
Abstract
Background
Immunotherapies targeting programmed cell death 1 (PD-1) and programmed death-ligand 1 (PD-L1) have been approved for gastric cancer (GC) patients. However, a large proportion of patients with T-cell-inflamed tumor microenvironment do not respond to the PD-1/PD-L1 blockade. The stromal component of the tumor microenvironment has been associated with immunotherapy. This study aims to explore the clinical significance of the non-immune cells in the tumor microenvironment and their potential as biomarkers for immunotherapy.
Methods
A total of 383 patients with GC from the Cancer Genome Atlas (TCGA) cohort, 300 patients with GC from the GSE62254 cohort in Gene Expression Omnibus (GEO) were included in the study. A stromal score was generated using the ESTIMATE algorithm, and the likelihood of response to PD-1/PD-L1 immunotherapy of GC patients was predicted using the TIDE algorithm. The prognostic value of the stromal score from GC cases was evaluated by the Kaplan–Meier method and Cox regression analysis. Gene set enrichment analysis (GSEA) was also conducted.
Results
The stromal score showed significant differences in different molecular subtypes and T stages. Multivariate analyses further confirmed that the stromal score was an independent indicator of overall survival (OS) in the two cohorts. The low stromal score group showed higher tumor mutation burden (TMB) and micro-satellite instability (MSI), and was more sensitive to immune checkpoint inhibitor according to the TIDE algorithm. Activation of the transforming growth factor and epithelial–mesenchymal transition were observed in the high stromal score subtype, which is associated with T-cell suppression, and may be responsible for resistance to PD-1/PD-L1 therapy. BPIFB2 was confirmed as a hub gene relevant to immunotherapy.
Conclusion
The stromal score was associated with cancer progression and molecular subtypes, and may serve as a novel biomarker for predicting the prognosis and response to immunotherapy in patients with GC.
Similar content being viewed by others


Background
Gastric cancer (GC) is one of the most common malignancies, which has the second-highest tumor-related mortality rate around the world [1, 2]. The overall survival (OS) of GC remains relatively poor because the majority of cases are detected only at an advanced stage with extensive node invasion and distant metastasis [3]. For patients with unresectable/metastatic disease, systemic chemotherapy and targeted therapies, namely, monoclonal antibodies targeting trastuzumab (HER2) [4] and VEGFR2 [5, 6], offer a limited survival advantage. Inhibition of PD-1 and its ligand PD-L1 using an immune-checkpoint inhibitor has emerged as a promising immunotherapy [7]. For the treatment of patients with recurrent, locally advanced, or metastatic gastric or gastroesophageal junction cancer (GC/EGJC), whose tumors express PD-L1 by immunohistochemistry (IHC), the Food and Drug Administration (FDA) approved the use of pembrolizumab (anti-PD-1 antibody) in September 2017 [8]. However, the response rates are relatively low, which highlights the need to better elucidate the mechanisms of treatment resistance and to identify patients who will benefit the most from immunotherapy.
The degree of tumor-infiltrating T-cells in the tumor microenvironment (TME) and PD-L1 expression has been reviewed extensively elsewhere [9]. Tumors with a high level of immune infiltrates and/or an Interferon (IFN) signature indicative of a T-cell-inflamed phenotype could benefit from anti-PD-L1/PD-1 therapies [10, 11]. However, a large proportion of patients with T-cell-inflamed TME do not respond to PD-1/PD-L1 blockade [12], suggesting the presence of an additional immunosuppressive mechanism.
The stromal elements of the TME include cancer-associated fibroblasts (CAFs), myofibroblasts, myeloid cells, endothelial cells, and mesenchymal stromal cells (MSCs) [13]. MSCs and differentiated MSCs, such as CAFs, one of the most abundant and critical components of the tumor mesenchyme, influence the phenotype of the immune cells and dramatically affect tumor progression, and thus, a better understanding of the precise mechanisms involved is critical to developing more efficacious immunotherapies [14,15,16,17,18,19,20].
The dynamic interplay between stromal cells and the innate and adaptive immune cells involves several cellular events and physiological processes [21]. Given that stromal cell depletion therapy proceeds with caution concerning of on-target/off-tumor effects [22], the exploration of targeting common stromal-dependent molecular pathways may represent an alternative approach.
Recently, the Estimation of stromal and immune cells in malignant tumor tissues (ESTIMATE) algorithm was introduced to quantify stromal and immune components in a tumor, reflecting the tumor microenvironment [23, 24]. Several studies have shown the effectiveness of such big-data-based algorithms in different types of malignancies, including prostate cancer [25], breast cancer [26], colon cancer [27], and glioblastoma multiforme [28].
In this study, we estimated the stromal elements of TME and established a robust prognostic biomarker and predictive factor for response to immune-checkpoint inhibitors based on the gene expression profiles of GC patients. The newly found targeted genes and signaling pathways can render tumor cells more sensitive to immunotherapy.
Materials and methods
Database
Two cohorts of patients with GC from two independent databases, 383 patients in the TCGA-STAD database (http://cancergenome.nih.gov/) and 300 in the GSE62254 database based on GPL570 platforms (Affymetrix Human Genome U133 Plus 2.0 Array) in Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/), were included in this study. RNA-sequencing and clinical data of the TCGA-STAD patients were acquired from the TCGA database. Updated clinical data, such as gender, age, TNM stage, and survival outcome of TCGA-STAD samples, were acquired from the Genomic Data Commons. The patient cohort from the GSE62254 database was used as a validating cohort. To make the samples between two databases more comparable, RNA-sequencing data (raw values) from the TCGA database were transformed into transcripts per kilobase million (TPM) values, which is similar to microarrays in the GSE62254 database [29].
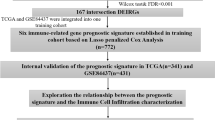
By applying the ESTIMATE, stromal score and immune score were calculated and were used to explore the crosstalk between the stromal and immune microenvironment as well as common mechanisms of immune modulation. The TMB was calculated from comprehensive genomic profiling data. The likelihood of response to immunotherapy for each sample was predicted based on the TIDE algorithm [30]. The research design was illustrated in Fig. 1.

Flow chart of study design
Identification of deferentially expressed genes (DEGs)
Deseq2 was used for performing data analysis. SurvivalRoc package was utilized to determine the best cutoff, and DEGs were identified between high stromal score and low stromal score group using Student’s t-test. A p-value < 0.05 and absolute log2 fold change (FC) > 1 were set as the cutoff criteria to screen for significant DEGs of interest.
Survival analysis
The Kaplan–Meier plots were generated to illustrate the relationship between patients’ OS and gene expression levels of DEGs or stromal scores. The log-rank test tested the association. Cox proportional hazards model was utilized to reveal the independent indicators related to OS.
Functional and pathway enrichment analysis
The gene ontology (GO) functional analysis was performed using R package clusterprofiler [31]. The GO terms were identified with a strict cutoff of p-value < 0.05 and a false discovery rate (FDR) of less than 0.05. To explore the potential function of the hub genes in GC, we performed GSEA of the adjusted expression data for all transcripts. Gene sets were downloaded from the MSigDB database of Broad Institute (http://www.broadinstitute.org/msigdb). Enrichment p values were based on 10,000 permutations and subsequently adjusted for multiple testing using the Benjamin–Hochberg procedure to control the FDR. The enrichment scores of molecular pathways and gene expression signatures were evaluated using single-sample gene set enrichment analysis (R package GSVA).
Construction of PPI network
We downloaded the integrated interaction information from the online database Search Tool for the Retrieval of Interacting Genes (STRING) (http://www.string-db.org/). Then we developed a protein–protein interaction (PPI) network by using Cytoscape software to further explore the relationships between DEGs at the protein level. DEGs pairs whose combined score was > 0.4 were mapped. The genes with the highest degree scores in the PPI network were selected as hub genes.
Results
Clinicopathological characteristics of stromal subtypes
ESTIMATE algorithm analysis showed that the stromal scores ranged from − 1957.19 to 2085.81 in the TCGA database and − 1098.03 to 5972.58 in the GEO database. Three hundred eighty-three samples from the TCGA database were separated into two subsets based on the optimal cut point of stromal score. Stromal score expression and clinicopathological factors are summarized in Table 1. Detailed clinical characteristics of the 184 patients with clinical data in the GEO database are shown in Additional file 1: Table S1.
The stromal score is involved in outcomes of GC subtypes and is associated with tumor progression
Notably, the patients staged in the early T group averaged a lower stromal score than those in the advanced T group in both cohorts (Fig. 2a; Additional file 2: Figure S1) (p < 0.001 in TCGA cohort, p < 0.0001 in GEO cohort). According to the report of Peter et al. [32], GC can be divided into five subtypes (CIN, EBV, GS, HM-SNV, and MSI subtype) based on the molecular parameters of histology. Our results showed that the average stromal score of the GS subtype was the highest rank of all of the five subtypes, followed by that of EBV subtype, MSI subtype, CIN subtype, and HM-SNV subtype (Fig. 2b) (p < 0.001). To investigate whether stromal scores predict the outcome of GC patients, we divided all of the patients into two groups based on the optimal cutoff and performed Kaplan–Meier survival analysis. The results showed that patients with low stromal score have a survival advantage over those with a high stromal score (Fig. 2c) (p < 0.01). These results demonstrated that the stromal score is correlated with subtype classification as well as the outcomes of GC patients.

Stromal scores are associated with T-stage/overall survival/subtypes/TMB of gastric cancer. a Compared to the early T-stage, the advanced T-stage is associated with a higher stromal score (p < 0.001). b Distribution of stromal scores of GC subtypes. The Volin-Box-plot shows that there is a significant association between GC subtypes and the level of stromal scores (p < 0.001). c The TCGA GC cases were divided into the high and low stromal score groups by the optimal cutoff. Kaplan–Meier survival analysis shows that there is a statistically significant survival advantage for the low stromal score group (p = 0.0042); and d TMB for the different stromal score subtypes
In addition, univariate and multivariate Cox regression analysis showed that the stromal score is an independent prognostic indicator of OS in patients from the TCGA database (Table 2) (p = 0.008). The GEO cohort further validated that the stromal score predicts the OS of GC patients using multivariate analysis (Additional file 3: Table S2) (p = 0.02).
Identification of DEGs based on the status of stromal scores
By comparing the global gene expression of samples from the TCGA cohort with high versus low stromal scores, we identified 119 DEGs, including 74 upregulated genes and 45 downregulated genes (p < 0.05, log2 fold change (FC) > 1). Volcano plot (Fig. 3) and Heatmap (Fig. 4) showed the representatives of the DEGs.

Volcano plot of the differentially expressed genes. Each dot represents a gene. Red dots indicate the upregulated genes. Green dots indicate downregulated genes. Black dots represent genes for which the differences are not significant

Heatmap of the representative DEGs of left half (low score) vs. right half (high score). The clustering of the DEGs is shown on the left, and gene symbols are shown on the right of the heatmap
Functional enrichment analysis of the DEGs
The identified DEGs were subjected to GO analysis. For enriched biological processes (BP), DEGs were mainly enriched in negative regulation of wound healing, negative regulation of hemostasis, negative regulation of endothelial cell apoptotic process, humoral immune response, fibrinolysis, extracellular structure organization, and acute inflammatory response. DEGs in the molecular function MF category were mainly associated with transforming growth factor beta receptor binding, extracellular matrix structural constituent. Concerning the cell component (CC), DEGs primarily clustered in the extracellular matrix (Fig. 5a; Additional file 4: Table S3) (p.adjust < 0.05).

Functional enrichment analysis of DEGs and relation between the stromal and immune scores. a GO analysis of the DEGs. The x-axis indicates the gene ratio, and the y-axis indicates the GO terms. p-value is shown in different colors. The dot size represents the number of genes. b The stromal score is significantly positively correlated with the immune score (p < 0.001, R = 0.65)
The low stromal score group may be more sensitive to immunotherapies
As shown in Fig. 5b, there was a significantly positive correlation between the stromal score and the immune score (p < 0.001), demonstrating the crosstalk between the stromal cells and immune cells in the TME. Besides, the high stromal score group was associated with high expression of immune and stromal-relevant signatures, whereas the expression of MSI-related mRNAs was relatively low (Fig. 6). Furthermore, the low stromal score group may be more likely (62.6% in the TCGA cohort and 49.3% in the GEO cohort) to respond to immunotherapy than the high stromal score group (13.2% in the TCGA cohort and 23.3% in the GEO cohort) according to the TIDE algorithm. Interestingly, in good agreement with the TIDE algorithm, the low stromal score group presented a significantly higher count of TMB, which is significantly associated with the efficacy of immunotherapy (Fig. 2d) (p < 0.01).

Characteristics of stromal subtype and functional annotation. The stromal subtypes are distinguished by different signatures (immune-relevant signature, mismatch-relevant signature, and stromal-relevant signature). Histological subtypes and the TME cluster are shown as patient annotations
Considering the associations between TMB, MSI relevant signatures, and immunotherapy, as well as results of TIDE algorithm, it can be referred that patients in the low stromal score group may be more sensitive to PD-1/PD-L1 checkpoint therapy than the high stromal score group patients. Interestingly, high stromal score group with high immunized terms was resistant to immunotherapy compared to low stromal score group, promoting stromal relevant pathways participate the process, consistent with studies [33,34,35], emphasizing that stromal activation is the core mechanism of resistance to checkpoint blockade.
Survival analysis of DEGs
The Kaplan–Meier survival analysis with log-rank test was used to evaluate the potential roles of individual DEGs for OS. The results showed that seven upregulated and three down regulated DEGs predict poorer OS with statistical significance (Fig. 7; Additional file 5: Figure S2) (p < 0.05).

Correlation of expression of individual DEGs in the OS of patients from the TCGA database. Kaplan–Meier survival analysis was performed to compare OS between high (grey line) and low (black line) gene expression of selected DEGs
Construction of protein–protein interaction (PPI) network
To better understand the interplay among the identified DEGs, we obtained protein-PPI networks using STRING tool, and the pairs whose combined score > 0.4 were extracted for visualization by Cytoscape (Fig. 8). The 12 genes with the highest degree scores were selected, and the expression of these genes was validated in the GEO cohort (Fig. 9; Additional file 6: Figure S3). The common genes included F2, AHSG, AFP, FGA, FGB, APOA2, PNLIP, and BPIFB2. All of these genes had degree scores > 10 and were upregulated genes. These hub genes might contribute to stromal cell modulation in gastric cancer patients.

The protein-PPI network for the DEGs. Each node represents a gene. The dotted line indicates the relationship between the two genes. The number of lines is correlated with the degree scores of the genes

Validation of the hub genes in the GEO cohort. a F2: coagulation factor II; b AHSG: alpha 2-HS Glycoprotein; c FGB: fibrinogen beta chain; d APOA2: apolipoprotein A-II
GSEA and functional annotation of HUB genes
To explore the potential function of hub genes in GC, we applied GSEA on the TCGA database using the KEGG gene sets. Interestingly, the most important enriched KEGG pathways of BPIFB2 included cell cycle, Human T-cell leukemia virus 1 infection, Th17 cell differentiation, human immunodeficiency virus 1 infection, Th1 and Th2 cell differentiation, PD-L1 expression and PD-1 checkpoint pathway in cancer (Fig. 10) (p.adjust < 0.05).

Enriched KEGG pathways of BPIFB2 by using GSEA. p-value is presented in different colors. The dot size represents the number of genes
Discussion
Immune-checkpoint inhibitors targeting PD-1 and PD-L1 have shown promising clinical results for GC [36,37,38]. The recent advances in the relationship between gene expression and TME in gastric cancer patients [39,40,41] highlight the link between the immune-cell infiltration and PD-1/PD-L1 expression. However, only a subset of patients that contain tumor-infiltrating immune cells respond to PD-1/PD-L1 inhibitors. The pivotal role of the stroma is now receiving wide attention. Emerging data suggest that targeting CAFs/anti-angiogenic therapy in combination with anti-PD-1 immunotherapy is an effective way to overcome GC [20, 42].
In this study, we investigated a study cohort of 383 gastric cancer patients in the TCGA database and 300 patients in the GEO cohort, and generated stromal scores using the ESTIMATE algorithm. Our results showed that the stromal score is a robust biomarker for predicting survival in GC and guiding more effective immunotherapy strategies. Patients in the low stromal score group have a survival advantage over those in the high stromal score group and would benefit more from anti-PD-1 therapy. Interestingly, this result is consistent with the finding that GC tumors classified as MSI, which is a relatively low stromal score subtype, show promising results for the use of PD-1/PD-L1 blockade [38,39,40]. In line with previous research [43], the high stromal score subtype showed activation of transforming growth factor and epithelial mesenchymal, which were considered T-cell suppressive [33,34,35] and to be involved with immunotherapy resistance.
We then identified 119 DEGs, and many of the DEGs were involved in immune and stromal modulation. Of these 119 DEGs, ten genes were found to be associated with outcomes in GC patients. F2, AHSG, AFP, FGA, FGB, APOA2, PNLIP, and BPIFB2 were selected as the hub genes. BPIFB2 was involved with PD-L1 expression and PD-1 checkpoint pathway in cancer and immune-cell modulation. To date, limited data are available on BPIFB2. At the ESMO Asia 2018 Congress, Z. Karim and colleagues showed that BPIFB2 leads to gene expression alteration in the EMT markers. Together, these results suggest that BPIFB2 may be an effective treatment for PD-1/PD-L1 resistance.
Of these hub genes, FGB and APOA2 were identified to predict poorer survival in this cohort. More recently, Moriggi et al. reported that fibrinogen, encoded by FGB, is involved in ECM remodeling, including the epithelial–mesenchymal transition (EMT) [44]. Matthew D. Galsky further confirmed that EMT-related gene expression and T-cell infiltration are positively correlated, and the balance of T-cell vs. EMT/stromal elements may have prognostic/predictive implications with the antitumor immune response [45].
We were also interested in other hub genes that may play an essential role in tumor stromal cell infiltration, although they are not associated with patients’ survival. F2, which encodes the coagulation factor II (also known as thrombin), is a pro-inflammatory and pro-coagulant molecule that is elevated in various cancers, including breast and gastric cancers [46, 47]. Recent studies showed that disorders of the coagulation-fibrinolysis system are associated with the efficacy of immune-checkpoint inhibitors [48]. Thrombin activates platelets in the tumor microenvironment and induces angiogenesis by various mechanisms, including the section of pro-angiogenic factors and the promotion of the EMT [49,50,51]. AHSG encodes fetuin-A (also known as alpha 2-HS Glycoprotein), a 59-kDa negative acute phase glycoprotein in humans that is predominantly synthesized by liver parenchymal cells [52]. Recent studies indicated that fetuin-A mediates the attachment and growth of tumor cells in an indirect way, which involves cellular exosomes [53]. Fetuin-A is also associated with dendritic cell and T-regulatory cells [54].
Conclusion
Conclusively, the evaluation of the stromal component in TME, and molecular, genetic factors associated with TME stromal infiltration, revealed that the stromal score may be a biomarker to predict response to immune checkpoint and prognosis of GC patients. The related genes and signal pathways, provide a new idea for novel drug combination strategies of GC. Further investigation should be performed to confirm the potential genes and signaling pathways.
Availability of data and materials
The data that support the findings of this study are available from TCGA and GEO databases.
Abbreviations
- PD-1:
-
Programmed cell death 1
- PD-L1:
-
Programmed death-ligand 1
- GC:
-
Gastric cancer
- GEO:
-
Gene Expression Omnibus
- GSEA:
-
Gene set enrichment analysis
- OS:
-
Overall survival
- TME:
-
Tumor microenvironment
- CAF:
-
Cancer-associated fibroblasts
- ESTIMATE:
-
Estimation of stromal and immune cells in malignant tumor tissues using expression data
- TCGA:
-
The Cancer Genome Atlas
- DEGs:
-
Deferentially expressed genes
- TMB:
-
Tumor mutation burden
- GO:
-
Gene ontology
- PPI:
-
Protein–protein interaction
- EMT:
-
Epithelial–mesenchymal transition
References
Cordero-García E, Ramos-Esquivel A, Alpízar-Alpízar W. Evaluation of pharmacological therapies used in Costa Rica in patients with metastatic gastric cancer: a retrospective study. J Gastrointest Oncol. 2019;10(3):523.
Kim HJ, Oh SC. Novel systemic therapies for advanced gastric cancer. J Gastric Cancer. 2018;18(1):1–19.
Ryu H, Baek SW, Moon JY, Jo IS, Kim N, Lee HJ. C-C motif chemokine receptors in gastric cancer. Mol Clin Oncol. 2018;8(1):3–8.
Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376(9742):687–97.
Fuchs CS, Tomasek J, Yong CJ, Dumitru F, Passalacqua R, Goswami C, Safran H, Dos Santos LV, Aprile G, Ferry DR, et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): an international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet. 2014;383(9911):31–9.
Wilke H, Muro K, Van Cutsem E, Oh SC, Bodoky G, Shimada Y, Hironaka S, Sugimoto N, Lipatov O, Kim TY, et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. Lancet Oncol. 2014;15(11):1224–35.
Nishino M, Ramaiya NH, Hatabu H, Hodi FS. Monitoring immune-checkpoint blockade: response evaluation and biomarker development. Nat Rev Clin Oncol. 2017;14(11):655.
Fashoyin-Aje L, Donoghue M, Chen H, He K, Veeraraghavan J, Goldberg KB, Keegan P, McKee AE, Pazdur R. FDA approval summary: pembrolizumab for recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma expressing PD-L1. Oncologist. 2019;24(1):103.
Rosenberg JE, Hoffman-Censits J, Powles T, Van Der Heijden MS, Balar AV, Necchi A, Dawson N, O’Donnell PH, Balmanoukian A, Loriot Y. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387(10031):1909–20.
Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying cancers based on T-cell infiltration and PD-L1. Cancer Res. 2015;75(11):2139–45.
Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4(127):127–37.
Fuchs CS, Doi T, Jang RW, Muro K, Satoh T, Machado M, Sun W, Jalal SI, Shah MA, Metges J-P. Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: phase 2 clinical KEYNOTE-059 trial. JAMA Oncol. 2018;4(5):e180013.
Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–22.
Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121(3):335–48.
Ziani L, Chouaib S, Thiery J. Alteration of the antitumor immune response by cancer-associated fibroblasts. Front Immunol. 2018;9:414.
Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, Bassez A, Decaluwe H, Pircher A, Van den Eynde K, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med. 2018;24(8):1277–89.
Chen Y, Ramjiawan RR, Reiberger T, Ng MR, Hato T, Huang Y, Ochiai H, Kitahara S, Unan EC, Reddy TP, et al. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology. 2015;61(5):1591–602.
Liu XD, Hoang A, Zhou L, Kalra S, Yetil A, Sun M, Ding Z, Zhang X, Bai S, German P, et al. Resistance to antiangiogenic therapy is associated with an immunosuppressive tumor microenvironment in metastatic renal cell carcinoma. Cancer Immunol Res. 2015;3(9):1017–29.
Chen Y, Liu YC, Sung YC, Ramjiawan RR, Lin TT, Chang CC, Jeng KS, Chang CF, Liu CH, Gao DY, et al. Overcoming sorafenib evasion in hepatocellular carcinoma using CXCR4-targeted nanoparticles to co-deliver MEK-inhibitors. Sci Rep. 2017;7:44123.
Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, Hernandez G, Mier J, He X, Hodi FS, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun. 2016;7:12624.
Lee K, Hwang H, Nam KT. Immune response and the tumor microenvironment: how they communicate to regulate gastric cancer. Gut Liver. 2014;8(2):131.
Puré E, Lo A. Can targeting stroma pave the way to enhanced antitumor immunity and immunotherapy of solid tumors? Cancer Immunol Res. 2016;4(4):269–78.
Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW, Levine DA. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612.
Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30(5):413.
Shah N, Wang P, Wongvipat J, Karthaus WR, Abida W, Armenia J, Rockowitz S, Drier Y, Bernstein BE, Long HW. Regulation of the glucocorticoid receptor via a BET-dependent enhancer drives antiandrogen resistance in prostate cancer. eLife. 2017;6:e27861.
Priedigkeit N, Watters RJ, Lucas PC, Basudan A, Bhargava R, Horne W, Kolls JK, Fang Z, Rosenzweig MQ, Brufsky AM. Exome-capture RNA sequencing of decade-old breast cancers and matched decalcified bone metastases. JCI Insight. 2017;2(17):e95703.
Alonso MH, Aussó S, Lopez-Doriga A, Cordero D, Guinó E, Solé X, Barenys M, de Oca J, Capella G, Salazar R. Comprehensive analysis of copy number aberrations in microsatellite stable colon cancer in view of stromal component. Br J Cancer. 2017;117(3):421.
Di Jia SL, Li D, Xue H, Yang D, Liu Y. Mining TCGA database for genes of prognostic value in glioblastoma microenvironment. Aging. 2018;10(4):592.
Wagner GP, Kin K, Lynch VJ. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012;131(4):281–5.
Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24(10):1550–8.
Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics J Integr Biol. 2012;16(5):284–7.
Liu Y, Sethi NS, Hinoue T, Schneider BG, Cherniack AD, Sanchez-Vega F, Seoane JA, Farshidfar F, Bowlby R, Islam M. Comparative molecular analysis of gastrointestinal adenocarcinomas. Cancer Cell. 2018;33(4):721–735.e728.
Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544.
Tauriello DV, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, Sevillano M, Ibiza S, Cañellas A, Hernando-Momblona X. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554(7693):538.
Ravi R, Noonan KA, Pham V, Bedi R, Zhavoronkov A, Ozerov IV, Makarev E, Artemov AV, Wysocki PT, Mehra R. Bifunctional immune checkpoint-targeted antibody-ligand traps that simultaneously disable TGFβ enhance the efficacy of cancer immunotherapy. Nat Commun. 2018;9(1):741.
Moehler M, Delic M, Goepfert K, Aust D, Grabsch HI, Halama N, Heinrich B, Julie C, Lordick F, Lutz MP, et al. Immunotherapy in gastrointestinal cancer: recent results, current studies and future perspectives. Eur J Cancer. 2016;59:160–70.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–54.
Bilgin B, Sendur MA, Bulent Akinci M, Sener Dede D, Yalcin B. Targeting the PD-1 pathway: a new hope for gastrointestinal cancers. Curr Med Res Opin. 2017;33(4):749–59.
Ma G, Miao Q, Liu Y, Gao H, Lian J, Wang Y, Zeng X, Luo T, Ma L, Shen Z. High FoxP3 expression in tumour cells predicts better survival in gastric cancer and its role in tumour microenvironment. Br J Cancer. 2014;110(6):1552.
Oshima H, Ishikawa T, Yoshida G, Naoi K, Maeda Y, Naka K, Ju X, Yamada Y, Minamoto T, Mukaida N. TNF-α/TNFR1 signaling promotes gastric tumorigenesis through induction of Noxo1 and Gna14 in tumor cells. Oncogene. 2014;33(29):3820.
Terai S, Fushida S, Tsukada T, Kinoshita J, Oyama K, Okamoto K, Makino I, Tajima H, Ninomiya I, Fujimura T. Bone marrow derived “fibrocytes” contribute to tumor proliferation and fibrosis in gastric cancer. Gastric Cancer. 2015;18(2):306–13.
Kobayashi H, Enomoto A, Woods SL, Burt AD, Takahashi M, Worthley DL. Cancer-associated fibroblasts in gastrointestinal cancer. Nat Rev Gastroenterol Hepatol. 2019;16(5):282–95.
Wu X, Tao P, Zhou Q, Li J, Yu Z, Wang X, Li J, Li C, Yan M, Zhu Z, et al. IL-6 secreted by cancer-associated fibroblasts promotes epithelial–mesenchymal transition and metastasis of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget. 2017;8(13):20741–50.
Moriggi M, Giussani M, Torretta E, Capitanio D, Sandri M, Leone R, De Palma S, Vasso M, Vozzi G, Tagliabue E. ECM remodeling in breast cancer with different grade: contribution of 2D-DIGE proteomics. Proteomics. 2018;18(24):1800278.
Wang L, Saci A, Szabo PM, Chasalow SD, Castillo-Martin M, Domingo-Domenech J, Siefker-Radtke A, Sharma P, Sfakianos JP, Gong Y, et al. EMT- and stroma-related gene expression and resistance to PD-1 blockade in urothelial cancer. Nat Commun. 2018;9(1):3503.
Bazzi ZA, Lanoue D, El-Youssef M, Romagnuolo R, Tubman J, Cavallo-Medved D, Porter LA, Boffa MB. Activated thrombin-activatable fibrinolysis inhibitor (TAFIa) attenuates breast cancer cell metastatic behaviors through inhibition of plasminogen activation and extracellular proteolysis. BMC Cancer. 2016;16(1):328.
Otsuki T, Fujimoto D, Hirono Y, Goi T, Yamaguchi A. Thrombin conducts epithelial–mesenchymal transition via protease-activated receptor-1 in human gastric cancer. Int J Oncol. 2014;45(6):2287–94.
Sato R, Imamura K, Sakata S, Ikeda T, Horio Y, Iyama S, Akaike K, Hamada S, Jodai T, Nakashima K, et al. Disorder of coagulation-fibrinolysis system: an emerging toxicity of anti-PD-1/PD-L1 monoclonal antibodies. J Clin Med. 2019;8(6):762.
Kanikarla-Marie P, Lam M, Menter DG, Kopetz S. Platelets, circulating tumor cells, and the circulome. Cancer Metastasis Rev. 2017;36(2):235–48.
Barrow AD, Edeling MA, Trifonov V, Luo J, Goyal P, Bohl B, Bando JK, Kim AH, Walker J, Andahazy M. Natural killer cells control tumor growth by sensing a growth factor. Cell. 2018;172(3):534–548.e519.
Jiang L, Luan Y, Miao X, Sun C, Li K, Huang Z, Xu D, Zhang M, Kong F, Li N. Platelet releasate promotes breast cancer growth and angiogenesis via VEGF–integrin cooperative signalling. Br J Cancer. 2017;117(5):695.
Brown W, Dziegielewska K, Saunders N, Møsllgård K. Fetui—an old friend revisited. BioEssays. 1992;14(11):749–55.
Watson K, Koumangoye R, Thompson P, Sakwe AM, Patel T, Pratap S, Ochieng J. Fetuin-A triggers the secretion of a novel set of exosomes in detached tumor cells that mediate their adhesion and spreading. FEBS Lett. 2012;586(19):3458–63.
Harris VK, Bell L, Langan RA, Tuddenham J, Landy M, Sadiq SA. Fetuin-A deficiency protects mice from Experimental Autoimmune Encephalomyelitis (EAE) and correlates with altered innate immune response. PLoS ONE. 2017;12(4):e0175575.
Acknowledgements
We sincerely thank the researchers for providing their TCGA and GEO databases information online, it is our pleasure to acknowledge their contributions.
Funding
This work was granted by the National Natural Science Foundation of China (Nos. 81301301; 81601579); Chen Xiao-ping Foundation for the Development of Science and Technology of Hubei Province (No. CXPJJH11800001-2018203).
Author information
Authors and Affiliations
Contributions
XX, ZG performed the design of the study. HZ, TY, and DL performed data acquisition. QR, PZ, and ZG carried out analysis and interpretation, and drafting of the manuscript. XX revised the manuscript critically for important intellectual contents. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Association between stromal score expression and the clinical parameters in the GEO cohort.
Additional file 2: Figure S1.
Stromal scores were associated with T-stage in the GEO cohort. A high stromal score is associated with advanced T-stage.
Additional file 3: Table S2.
Univariable and multivariable analyses of the stromal score and clinical variables in the GEO cohort.
Additional file 4: Table S3.
Go terms of DEGs.
Additional file 5: Figure S2.
Kaplan–Meier survival analysis of C8B, GFL1, INHBB, PRSS1, RHOXF2, SLC38A4, VTN, and Z1C1.
Additional file 6: Figure S3.
Comparison of AFP, FGA, PNLIP, and BPIFB2 in different groups of the stromal score.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Ren, Q., Zhu, P., Zhang, H. et al. Identification and validation of stromal-tumor microenvironment-based subtypes tightly associated with PD-1/PD-L1 immunotherapy and outcomes in patients with gastric cancer. Cancer Cell Int 20, 92 (2020). https://doi.org/10.1186/s12935-020-01173-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-020-01173-3