Rashid A, Hamilton SR, Wu TT. Frequent CpG island methylation in precursor lesions and early gastric adenocarcinomas. Oncogene. 2004;23(26):4646–54. https://doi.org/10.1038/sj.onc.1207588.
Niwa T, Toyoda T, Tsukamoto T, Mori A, Tatematsu M, Ushijima T. Prevention of Helicobacter pylori-induced gastric cancers in gerbils by a DNA demethylating agent. Cancer Prev Res (Phila). 2013;6(4):263–70. https://doi.org/10.1158/1940-6207.CAPR-12-0369.
Yang W, Mok MT, Li MS, Kang W, Wang H, Chan AW, Chou JL, Chen J, Ng EK, To KF, et al. Epigenetic silencing of GDF1 disrupts SMAD signaling to reinforce gastric cancer development. Oncogene. 2016;35(16):2133–44. https://doi.org/10.1038/onc.2015.276.
Vedeld HM, Goel A, Lind GE. Epigenetic biomarkers in gastrointestinal cancers: the current state and clinical perspectives. Semin Cancer Biol. 2018;51:36–49. https://doi.org/10.1016/j.semcancer.2017.12.004.
Drilon A, Sugita H, Sima CS, Zauderer M, Rudin CM, Kris MG, Rusch VW, Azzoli CG. A prospective study of tumor suppressor gene methylation as a prognostic biomarker in surgically resected stage I to IIIA non-small-cell lung cancers. J Thorac Oncol. 2014;9(9):1272–7. https://doi.org/10.1097/JTO.0000000000000256.
Guo W, Zhu L, Yu M, Zhu R, Chen Q, Wang Q. A five-DNA methylation signature act as a novel prognostic biomarker in patients with ovarian serous cystadenocarcinoma. Clin Epigenetics. 2018;10(1):142. https://doi.org/10.1186/s13148-018-0574-0.
Okugawa Y, Grady WM, Goel A. Epigenetic alterations in colorectal cancer: emerging biomarkers. Gastroenterology. 2015;149(5):1204–0100. https://doi.org/10.1053/j.gastro.2015.07.011.
Kneip C, Schmidt B, Seegebarth A, Weickmann S, Fleischhacker M, Liebenberg V, Field JK, Dietrich D. SHOX2 DNA methylation is a biomarker for the diagnosis of lung cancer in plasma. J Thorac Oncol. 2011;6(10):1632–8. https://doi.org/10.1097/JTO.0b013e318220ef9a.
Visvanathan K, Fackler MS, Zhang Z, Lopez-Bujanda ZA, Jeter SC, Sokoll LJ, Garrett-Mayer E, Cope LM, Umbricht CB, Euhus DM, et al. Monitoring of serum DNA methylation as an early independent marker of response and survival in metastatic breast cancer: TBCRC 005 prospective biomarker study. J Clin Oncol. 2017;35(7):751–8. https://doi.org/10.1200/JCO.2015.66.2080.
Wouters J, Vizoso M, Martinez-Cardus A, Carmona FJ, Govaere O, Laguna T, Joseph J, Dynoodt P, Aura C, Foth M, et al. Comprehensive DNA methylation study identifies novel progression-related and prognostic markers for cutaneous melanoma. BMC Med. 2017;15(1):101. https://doi.org/10.1186/s12916-017-0851-3.
Ma G, Liu H, Hua Q, Wang M, Du M, Lin Y, Ge Y, Gong W, Zhao Q, Qiang F, et al. KCNMA1 cooperating with PTK2 is a novel tumor suppressor in gastric cancer and is associated with disease outcome. Mol Cancer. 2017;16(1):46. https://doi.org/10.1186/s12943-017-0613-z.
Gampenrieder SP, Rinnerthaler G, Hackl H, Pulverer W, Weinhaeusel A, Ilic S, Hufnagl C, Hauser-Kronberger C, Egle A, Risch A, et al. DNA methylation signatures predicting bevacizumab efficacy in metastatic breast cancer. Theranostics. 2018;8(8):2278–88. https://doi.org/10.7150/thno.23544.
Li X, Zhang Y, Zhang Y, Ding J, Wu K, Fan D. Survival prediction of gastric cancer by a seven-microRNA signature. Gut. 2010;59(5):579–85. https://doi.org/10.1136/gut.2008.175497.
Zhang J, Zhu Z, Wu H, Yu Z, Rong Z, Luo Z, Xu Y, Huang K, Qiu Z, Huang C. PODXL, negatively regulated by KLF4, promotes the EMT and metastasis and serves as a novel prognostic indicator of gastric cancer. Gastric Cancer. 2019;22(1):48–59. https://doi.org/10.1007/s10120-018-0833-y.
Ikoma H, Ichikawa D, Koike H, Ikoma D, Tani N, Okamoto K, Ochiai T, Ueda Y, Otsuji E, Yamagishi H. Correlation between serum DNA methylation and prognosis in gastric cancer patients. Anticancer Res. 2006;26(3b):2313–6.
Yu QM, Wang XB, Luo J, Wang S, Fang XH, Yu JL, Ling ZQ. CDH1 methylation in preoperative peritoneal washes is an independent prognostic factor for gastric cancer. J Surg Oncol. 2012;106(6):765–71. https://doi.org/10.1002/jso.23116.
Graziano F, Arduini F, Ruzzo A, Bearzi I, Humar B, More H, Silva R, Muretto P, Guilford P, Testa E, et al. Prognostic analysis of E-cadherin gene promoter hypermethylation in patients with surgically resected, node-positive, diffuse gastric cancer. Clin Cancer Res. 2004;10(8):2784–9. https://doi.org/10.1158/1078-0432.ccr-03-0320.
Ned RM, Melillo S, Marrone M. Fecal DNA testing for Colorectal Cancer Screening: the ColoSure™ test. PLoS Curr. 2011;3:RRN1220-RRN1220. https://doi.org/10.1371/currents.rrn1220.
Huang LY, Wang X, Cui XF, Li H, Zhao J, Wu CC, Min L, Zhou Z, Wan L, Wang YP, et al. IRTKS is correlated with progression and survival time of patients with gastric cancer. Gut. 2018;67(8):1400–9. https://doi.org/10.1136/gutjnl-2016-313478.
Gu Z, Li Y, Yang X, Yu M, Chen Z, Zhao C, Chen L, Wang L. Overexpression of CLC-3 is regulated by XRCC5 and is a poor prognostic biomarker for gastric cancer. J Hematol Oncol. 2018;11(1):115. https://doi.org/10.1186/s13045-018-0660-y.
Yang XZ, Cheng TT, He QJ, Lei ZY, Chi J, Tang Z, Liao QX, Zhang H, Zeng LS, Cui SZ. LINC01133 as ceRNA inhibits gastric cancer progression by sponging miR-106a-3p to regulate APC expression and the Wnt/beta-catenin pathway. Mol Cancer. 2018;17(1):126. https://doi.org/10.1186/s12943-018-0874-1.
Liu H, Liu Y, Bian Z, Zhang J, Zhang R, Chen X, Huang Y, Wang Y, Zhu J. Circular RNA YAP1 inhibits the proliferation and invasion of gastric cancer cells by regulating the miR-367-5p/p27 (Kip1) axis. Mol Cancer. 2018;17(1):151. https://doi.org/10.1186/s12943-018-0902-1.
Gordon MA, Gundacker HM, Benedetti J, Macdonald JS, Baranda JC, Levin WJ, Blanke CD, Elatre W, Weng P, Zhou JY, et al. Assessment of HER2 gene amplification in adenocarcinomas of the stomach or gastroesophageal junction in the INT-0116/SWOG9008 clinical trial. Ann Oncol. 2013;24(7):1754–61. https://doi.org/10.1093/annonc/mdt106.
Begnami MD, Fukuda E, Fregnani JH, Nonogaki S, Montagnini AL, da Costa WL Jr, Soares FA. Prognostic implications of altered human epidermal growth factor receptors (HERs) in gastric carcinomas: HER2 and HER3 are predictors of poor outcome. J Clin Oncol. 2011;29(22):3030–6. https://doi.org/10.1200/JCO.2010.33.6313.
Barria A. Dangerous liaisons as tumour cells form synapses with neurons. Nature. 2019;573(7775):499–501. https://doi.org/10.1038/d41586-019-02746-7.
Venkatesh HS, Morishita W, Geraghty AC, Silverbush D, Gillespie SM, Arzt M, Tam LT, Espenel C, Ponnuswami A, Ni L, et al. Electrical and synaptic integration of glioma into neural circuits. Nature. 2019;573(7775):539–45. https://doi.org/10.1038/s41586-019-1563-y.
Zeng Q, Michael IP, Zhang P, Saghafinia S, Knott G, Jiao W, McCabe BD, Galvan JA, Robinson HPC, Zlobec I, et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature. 2019;573(7775):526–31. https://doi.org/10.1038/s41586-019-1576-6.
Venkataramani V, Tanev DI, Strahle C, Studier-Fischer A, Fankhauser L, Kessler T, Korber C, Kardorff M, Ratliff M, Xie R, et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature. 2019;573(7775):532–8. https://doi.org/10.1038/s41586-019-1564-x.
Zhang C, Ge C. A simple competing endogenous RNA network identifies novel mRNA, miRNA, and lncRNA markers in human cholangiocarcinoma. Biomed Res Int. 2019;2019:3526407. https://doi.org/10.1155/2019/3526407.
Rajurkar M, De Jesus-Monge WE, Driscoll DR, Appleman VA, Huang H, Cotton JL, Klimstra DS, Zhu LJ, Simin K, Xu L, et al. The activity of Gli transcription factors is essential for Kras-induced pancreatic tumorigenesis. Proc Natl Acad Sci USA. 2012;109(17):E1038–47. https://doi.org/10.1073/pnas.1114168109.
Al Saleh S, Al Mulla F, Luqmani YA. Estrogen receptor silencing induces epithelial to mesenchymal transition in human breast cancer cells. PLoS ONE. 2011;6(6):e20610. https://doi.org/10.1371/journal.pone.0020610.
Yeo CW, Ng FS, Chai C, Tan JM, Koh GR, Chong YK, Koh LW, Foong CS, Sandanaraj E, Holbrook JD, et al. Parkin pathway activation mitigates glioma cell proliferation and predicts patient survival. Cancer Res. 2012;72(10):2543–53. https://doi.org/10.1158/0008-5472.CAN-11-3060.
Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481(7380):157–63. https://doi.org/10.1038/nature10725.
Yang YC, Chang TY, Chen TC, Lin WS, Chang SC, Lee YJ. Genetic susceptibility to cervical squamous cell carcinoma is associated with HLA-DPB1 polymorphisms in Taiwanese women. Cancer Immunol Immunother. 2015;64(9):1151–7. https://doi.org/10.1007/s00262-015-1721-5.
Alexander FE, Jarrett RF, Cartwright RA, Armstrong AA, Gokhale DA, Kane E, Gray D, Lawrence DJ, Taylor GM. Epstein-Barr Virus and HLA-DPB1-*0301 in young adult Hodgkin’s disease: evidence for inherited susceptibility to Epstein-Barr Virus in cases that are EBV(+ve). Cancer Epidemiol Biomarkers Prev. 2001;10(6):705–9.
Tang J, Fang F, Miller DF, Pilrose JM, Matei D. Huang TH-M, Nephew KP, Global DNA methylation profiling technologies and the ovarian cancer methylome. In: Verma M, editor. Cancer epigenetics: risk assessment, diagnosis, treatment, and prognosis. New York: Springer; 2015. p. 653–75.
Stirzaker C, Taberlay PC, Statham AL, Clark SJ. Mining cancer methylomes: prospects and challenges. Trends Genet. 2014;30(2):75–84. https://doi.org/10.1016/j.tig.2013.11.004.
Staneva R, Toncheva D. Methylation analysis on whole genome level. What did we learn from BEN studies? Prilozi (Makedonska akademija na naukite i umetnostite Oddelenie za medicinski nauki). 2014;35(1):17–24.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural Science Foundation of China (71964021, 81570783), National Key R&D Program of China (2016YFC1302201, 2016YFC0107006), the Key Program of the Natural Science Foundation of Gansu Province, China (18JR3RA366), the Open Fund of State Key Laboratory of Cancer Biology, China (CBSKL201718).
Author information
Authors and Affiliations
Contributions
CML: study concept and design, title, abstract, full-text screening, data abstraction, statistical analysis, data interpretation, and drafting the article. YZ and KP: study design, data interpretation, critical revision of the manuscript. DZ, QLG, YPW and YNZ: data interpretation, revision of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participant
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, C., Zheng, Y., Pu, K. et al. A four-DNA methylation signature as a novel prognostic biomarker for survival of patients with gastric cancer. Cancer Cell Int 20, 88 (2020). https://doi.org/10.1186/s12935-020-1156-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-020-1156-8