
Abstract
Background
Cancer has been a worldwide health problem with a high risk of morbidity and mortality, however ideal biomarkers for effective screening and diagnosis of cancer patients are still lacking. Small nucleolar RNA host gene 16 (SNHG16) is newly identified lncRNA with abnormal expression in several human malignancies. However, its prognostic value remains controversial. This meta-analysis aimed to synthesize available data to clarify the association between SNHG16 expression levels and clinical prognosis value in multiple cancers.
Methods
Extensive literature retrieval was conducted to identify eligible studies, and data regarding SNHG16 expression levels on survival outcomes and clinicopathological features were extracted and pooled for calculation of the hazard ratios (HRs) or odds ratios (ORs) with 95% confidence intervals (CIs). Forest plots were applied to show the association between SNHG16 expression and survival prognosis. Additionally, The Cancer Genome Atlas (TCGA) dataset was screened and extracted for validation of the results in this meta-analysis.
Results
A total of eight studies comprising 568 patients were included in the final meta-analysis according to the inclusion and exclusion criteria. In the pooled analysis, high SNHG16 expression significantly predicted worse overall survival (OS) in various cancers (HR = 1.87, 95% CI 1.54–2.26, P < 0.001), and recurrence-free survival (RFS) in bladder cancer (HR = 1.68, 95% CI 1.01–2.79, P = 0.045). Meanwhile, stratified analyses revealed that the survival analysis method, tumor type, sample size, and cut-off value did not alter the predictive value of SNHG16 for OS in cancer patients. In addition, compared to the low SNHG16 expression group, patients with high SNHG16 expression were more prone to worse clinicopathological features, such as larger tumor size, advanced clinical stage, lymph node metastasis (LNM) and distant metastasis (DM). Exploration of TCGA dataset further validated that the upregulated SNHG16 expression predicted unfavorable OS and disease-free survival (DFS) in cancer patients.
Conclusions
The present study implicated that aberrant expression of lncRNA SNHG16 was strongly associated with clinical survival outcomes in various cancers, and therefore might serve as a promising biomarker for predicting prognosis of human cancers.
Similar content being viewed by others
Background
Nowadays, cancer has become one of the most prevalent causes of mortality worldwide [1]. Over the past century, there has been a dramatic improvement in modern treatments for cancer including surgery, adjuvant therapy and supportive therapy [2,3,4]. Despite this, the patients’ survival rate are still unsatisfied and quality of life remains largely to be improved, especially for those with advanced clinical stage or metastasis [5]. It has been well established that early diagnosis and treatment of cancer could greatly reduce its mortality. However, the insufficiency of suitable biomarkers presents a major obstacle to this issue [6]. Consequently, there is an urgent need to find new biological targets in the carcinogenesis of tumors.
LncRNAs is a class of RNAs with a length of more than 200 nucleotides (nt) [7]. Previous evidences suggest that lncRNAs can regulate gene expression at all levels-transcriptional, translational, and post-translational-by interacting with DNA, RNA and protein [8], and subsequently accomplish a remarkable variety of biological processes [8, 9]. In recent years, an increasing number of lncRNAs have been revealed to be aberrantly expressed in human cancers [10]. Moreover, dysregulation of lncRNAs is significantly correlated with cancer cell proliferation, migration, metastasis and recurrence, implicating a crucial role of lncRNAs in regulation of carcinogenesis and cancer progression [7].
Small nucleolar RNA host gene 16 (SNHG16) is a recently identified lncRNA with abnormal expression in multiple cancers [11,12,13,14]. Increased expression of SNHG16 usually predicted poor prognosis in multiple cancers including osteosarcoma [15], bladder cancer [16,17,18], esophagus cancer [19], non-small cell lung cancer (NSCLC) [11], glioma [20, 21], oral squamous cell carcinoma [13], hepatocellular carcinoma (HCC) [22], breast cancer [23], and ovarian cancer [24]. In these cancers, high expression levels of SNHG16 were usually correlated with worse clinicopathological features, such as tumor size, clinical stage, lymph nodes metastasis (LNM), distant metastasis (DM), and drug resistance. For incidence, in HCC, SNHG16 predicted portal vein tumor thrombus and sorafenib resistance [25]. Moreover, SNHG16 may be also engaged in the pathogenesis and progression of cancers, including proliferation, migration and invasion [26]. Furthermore, emerging studies have demonstrated and emphasized the importance of lncRNA SNHG16 in regulation of cancer-related signaling pathways, including Wnt/β-catenin, PI3K/Akt, and JAK2/STAT3 pathway [21, 27,28,29]. Collectively, SNHG16 may serve as a risk factor and therapeutic target for several types of human malignancies. However, most researches evaluating the prognostic value of SNHG16 in cancer survivals are limited due to small sample size and the contentious outcomes in clinical settings. In this meta-analysis, we report, for the first time, the comprehensive role of SNHG16 expression in human pan-cancers, which may provide promising targets for the development of novel diagnostic and therapeutic strategies against cancers.
Methods
Publication search strategy
The present study was rigorously projected, reviewed and reported in accordance with the PRISMA checklist [30,31,32]. We searched numerous electronic databases, including MEDLINE, Web of Science, Scopus, the Cochrane Library, EmBase, and China National Knowledge Infrastructure (CNKI) for eligible studies from their inceptions up to Jan 1st, 2019. The following search items were used: “small nucleolar RNA host gene 16 OR SNHG16” AND “tumor OR cancer OR carcinoma OR sarcoma” with language limitation to English and Chinese. Additionally, the citation lists in these retrieved articles were manually searched for identification of other relevant studies to ensure sensitivity of the search strategy.
Inclusion and exclusion criteria
Two investigators (CHZ and XLR) critically reviewed and assessed all eligible studies independently. Studies for inclusion should meet the following criteria: (a) SNHG16 expression level was examined in human cancer tissues and adjacent normal tissues; (b) patients were separated into high and low expression groups based on the cut-off value of SNHG16 expression; (c) sufficient data regarding association between SNHG16 expression and survival outcomes or clinicopathological features; and (d) estimated hazard ratios (HRs) or odds ratios (ORs) with corresponding 95% confidence intervals (CIs) for survival outcomes could be extracted directly or indirectly.
While those studies should be excluded if meet any one of the following criteria: (a) irrelevant to cancer and SNHG16; (b) focused on the molecular structure or functions of SNHG16 rather than its correlation with survival outcomes; (c) duplicate publications; (d) animal studies; and (e) publications without usable data, such as reviews, letters to the editor, and abstracts.
Data extraction and quality control
The following information was extracted by two independent investigators (CHZ and XLR) from each selected study: Surname of first author, publication year, country of origin, tumor type, sample type and size, follow-up months, detection assay, clinical stage, metastasis, cut-off value, survival outcome including overall survival (OS), disease-free survival (DFS), progression-free survival (PFS), and recurrence-free survival (RFS). If data of interest were not accessible, we obtained the missing data by contacting the corresponding author of enrolled articles. If only Kaplan-Meier (K–M) curves were provided in some studies, we used the Engauge Digitizer (Version 4.1) to calculate the pooled HRs and 95% CIs through indirect extraction from the plots [33].
Since all studies included in our meta-analysis were cohort studies, Newcastle–Ottawa Scale (NOS) with score ranging from 0 to 9 was utilized to carry out the quality assessment by two investigators (JYH and WCW) [34]. Included studies with NOS score ≥ 7 were considered of high methodological quality.
Validation by reviewing public data
This study meets the publication guidelines provided by The Cancer Genome Atlas (TCGA). Gene Expression Profiling Interactive Analysis (GEPIA) was used in this meta-analysis to verify the correlations with OS and DFS and to detect the difference in expression levels of SNHG16 between tumor and normal tissues [35]. The survival analysis was calculated by K–M method and logrank test, and the HRs and p value were shown in the figure of K–M curves as previously described [36].
Statistical analysis
Analyses were conducted by using STATA software (Version 12.0) and Review Manager (RevMan 5.3). Pooled HRs (ORs) and 95% CIs were extracted from the enrolled studies. Chi square-based Q test and I2 statistics were used to determine the heterogeneity across the eligible studies. If I2 > 50% or p-value < 0.05, we considered the heterogeneity was significant and the random-effect model was adopted. On the contrary, the fixed-effect model was applied. Publication bias was evaluated by Egger’s test as well as visual inspection of the symmetry of funnel plot. Sensitivity analysis was performed by sequential omission of each individual study so as to testify the stability of results as previously described [37].
Results
Characteristics and eligible studies
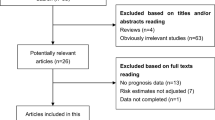
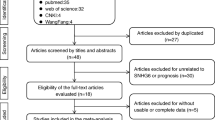
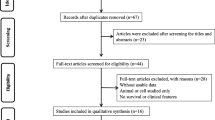
A total of 147 publications were initially identified as potential articles, of which 66 were excluded as duplicates. Afterwards, 81 publications were screened via their titles and abstracts, and 58 studies were further excluded since they were case reports, reviews, meeting abstracts, or irrelevant topics. Consequently, 23 full-text articles were thoroughly evaluated. Among them, fifteen studies lacking of survival data were excluded. Thus, eight studies comprising 568 patients were included in the final meta-analysis. The selection procedure was concisely demonstrated by a flow diagram in Fig. 1.

Flow diagram of study selection procedure
The characteristics of the included studies are presented in Table 1. These articles were published between 2016 and 2019 with the sample size ranging from 38 to 128. All studies were carried out in China and quantitative real-time polymerase chain reaction (qRT-PCR) was applied as the detecting method. Six studies containing five different tumor types utilized tissue samples to detect SNHG16 expression, of which four studies used the median value and the other two studies used the mean value. Association between expression level of SNHG16 with OS were included in the abovementioned studies, including two bladder cancer, one cervical cancer, one esophageal squamous cell carcinoma (ESCC), one glioma, one NSCLC, and one ovarian cancer. The rest two studies used serum samples to detect SNHG16 expression and reported RFS, both of which used the median value as the cut-off value. In all eligible studies, patients were divided into high or low SNHG16 expression groups based on the cut off value. The follow-up months ranged from 60 to 80 months. Univariate and multivariate analysis were both adopted in four studies respectively. As for clinical stage, there were two studies used tumor node metastasis (TNM) classification system, while two studies adopted the International Federation of Gynecology and Obstetrics (FIGO) staging. All of these included studies were cohort studies and of high quality with their NOS scores ≥ 7.
Association between SNHG16 and OS
Six studies using tissue samples were included for OS analysis. In the absence of obvious heterogeneity among these studies (I2 = 9.2%, p = 0.357), fixed-effects model was used to calculate the HR and 95% CI. The pooled result demonstrated that high SNHG16 expression significantly associated with worse OS in cancers (HR = 1.87, 95% CI 1.54–2.26, P < 0.001) (Fig. 2a).

Forest plots of studies evaluating the hazard ratios of high SNHG16 expression in cancers for a overall survival and b recurrence-free survival
Association between SNHG16 and RFS
Two studies utilizing serum samples provided suitable data for RFS analysis. The fixed-effects model was applied to analyze the pooled HR and its 95% CI since no apparent heterogeneity was observed (I2 = 0.0%, p = 0.918). As shown in Fig. 2b, the results indicated that high SNHG16 expression in serum predicted unfavorable RFS in bladder cancer (HR = 1.68, 95% CI 1.01–2.79, P = 0.045).
Subgroup analysis of association between SNHG16 and OS
Besides, we performed stratified analyses to investigate the association between SNHG16 expression level and OS in divergent subgroups in the light of survival analysis method (univariate or multivariate analysis), tumor type (gynecologic tumor or others), sample size (more or less than 60), and cut-off value (mean or median). As depicted in Fig. 3 and Table 2, all stratified analyses did not alter the predictive value of SNHG16 for OS in several kinds of cancers.

Forest plots evaluating the stratified analyses of SNHG16 expression on OS in regard to subgroup including a survival analysis method, b tumor type, c sample size and d cut-off value
Association between SNHG16 and clinicopathologic parameters
ORs and its 95% CIs were utilized to investigate the correlation between SNHG16 expression level and clinicopathologic parameters including age, gender, smoking history, tumor size, clinical stage, LNM and DM. The results of these analyses were presented in Fig. 4 and Table 3. The fixed-effect model was applied in all analyses. From the pooled ORs, no significant association was detected between SNHG16 expression and age, gender and smoking history. Notably, high SNHG16 expression was significantly correlated with larger tumor size (OR = 6.36, 95% CI 2.43–16.60, P = 0.0002), poor clinical stage (OR = 2.91, 95% CI 1.60–5.28, P = 0.005), LNM (OR = 4.42, 95% CI 2.66–7.35, P = 0.0001) and DM (OR = 3.86, 95% CI 1.92–7.77, P = 0.002).

Forest plots evaluating the relationship between SNHG16 expression and clinicopathologic features, including a age (≥ 60/< 60), b gender, c smoking history, d tumor size (≥ 5 cm/< 5 cm), e clinical stage, f lymph node metastasis and g distant metastasis
Sensitivity analysis
In order to test the stability of the pooled result of the association between SNHG16 expression and OS, sensitivity analysis was conducted by removing each eligible study. As demonstrated in Fig. 5a, when “Lu 2018” [21] was removed, the pooled result fluctuated. Subsequently, the pooled HR was calculated again after removing “Lu 2018”, and the result showed that high SNHG16 expression still predicted worse OS in multiple cancers (HR = 2.31, 95% CI 1.71–3.13, P < 0.001), which meant that the significance of the pooled result was not altered by the influential study. Therefore, our pooled result of SNHG16 expression on prediction of OS was reliable.

a Sensitivity analysis of pooled HR for overall survival. b Begg’s funnel plot of SNHG16 for overall survival. c Funnel plot of “Trim and Fill analysis” (The trim and fill adjusted HR = 1.69, 95% CI 1.41–2.01, fixed-effects model)
Publication bias
For meta-analysis of the association between SNHG16 expression and OS, Begg’s funnel plot and Egger’s regression test were performed to test for publication bias. There was evidence of publication bias based on asymmetry in the Begg’s funnel plot (Fig. 5b) as well as the result of Egger’s regression test (P = 0.025). Later, “Trim and Fill analysis” was adopted to evaluate the influence of publication bias as previously described [38]. As depicted in Fig. 5c, the adjusted HR and 95% CI was 1.69 (1.41–2.01), indicating that the publication bias did not have significant influence on the pooled result, and thus our result was credible.
Validation of the results in TCGA dataset
In order to further validate our results, we utilized TCGA dataset to investigate SNHG16 expression level in various cancers. As shown in Fig. 6, SNHG16 was aberrantly expressed in sarcoma, breast invasion carcinoma, bladder urothelial carcinoma, esophageal carcinoma, stomach adenocarcinoma, liver hepatocellular carcinoma, colon adenocarcinoma, rectum adenocarcinoma, lung adenocarcinoma, and lung squamous cell carcinoma when compared with normal control. In addition, the violin plot showed that SNHG16 expression level was significantly associated with clinical stage in human pan-cancers. Moreover, we adopted survival plots in GEPIA via merging SNHG16 expression data and OS (DFS) data of malignancies from all of the TCGA dataset, which containing 9502 patients divided into high or low expression group based on SNHG16 expression. The results indicated that the upregulated SNHG16 expression predicted unfavorable OS and DFS, which certified our results in this meta-analysis.

Validation of SNHG16 expression in various cancers in TCGA cohort. a The expression levels of SNHG16 in SARC (sarcoma), BRCA (breast invasion carcinoma), BLCA (bladder urothelial carcinoma), and CESC (cervical squamous cell carcinoma and endocervical adenocarcinoma). b The expression levels of SNHG16 in ESCA (esophageal carcinoma), STAD (stomach adenocarcinoma), LIHC (liver hepatocellular carcinoma), COAD (colon adenocarcinoma), READ (rectum adenocarcinoma), LUAD (lung adenocarcinoma), and LUSC (lung squamous cell carcinoma). c Violin plot showing SNHG16 expression in different major clinical stage of pan-cancers in TCGA cohort. d Overall survival plot of SNHG16 in TCGA cohort (n = 9502). e Disease-free survival plot of SNHG16 in TCGA cohort (n = 9502)
Discussion
LncRNAs were previously recognized as “junk DNA” or “transcriptional noise” and did not attract too much attention among researchers in the past decades [39]. Recently, role of lncRNAs in human diseases has grown in importance due to advancement in the application of next-generation genome wide sequencing and microarray [40,41,42,43]. Previous studies have demonstrated that lncRNAs are abnormally expressed in cancer and acted as oncogenes or tumor suppressors, revealing an important role in cancer prognosis [44,45,46]. For instance, tubulin alpha 4b (TUBA4B) was significantly decreased in cancer tissues compared with adjacent normal specimens [47]. Low TUBA4B expression was closely correlated with pathological grade, LNM, OS, DFS, and RFS in cancer patients and can be a novel biomarker for the prognosis of various cancers [47, 48]. Likewise, plasmacytoma variant translocation gene 1 (PVT1) expressions in cancer tissues were higher than paired healthy controls. Overexpression of serum PVT1 was markedly associated with larger tumor size, advanced clinical stage, and accurately predicted the disease and poor prognosis [49,50,51]. Moreover, PVT1 could engage in multiple signaling pathways or act as competitive endogenous RNA (ceRNA) to affect the biological function of cancer cells via interacting with miRNAs and target genes, indicating a novel perspective for therapeutic strategies of human cancers [51, 52].
SNHG16 is a newly identified lncRNA and has been reported to be aberrantly expressed in multiple malignancies. For instance, SNHG16 expression levels were significantly upregulated in osteosarcoma [15], glioma [20, 21], colorectal adenocarcinoma [27], breast cancer [23], cervical cancer [26], ovarian cancer [24], bladder cancer [17, 18], ESCC [19, 28], NSCLC [11], oral squamous cell carcinoma [13], and acute lymphoblastic leukemia [53], but downregulated in HCC [22]. However, data in TCGA showed that SNHG16 was overexpressed in liver hepatocellular carcinoma (LIHC), which is contradictory to the study reported by Xu et al. [22]. Additionally, a strong correlation between SNHG16 and cancer biological functions has been well illustrated in the literature. Overexpression of SNHG16 could lead to changes in cancer cell proliferation [18, 19, 24], apoptosis [21], migration [23], invasion [11, 26], epithelial-mesenchymal transition (EMT) [19] and chemoresistance in a majority of cancers [22]. In our meta-analysis, we explored the correlation between SNHG16 expression levels and cancer prognostic parameters. The pooled results revealed that high expression levels of SNHG16 predicted unfavorable OS. Furthermore, a shorter RFS was observed in bladder cancer patients with high expression of SNHG16 in serum, implying that expression of SNHG16 in serum was a hazardous factor for the recurrence of bladder cancer. Besides, SNHG16 was also correlated with PFS and DFS in two studies. Glioma patients with higher SNHG16 expression had a significantly poorer PFS based on the result of multivariate analysis (HR = 3.167, 95% CI 1.552–6.231) [21]. SNHG16 expression could serve as independent predictor for DFS in NSCLC patients (HR = 2.641, 95% CI 1.394–5.002) [11]. Therefore, SNHG16 upregulation was closely associated with poor prognosis. Our pooled results also showed that patients with high SNHG16 expression were more prone to worse clinicopathological features including larger tumor size, advanced clinical tumor stage, LNM and DM. Besides, there were other parameters that cannot be included in the meta-analysis since they were only reported in a single study. For instance, cervical patients with high SNHG16 expression had poorer differentiation (P = 0.047) and worse FIGO stage (P = 0.008). In ovarian cancer, higher SNHG16 expression predicted higher histological grade (P = 0.002) [24]. Moreover, subgroup analyses showed that survival analysis method, tumor type, sample size and cut-off value did not alter the predictive value of SNHG16 on OS. Taken together, SNHG16 could serve as a functional regulator and potential biomarker of poor prognosis in pan-cancer patients.
Further mechanism studies highlighted that SNHG16 may function as ceRNA by directly sponge to miRNA and thereby regulating target genes in cancers, such as miR-205/ZEB1 in osteosarcoma [15], hsa-miR-93 in HCC [22], miR-4518/PRMT5 or miR-20a-5p/E2F1 in glioma [20, 21], miR-146a/MUC5AC in NSCLC [11], miR-98/E2F5 in breast cancer [23], miR-216A-5p/ZEB1 in cervical cancer [26], miR-140-5p/ZEB1 in ESCC [19], miR-98/STAT3 in bladder cancer [17], and hsa-miR-124-3p in acute lymphoblastic leukemia [53]. Besides, SNHG16 could interact with a variety of signaling pathways including Wnt/β-catenin [17, 27] and PI3 K/Akt [21, 24] in the pathogenesis of cancers. A brief summary of SNHG16 with their potential targets, functional roles, signaling pathways, and relevant miRNAs was presented in Table 4 and illustrated in Fig. 7.

Summary of aberrant expression of SNHG16 in various types of human malignancies (a). A comprehensive biological role of SNHG16 in carcinogenesis of human cancers, including proliferation, apoptosis, migration, invasion, epithelial-mesenchymal transition (EMT) and chemoresistance regulation. SNHG16 may function as competitive endogenous RNA (ceRNA) by directly sponge to miRNA and subsequently regulating target geness, or interact with several signaling pathways in the pathogenesis of cancers (b)
Of note, several limitations should be addressed in current meta-analysis. First, all the included studies were performed within Chinese populations, thus caution must be applied, as the findings might not be able to generalize to other populations. Second, some of the HR values had been computed through reconstruction of K–M curves instead of directly obtaining from the original studies, which inevitably could generate possible bias. Third, all included studies set the cut-off value as mean or median value without detailed description on the calculation process or providing original data. Therefore, the real cut-off value for each study was unknown, and the different cut-off values across the selected studies may lead to potential bias. Fourth, heterogeneity may exist in different treatments for diverse cancer patients, which may contribute to the bias. Fifth, most included articles reported positive results rather than negative results, which may introduce publication bias.
Conclusions
In conclusion, our results infer that high SNHG16 expression was strongly associated with unfavorable survival outcome of several cancers and therefore might serve as a novel prognostic biomarker and potential therapeutic target in cancer patients. However, studies with a larger sample size on the current topic are still needed to substantiate these results.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- Akt:
-
protein kinase B
- ceRNA:
-
competitive endogenous RNA
- CI:
-
confidence interval
- CNKI:
-
China National Knowledge Infrastructure
- DFS:
-
disease-free survival
- DM:
-
distant metastasis
- E2F1:
-
E2F transcription factor 1
- E2F5:
-
E2F transcription factor 5
- EMT:
-
epithelial–mesenchymal transition
- ESCC:
-
esophageal squamous cell carcinoma
- FIGO:
-
the International Federation of Gynecology and Obstetrics
- GEPIA:
-
Gene Expression Profiling Interactive Analysis
- HCC:
-
hepatocellular carcinoma
- HR:
-
hazard ratio
- JAK2:
-
Janus kinase 2
- K–M curve:
-
Kaplan–Meier curve
- LNM:
-
lymph node metastasis
- MMP-2/-9:
-
matrix metalloproteinase 2/9
- MUC5AC:
-
mucin 5AC
- NOS:
-
Newcastle–Ottawa Scale
- NSCLC:
-
non-small cell lung cancer
- OR:
-
odd ratio
- OS:
-
overall survival
- PFS:
-
progression-free survival
- PI3 K:
-
phosphoinositide 3-kinase
- PRMT5:
-
protein arginine methyltransferase 5
- PVT1:
-
plasmacytoma variant translocation gene 1
- qRT-PCR:
-
quantitative real-time polymerase chain reaction
- RFS:
-
recurrence-free survival
- SNHG16:
-
small nucleolar RNA host gene 16
- STAT3:
-
signal transducer and activator of transcription 3
- TCGA:
-
The Cancer Genome Atlas
- TNM:
-
tumor node metastasis
- TUBA4B:
-
tubulin alpha 4b
- ZEB1:
-
zinc finger E-box-binding homeobox 1
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108.
Wright AA, Keating NL, Ayanian JZ, Chrischilles EA, Kahn KL, Ritchie CS, et al. Family perspectives on aggressive cancer care near the end of life. JAMA. 2016;315(3):284–92.
Wu X, Chung VC, Hui EP, Ziea ET, Ng BF, Ho RS, et al. Effectiveness of acupuncture and related therapies for palliative care of cancer: overview of systematic reviews. Sci Rep. 2015;5:16776.
Jiang J, Zhou H, Ni C, Hu X, Mou Y, Huang D, et al. Immunotherapy in pancreatic cancer: new hope or mission impossible? Cancer Lett. 2019;445:57–64.
Binabaj MM, Bahrami A, Bahreyni A, Shafiee M, Rahmani F, Khazaei M, et al. The prognostic value of long noncoding RNA MEG3 expression in the survival of patients with cancer: a meta-analysis. J Cell Biochem. 2018;119(11):9583–90.
Zhu HE, Zhang LY, Yan S, Li WM, Cui JT, Zhu M, et al. LncRNA16 is a potential biomarker for diagnosis of early-stage lung cancer that promotes cell proliferation by regulating the cell cycle. Oncotarget. 2017;8(5):7867–77.
Schmitt AM, Chang HY. Long noncoding RNAs in cancer pathways. Cancer Cell. 2016;29(4):452–63.
He J, Tu C, Liu Y. Role of lncRNAs in aging and age-related diseases. Aging Med. 2018;1(2):158–75.
Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 2014;157(1):77–94.
Liu J-L, Zhang W-Q, Zhao M, Huang M-Y. Upregulation of long noncoding RNA XIST is associated with poor prognosis in human cancers. J Cell Physiol. 2018;234(5):6594–600.
Han W, Du X, Liu M, Wang J, Sun L, Li Y. Increased expression of long non-coding RNA SNHG16 correlates with tumor progression and poor prognosis in non-small cell lung cancer. Int J Biol Macromol. 2019;121:270–8.
Wen Q, Zhao L, Wang T, Lv N, Cheng X, Zhang G, et al. LncRNA SNHG16 drives proliferation and invasion of papillary thyroid cancer through modulation of miR-497. Onco Targets Ther. 2019;12:699–708.
Li S, Zhang S, Chen J. c-Myc induced upregulation of long non-coding RNA SNHG16 enhances progression and carcinogenesis in oral squamous cell carcinoma. Cancer Gene Ther. 2019. https://doi.org/10.1038/s41417-018-0072-8.
Liu S, Zhang W, Liu K, Liu Y. LncRNA SNHG16 promotes tumor growth of pancreatic cancer by targeting miR-218-5p. Biomed Pharmacother. 2019;114:108862.
Zhu C, Cheng D, Qiu X, Zhuang M, Liu Z. Long noncoding RNA SNHG16 promotes cell proliferation by sponging microRNA-205 and upregulating ZEB1 expression in osteosarcoma. Cell Physiol Biochem. 2018;51(1):429–40.
Zhang S, Du L, Wang L, Jiang X, Zhan Y, Li J, et al. Evaluation of serum exosomal LncRNA-based biomarker panel for diagnosis and recurrence prediction of bladder cancer. J Cell Mol Med. 2019;23(2):1396–405.
Feng F, Chen A, Huang J, Xia Q, Chen Y, Jin X. Long noncoding RNA SNHG16 contributes to the development of bladder cancer via regulating miR-98/STAT3/Wnt/beta-catenin pathway axis. J Cell Biochem. 2018;119(11):9408–18.
Cao X, Xu J, Yue D. LncRNA-SNHG16 predicts poor prognosis and promotes tumor proliferation through epigenetically silencing p21 in bladder cancer. Cancer Gene Ther. 2018;25(1–2):10–7.
Zhang K, Chen J, Song H, Chen LB. SNHG16/miR-140-5p axis promotes esophagus cancer cell proliferation, migration and EMT formation through regulating ZEB1. Oncotarget. 2018;9(1):1028–40.
Yang BY, Meng Q, Sun Y, Gao L, Yang JX. Long non-coding RNA SNHG16 contributes to glioma malignancy by competitively binding miR-20a-5p with E2F1. J Biol Regul Homeost Agents. 2018;32(2):251–61.
Lu YF, Cai XL, Li ZZ, Lv J, Xiang Y, Chen JJ, et al. LncRNA SNHG16 functions as an oncogene by sponging MiR-4518 and up-regulating PRMT5 expression in glioma. Cell Physiol Biochem. 2018;45(5):1975–85.
Xu F, Zha G, Wu Y, Cai W, Ao J. Overexpressing lncRNA SNHG16 inhibited HCC proliferation and chemoresistance by functionally sponging hsa-miR-93. Onco Targets Ther. 2018;11:8855–63.
Cai C, Huo Q, Wang X, Chen B, Yang Q. SNHG16 contributes to breast cancer cell migration by competitively binding miR-98 with E2F5. Biochem Biophys Res Commun. 2017;485(2):272–8.
Yang XS, Wang GX, Luo L. Long non-coding RNA SNHG16 promotes cell growth and metastasis in ovarian cancer. Eur Rev Med Pharmacol Sci. 2018;22(3):616–22.
Guo Z, Zhang J, Fan L, Liu J, Yu H, Li X, et al. Long noncoding RNA (lncRNA) small nucleolar RNA Host gene 16 (SNHG16) predicts poor prognosis and sorafenib resistance in hepatocellular carcinoma. Med Sci Monit. 2019;25:2079–86.
Zhu H, Zeng Y, Zhou C-c, Ye W. SNHG16/miR-216-5p/ZEB1 signal pathway contributes to the tumorigenesis of cervical cancer cells. Arch Biochem Biophys. 2018;637:1–8.
Christensen LL, True K, Hamilton MP, Nielsen MM, Damas ND, Damgaard CK, et al. SNHG16 is regulated by the Wnt pathway in colorectal cancer and affects genes involved in lipid metabolism. Mol Oncol. 2016;10(8):1266–82.
Han GH, Lu KJ, Wang P, Ye J, Ye YY, Huang JX. LncRNA SNHG16 predicts poor prognosis in ESCC and promotes cell proliferation and invasion by regulating Wnt/beta-catenin signaling pathway. Eur Rev Med Pharmacol Sci. 2018;22(12):3795–803.
Wang X, Kan J, Han J, Zhang W, Bai L, Wu H. LncRNA SNHG16 functions as an oncogene by sponging MiR-135a and promotes JAK2/STAT3 signal pathway in gastric cancer. J Cancer. 2019;10(4):1013–22.
Moher D, Liberati A, Tetzlaff J, Altman DG, Group P. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ. 2009;339:b2535.
Moher D, Shamseer L, Clarke M, Ghersi D, Liberati A, Petticrew M, et al. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst Rev. 2015;4:1.
Shamseer L, Moher D, Clarke M, Ghersi D, Liberati A, Petticrew M, et al. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015: elaboration and explanation. BMJ. 2015;350:g7647.
Xu L, Yan N, Li Z, Luo L, Wu X, Liu Q, et al. A comparison of fulvestrant plus a targeted agent with fulvestrant alone in hormone receptor-positive advanced breast cancer that progressed on prior endocrine therapy: a meta-analysis. Onco Targets Ther. 2018;11:8389–98.
Stang A. Critical evaluation of the Newcastle–Ottawa scale for the assessment of the quality of nonrandomized studies in meta-analyses. Eur J Epidemiol. 2010;25(9):603–5.
Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–102.
Zhao W, Wang Z, Fang X, Li N, Fang J. Long noncoding RNA Breast cancer antiestrogen resistance 4 is associated with cancer progression and its significant prognostic value. J Cell Physiol. 2019;234(8):12956–63.
Nie D, Yang E, Li Z. Pretreatment thrombocytosis predict poor prognosis in patients with endometrial carcinoma: a systematic review and meta-analysis. BMC Cancer. 2019;19(1):73.
Liu W, Zhou Z, Dong D, Sun L, Zhang G. Sex differences in the association between night shift work and the risk of cancers: a meta-analysis of 57 articles. Dis Markers. 2018;2018:7925219.
Adams BD, Parsons C, Walker L, Zhang WC, Slack FJ. Targeting noncoding RNAs in disease. J Clin Invest. 2017;127(3):761–71.
Muller S, Raulefs S, Bruns P, Afonso-Grunz F, Plotner A, Thermann R, et al. Next-generation sequencing reveals novel differentially regulated mRNAs, lncRNAs, miRNAs, sdRNAs and a piRNA in pancreatic cancer. Mol Cancer. 2015;14:94.
Serrati S, De Summa S, Pilato B, Petriella D, Lacalamita R, Tommasi S, et al. Next-generation sequencing: advances and applications in cancer diagnosis. Onco Targets Ther. 2016;9:7355–65.
Zhang H, Feng X, Zhang M, Liu A, Tian L, Bo W, et al. Long non-coding RNA CASC2 upregulates PTEN to suppress pancreatic carcinoma cell metastasis by downregulating miR-21. Cancer Cell Int. 2019;19:18.
Bin X, Hongjian Y, Xiping Z, Bo C, Shifeng Y, Binbin T. Research progresses in roles of LncRNA and its relationships with breast cancer. Cancer Cell Int. 2018;18:179.
Grelet S, Link LA, Howley B, Obellianne C, Palanisamy V, Gangaraju VK, et al. A regulated PNUTS mRNA to lncRNA splice switch mediates EMT and tumour progression. Nat Cell Biol. 2017;19(9):1105–15.
Guo L, Gu J, Hou S, Liu D, Zhou M, Hua T, et al. Long non-coding RNA DANCR promotes the progression of non-small-cell lung cancer by inhibiting p21 expression. Onco Targets Ther. 2019;12:135–46.
Gu H, Chen J, Song Y, Shao H. Gastric adenocarcinoma predictive long intergenic non-coding RNA promotes tumor occurrence and progression in non-small cell lung cancer via regulation of the miR-661/eEF2K signaling pathway. Cell Physiol Biochem. 2018;51(5):2136–47.
Zhu FF, Zheng FY, Wang HO, Zheng JJ, Zhang Q. Downregulation of lncRNA TUBA4B is associated with poor prognosis for epithelial ovarian cancer. Pathol Oncol Res. 2018;24(2):419–25.
Zhang T, Wu DM, Deng SH, Han R, Liu T, Li J, et al. Integrated analysis reveals that long non-coding RNA TUBA4B can be used as a prognostic biomarker in various cancers. Cell Physiol Biochem. 2018;49(2):530–44.
Wang X, Wang G, Zhang L, Cong J, Hou J, Liu C. LncRNA PVT1 promotes the growth of HPV positive and negative cervical squamous cell carcinoma by inhibiting TGF-beta1. Cancer Cell Int. 2018;18:70.
Guo J, Hao C, Wang C, Li L. Long noncoding RNA PVT1 modulates hepatocellular carcinoma cell proliferation and apoptosis by recruiting EZH2. Cancer Cell Int. 2018;18:98.
Zhang SR, Zhang GL, Liu JY. Long noncoding RNA PVT1 promotes cervical cancer progression through epigenetically silencing miR-200b. APMIS. 2016;124(8):649–58.
Zhang Y, Wen DY, Zhang R, Huang JC, Lin P, Ren FH, et al. A preliminary investigation of PVT1 on the effect and mechanisms of hepatocellular carcinoma: evidence from clinical data, a meta-analysis of 840 cases, and in vivo validation. Cell Physiol Biochem. 2018;47(6):2216–32.
Yang T, Jin X, Lan J, Wang W. Long non-coding RNA SNHG16 has Tumor suppressing effect in acute lymphoblastic leukemia by inverse interaction on hsa-miR-124-3p. IUBMB Life. 2019;71(1):134–42.
Acknowledgements
We would like to thank the researchers and study participants for their contributions.
Funding
This work was financially supported by the Natural Science Foundation of Hunan Province, China (2017DK2013, 2018JJ3716, and 2018JJ3759), China Scholarship Council (201806375067, 201806375068), National Natural Science Foundation of China (81372180) and Central South University Innovation Foundation for Postgraduate Studies (NO. 2017zzts231).
Author information
Authors and Affiliations
Contributions
ZHL and CT designed the project. CHZ and XLR performed data extraction and analysis. JYH and WCW performed the quality assessment. JYH and CT contributed to the article drafting. ZHL and CT revised the manuscript critically and supervised the project. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, C., Ren, X., He, J. et al. The prognostic value of long noncoding RNA SNHG16 on clinical outcomes in human cancers: a systematic review and meta-analysis. Cancer Cell Int 19, 261 (2019). https://doi.org/10.1186/s12935-019-0971-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-019-0971-2