
Abstract
Background
Cervical cancer is a major cause of death in women worldwide. Interferon-induced transmembrane protein 1 (IFITM1) is involved in antivirus defense, cell adhesion, and carcinogenesis in different tissues. However, the role of IFITM1 gene in cervical squamous cell cancer is unclear.
Methods
To explore the role of IFITM1 in carcinogenesis of cervical cancer, we investigated the expression of IFITM1 gene in cervical squamous cell carcinoma. IFITM1 mRNA level was measured by real-time quantitative RT-PCR in cervical cancer tissues and their adjacent normal tissues. IFITM1 protein level was measured by immunohistochemistry. Methylation in the IFITM1 gene promoter was detected by methylation-specific PCR. We then transfected HeLa cells with IFITM1 expression vector or control vector. IFITM1 expression was examined; cell migration and invasion were analyzed by wound healing assay and matrigel-coated transwell migration assays, respectively. HeLa cell proliferation was measured by cell counting kit-8 assay and cell cycle analysis. Cell apoptosis was analyzed by Annexin V/propidium iodide double staining assay.
Results
The difference in IFITM1 protein expression between samples from chronic cervicitis and cervical carcinoma was statistically significant (P < 0.01). Ki-67 and PCNA protein expression levels were significantly higher in cervical cancer tissues than in their corresponding cervicitis tissues (P < 0.05 and P < 0.001, respectively). IFITM1 mRNA level was significantly lower in cervical cancer tissues than in normal cervical tissues (P < 0.05). Methylation of the IFITM1 gene promoter was significantly higher in cervical cancer than in normal cervical tissues (P < 0.05). Transfection of the IFITM1 pcDNA3.1 construct decreased cell migration and invasion of HeLa cells, inhibited cell proliferation, and increased cell apoptosis.
Conclusion
IFITM1 gene expression may reduce the proliferation, migration, and invasion of cervical squamous cancer cells.
Similar content being viewed by others

Background
Cervical cancer is a major cause of death in women worldwide, with approximately 500,000 new cases and 280,000 deaths reported each year [1]. In China, 75,000 new cases are diagnosed every year; 35% of these patients have recurrent diseases. Multidrug resistance and resistance to radiotherapy are the main causes of recurrent cervical cancer cases, in which conventional treatment methods are ineffective [2]. Although much progress has been made in cervical cancer research, reliable biomarkers to predict the development of cervical cancer tumors are still lacking [3].
Developed technologies, such as gene expression analysis, can be used to identify genetic alterations related to the development of cervical cancer; such alterations are potential biomarkers for the diagnosis and prognosis of cervical cancer patients [4,5,6]. Previous studies demonstrated the overexpression of the DeltaNp63alpha gene together with p53 gene inactivation in squamous cell cancer (SCC) and down-regulation of the expression of the C/EBPα gene in cervical SCC [7, 8]. Overexpression of the Beclin1 gene in CaSki cells may enhance apoptosis signaling induced by anticancer drugs [9]. Moreover, epigenetic modifications are involved in cervical tumorigenesis; for example, methylation of CpGs, especially in the promoter region of genes, has been suggested as a possible factor influencing the activity of cervical cancer-related genes [10, 11].
We compared the gene expression profiles between cervical cancer tissues and their corresponding normal cervical tissues in our previous study [12]. We found that the mRNA expression level of the interferon-induced transmembrane gene (IFITM1) is reduced in cancer tissues [12]. The IFITM protein is a superfamily with a transmembrane domain and an inserting conserved intracellular loop [13]. Over 30 superfamily members of IFITM are involved in antivirus defense, immune cell signaling transduction, cell adhesion, carcinogenesis, and germ cell maturation [14].
IFITM1 is involved in several types of cancers, e.g. it promotes the aggressiveness of colorectal cancer cells [15]. Over expression of IFITM1 enhances the aggressive phenotype of triple-negative SUM149 of inflammatory breast cancer cells [16]. However, over expression of IFITM1 negatively regulated cell growth, whereas suppression of IFITM1 blocked the antiproliferative effect of IFN-gamma, accelerated the cell growth rate and conferred tumorigenicity to a non-malignant hepatocyte in nude mice [17]. In addition, higher IFITM1 expression correlated with improved survival in chronic myeloid leukemia patients [18]. The present study focused on the effect of IFITM1 gene on the carcinogenesis and development of cervical cancer.
Methods
Tissue samples
Between 2008 and 2014, clinical data and cervical SCC samples from patients were collected at the First Affiliated Hospital and the Third Affiliated Hospital of the Medical College of Shihezi University in Xinjiang, China, with the approval of the ethical committee of each hospital. Written informed consent was obtained from patients. Patients received neither chemotherapy nor radiotherapy before sample collection. The diagnoses were confirmed independently by two experienced pathologists. Cervical SCC tissues and adjacent normal cervical tissues were collected from each patient. Tissue samples were frozen immediately after removal and stored at − 80 °C.
Immunohistochemical staining
Tumor tissues were fixed in 10% formalin, embedded in paraffin, and cut into 4 μm-thick sections. For immunohistochemical staining, tissue sections were deparaffinized in xylene and rehydrated in descending grades of ethanol. Endogenous peroxidase activity was blocked with methanol containing 0.3% H2O2 for 30 min and then washed in PBS. Antigen retrieval was performed by microwaving with citrate phosphate buffer (pH 6.0). The sections were then placed with the primary antibodies at 4 °C overnight. After incubation, the sections were washed in PBS for 3 min. The sections were then washed five times with PBS for several seconds, incubated with secondary antibodies at 37 °C for 30 min, and washed twice with PBS. After incubation with the secondary antibodies, staining was completed using anti-mouse peroxidase and DAB substrate. Tissue sections were counterstained with hematoxylin. IFITM1, Ki-67, and PCNA protein signals were scored on the following scale considering both the proportion of cells stained and the intensity of staining in those cells: score 0, no cells stained; score 1, weak or absent nuclear staining and < 5% of cells stained; score 2, nuclear staining and between 5 and 25% of the cells stained; score 3, nuclear staining and between 26 and 50% of the cells stained; and score 4, nuclear staining and more than 50% of the cells stained. Two observers scored independently using this scale.
Real-time RT-PCR
Total RNA was extracted from cell or tissue samples using TRIzol reagent according to the manufacturer’s protocol (Invitrogen). The RNA concentration was determined by agarose gel electrophoresis or absorbance at 260 nm. cDNA was synthesized with Invitrogen’s SuperScript One-Step RT-PCR Kit; each reaction contained 2 μg of total RNA, 2 μL of Oligo(dT) (500 μg/mL), and 7.5 μL of DEPC water. Reactions were heated for denaturation at 65 °C for 5 min and then quenched on ice for 5 min. The following reagents were added to each reaction: 4 µL of 5 × first buffer, 2 µL of 0.1 M DTT, 1 µL of dNTPs (10 mM each), 0.5 μL of RNase inhibitor (40 U/μL), and 1 μL of M-MLV (200 U/μL); the total volume of each reaction was 20 μL. The reactions were maintained at 25 °C for 10 min and 37 °C for 1 h. The reactions were terminated at 70 °C for 10 min. IFITM1 mRNA quantitative PCR amplification was performed on Light Cycler 480 (Roche Diagnostics) with the forward primer 5′-ACTCAACACTTCCTTCCCCAA-3′ and the reverse primer 5′-CTTCCTGTCCCTAGACTTCACG-3′. The amplicons were 231 bp in size. To normalize the amount of cDNA in each sample, the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was quantified in the control experiment with specific primers (forward: 5′-TGTTGCCATCAATGACCCCTT-3′; reverse: 5′-CTCCACGACGTACTCAGCG-3′); the amplicons were 202 bp in size. PCR reactions were performed in a volume of 20 μL, consisting of 10 μL of 2 × PCR buffer, 500 ng of cDNA, 10 μL of 2 × PCR buffer for EvaGreen, 0.6 μL of 20 × EvaGreen, 0.6 μL of forward primer and reverse primer (10 μM) each, and 0.3 μL of Cap Taq polymerase (5 U/μL). DEPC water was added to bring the volume to 20 μL. Reaction conditions were as follows: initial denaturation for 5 min at 95 °C, followed by 40 cycles of denaturation for 15 s at 95 °C, primer annealing for 15 s at 55 °C, extension for 20 s at 72 °C, and UPL fluorescence measurement for 3 s at 76 °C. The GAPDH gene was used as an endogenous control to normalize the difference in the amount of cDNA in each sample.
DNA preparation and detection of HPV
DNA was extracted with an SK1252 genomic DNA isolation mini kit (Shanghai Sangon Biological Engineering Technology and Services Company) according to the manufacturer’s protocol. HPV detection and typing were performed in all samples; HPV16 and HPV18 were analyzed by PCR. The primers used were pHPV16-F (5′-GACCCAGAAAGTTACCACAG-3′) with pHPV16-R (5′-CACAACGGTTTGTTGTATTG-3′) for HPV16 virus detection, as well as pHPV18-F (5′-TGCCAGAAACCGTTGAATCC-3′) with pHPV18-R (5′-TCTGAGTCGCTTAATTGCTC-3′) for HPV18 virus detection. Both HPV16 and HPV18 PCR amplicons were 268 bp in size.
DNA modification by bisulfite treatment and methylation-specific PCR (MSP)
The DNA samples were subjected to bisulfite treatment using CpGenome™ DNA modification kit S7820 (CHEMICON, Temecula, CA, USA) according to the manufacturer’s protocol. The modified DNA was purified, followed by ethanol precipitation and then stored at − 80 °C. Methylated primers were pM-IFITM1-F (5′-GAGATTTTCGTGTTCGATTATGTC-3′) and pM-IFITM1-R (5′-ATAAAACCCCAAACTCACCG-3′), whereas unmethylated primers were pUM-IFITM1-F (5′-AGATTTTTGTGTTTGATTATGTTGT-3′) and pUM-IFITM1-R (5′-ATAAAACCCCAAACTCACCAAC-3′). Each reaction was 25 μL containing 1 μL of modified DNA template, 0.5 μL of forward and reverse primer each (0.5 μmol/L), 12.5 μL of Taq DNA Polymerase Mix, and 10.5 μL of H2O. The reaction conditions were as follows: 34 cycles at 95 °C for 5 min, 94 °C for 1 min, 58 °C for 1 min, and 72 °C for 1 min, and an extension step at 72 °C for 10 min. PCR results were checked by agarose gel (2%) electrophoresis.
RT-PCR
mRNA expression in the 10 methylated and 10 unmethylated cervical tissues was measured by RT-PCR. Total RNA was extracted with TRIzol Reagent (Life Technologies) according to the manufacturer’s instructions. RNA was subjected to cDNA synthesis using an oligo(dt) primer and reverse transcriptase (Fermentas). About 2 μL of cDNA products was then PCR-amplified using IFITM1 gene primers and Taq DNA Polymerase Mix according to the manufacturer’s instructions. RT-PCR primers were pRT-IFITM1-F (forward: 5′-ATGTCGTCTGGTCCCTGTTC-3′) and pRT-IFITM1-R (reverse: 5′-GTCATGAGGATGCCCAGAAT-3′). GAPDH served as the internal control; the primers were GAPDH-F (forward, 5′-GCCAAAAGGGTCATCATCTC-3′) and GAPDH-R (reverse, 5′-GTAGAGGCAGGGATGATGTTC-3′). 5 μL of PCR products was run on 1% agarose gel to determine IFITM1 gene expression.
Cell culture and transfection
cDNA of IFITM1 gene was cloned into pcDNA3.1(−) expression vector and confirmed by gene sequencing. In addition, pcDNA3.1(−) vectors were used as the control. HeLa cells were plated at 2 × 105 cells per well in a six-well cell culture plate at 24 h before transfection. Subsequently, 2 μg of IFITM1 constructs and the control vector were mixed with 6 μL of Lipofectamine 2000 (Invitrogen). The mixture was incubated at room temperature for 10 min. After washing the cells with 1 × PBS, the DNA/Superfect mixtures were transferred to HeLa cells. The control pcDNA3.1 vector was transferred into HeLa cells separately. Transfected HeLa cells were then incubated at 37 °C in 5% CO2 for 24 h. The effect of IFITM1 overexpression was determined by measuring immunofluorescence luciferase activity using an assay system according to the manufacturer’s protocol (Promega). Each experiment was repeated with multiple batches of cells.
Wound healing assay
Cells were seeded onto six-well plates (1 × 105 cells/dish). When cells reached over 90% confluence, a scratch was made across the cell monolayer with a tip. Cells were gently washed with PBS three times and maintained in a fresh medium. Cells were incubated for 24 h and photographed using an inverted tissue culture microscope at 100 × magnification. Assays were performed at least three times, and data are presented as the mean ± SD. The migration potential between IFITM1 expressing cells and control cells was compared by relative gap distance. P < 0.05 was considered statistically significant.
Cell migration and invasion assays
Cells were serum-starved overnight. The top chambers of 6.5 mm Corning Costar transwells (Corning, USA) were loaded with 0.2 mL of cells (5 × 105 cells/mL) in serum-free media. Complete media (0.6 mL) were added to the bottom wells, and cells were incubated at 37 °C overnight. Cells that migrated through the membrane were fixed, stained with 0.1% crystal violet, and examined under a light microscope. Images of the cells at the bottom of the membrane were captured using a Canon camera and a Zeiss microscope. The mean values were obtained from three individual experiments using Student’s t tests. For cell invasion assay, cells were serum-starved overnight. The 24-well cell culture inserts (8 mm pore size, BD Biosciences, San Jose, USA) were loaded with 0.5 mL of cells (5 × 105 cells/mL) in serum-free media. Complete media (0.5 mL) were added to the bottom wells, and cells were incubated at 37 °C for 24 h. Cells were fixed, stained, and analyzed as described above.
Cell proliferation determined by cell counting kit-8 assay
Proliferation of HeLa cells transfected by the IFITM1 pcDNA3.1 construct and pcDNA3.1 was determined using cell counting kit-8 reagents (Dojindo Laboratories, Japan). HeLa cells were seeded in 96-well plates (Falcon; Becton–Dickinson Labware) at 2 × 105 cells per well in DMEM containing 10% FBS. The cells were incubated for 24, 48, 72 h at 37 °C. Spectrometric absorbance at 450 nm was measured with a microplate reader (X-Mark; Bio-Rad, USA); the quantification was repeated five times.
Cell cycle analysis
IFITM1—pcDNA3.1 construct- or pcDNA3.1 vector-transfected cells were digested by trypsin, fixed with 70% ethanol, and stored at 4 °C until analysis. The cells were suspended in 100 μL of 180 μg/mL RNA enzyme A in and then incubated for 30 min at room temperature. Propidium iodide (PI) solution (50 μg/mL; Merck, Darmstadt, Germany) was added to the cells. The cells were then kept in a dark room for 15 min. The DNA content of cells was measured by flow cytometry (FACScan system, Becton–Dickinson). The G0/G1 phase ratio, as well as S and G2/M phase ratio, was determined from flow cytometry data.
Cell apoptosis detected by flow cytometry
Apoptosis was analyzed by Annexin V-fluorescein isothiocyanate staining using an Alexa Fluor 488 Annexin V kit (Invitrogen). In total, 1.0 × 106 of IFITM1—pcDNA3.1 construct-transfected cells, as well as the same amount of pcDNA3.1 vector-transfected cells, were washed twice with cold PBS. Cells were then resuspended in 1 × Annexin-binding buffer for 30 min. Cell apoptosis was determined by Annexin V-fluorescein isothiocyanate/PI double staining. Cell transfection was analyzed on FACS calibration with the software Cell Quest (Becton–Dickinson). All experiments were carried out in triplicate.
Statistical analysis
SPSS17 software was used in all statistical analyses. IFITM1 protein levels were compared between cervical carcinoma and chronic cervicitis using non-parametric test. Results of the triplicates were represented as the mean ± standard deviation (if applicable). Results were considered statistically significant if P < 0.05 in two-tailed Student’s t tests.
Results
IFITM1, Ki-67, and PCNA protein expression in cervical cancer and chronic cervicitis tissues measured by immunohistochemistry
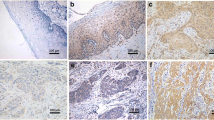
A total of 55 cervical cancer and 40 chronic cervicitis paraffin tissues were cut into triplicate sections to analyze the expression of IFITM1, Ki-67, and PCNA proteins. IFITM1 protein was stained brown in the cell membrane and cytosol of cervical tissue cells by immunohistochemistry (Fig. 1a, b). Ki-67 and PCNA protein expression was stained brown in the nucleus of cervical tissue cells by immunohistochemistry (Fig. 1c–f). IFITM1 protein expression in chronic cervicitis tissues was higher than that in cervical cancer tissues. However, in the corresponding chronic cervicitis tissues, the protein expression levels of Ki-67 and PCNA were both lower than those in the corresponding cervical cancer tissues.

IFITM1, Ki-67, and PCNA protein expression level in cervical cancer tissues and cervicitis tissues. a IFITM1 protein expression in chronic cervicitis tissues (× 200); b IFITM1 protein expression in cervical cancer tissues (× 200); c Ki-67 protein expression in chronic cervicitis tissues (× 200); d Ki-67 protein expression in cervical cancer tissues (× 200); e PCNA protein expression in chronic cervicitis tissues (× 200); f PCNA protein expression in cervical cancer tissues (× 200)
IFITM1 protein expression significantly decreased in cervical cancer tissues than in chronic cervicitis tissues (P < 0.01; Table 1). By contrast, Ki-67 protein level significantly increased in cervical cancer tissues than in chronic cervicitis tissues (P < 0.01; Table 1). In addition, PCNA protein level was significantly higher in cervical cancer tissues than in the corresponding chronic cervicitis tissues (P < 0.001; Table 1).
Methylation of the IFITM1 gene promoter in cervical cancer and normal cervical tissues
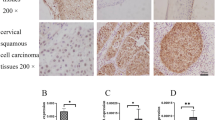
IFITM1 mRNA expression in 24 cervical cancer tissues was 0.39 ± 0.088, which was significantly lower than that in 15 adjacent normal cervical tissues at 1.295 ± 0.276 (t = − 2.401, P < 0.05) (Fig. 2a). Methylation analysis of the IFITM1 gene promoter by MSP revealed 47 samples of IFITM1 gene methylation in 60 cervical cancer tissues and five samples of IFITM1 gene methylation in 60 normal cervical tissues (Fig. 2b); the difference was statistically significant (P < 0.001; Table 2). HPV16 and HPV18 infections in cervical cancer and normal cervical tissues were detected by PCR. A total of 44 HPV16 and HPV18 infections were found in 60 cervical cancer tissues. By contrast, seven HPV16 and HPV18 infections were noted in 60 normal cervical tissues; the difference was statistically significant (P < 0.05; Table 2). The IFITM1 gene methylation rate in 44 HPV16 and HPV18 infection-positive samples was higher than that in 16 HPV16 and HPV18 infection-negative samples; the difference was statistically significant (P < 0.05; Table 3).

IFITM1 gene methylation analysis and mRNA expression analysis. a IFITM1 mRNA expression level in 24 cervical cancer tissues significantly decreased than that in 15 adjacent normal cervical tissues by real-time quantitative RT-PCR (P < 0.05). b Methylation of the IFIMT1 gene promoter was analyzed by MSP: 1–3 cervical cancer tissues; 4–6 normal cervical tissues; M: Marker (100–600 bp); m: production of methylation via MSP; u: production of non-methylation via MSP. c IFITM1 mRNA expression in the IFITM1 gene promoter of methylated and unmethylated cervical tissues: 1–4 the IFITM1 gene in unmethylated normal cervical tissues; 5–8 the IFITM1 gene promoter in methylated samples; M: Marker (100–600 bp); GAPDH as an internal control
In addition, the IFITM1 gene promoter in methylated cervical cancer tissues at the mRNA expression level was lower than that of the IFITM1 gene promoter in unmethylated normal cervical tissues (Fig. 2c).
Effect of the IFITM1 gene on HeLa cell migration
The IFITM1 gene recombinant construct and pcDNA3.1 vector were transfected into HeLa cells. The gap of wound width was 500 µm, and wound healing was observed at 0, 24, and 48 h. The images taken at these time points showed that the wound width of relative gap rates at 48 h of the IFITM1 gene recombinant group and pcDNA3.1 vector-transfected group were 175 ± 26.81 and 81 ± 43.25, respectively. The difference was statistically significant (P < 0.05). This result indicated that the IFITM1 gene could inhibit the migration of HeLa cells (Fig. 3).

Wound healing assay for analysis of overexpression of the IFITM1 gene on HeLa cell migration. a Overexpression of the IFITM1 gene in HeLa cells inhibited wound healing capabilities compared with that in pcDNA3.1-transfected HeLa cells. b Overexpression of the IFITM1 gene in HeLa cells inhibited migration compared with that in pcDNA3.1-transfected HeLa cells. The relative gap rate in the IFITM1 gene-transfected group was significantly greater than that in the pcDNA3.1-transfected group (n = 3, P < 0.05)
Effect of IFITM1 gene on the migration and invasion of HeLa cells by transwell gel assay
Cells of the IFITM1 gene construct-transfected group and pcDNA3.1 vector-transfected group were seeded in transwells. Cells moved from low nutrition to high nutrition through an 8 µm hole. After 24 h, HeLa cells were stained by crystal violet, and images of the number of HeLa cells were obtained. The numbers of HeLa cells through the micropore in the IFITM1 gene construct group and pcDAN3.1 control group were 82 ± 4.04 and 122 ± 7.23, respectively. The difference was statistically significant (P < 0.05). This finding indicated that overexpression of IFITM1 gene inhibited the migration of HeLa cells (Fig. 4a).

HeLa cell migration and invasion were inhibited by the IFITM1 gene. a HeLa cell migration in the IFITM1 gene-transfected group was lower than that in the pcDNA3.1 gene-transfected group (× 100). b HeLa cell migration in the IFITM1 recombinant construct was lower than that in the control group; the difference was statistically significant (P < 0.05). c Invasion of HeLa cells in the IFITM1 gene-transfected group was lower than that in the pcDNA3.1-transfected group (× 10). d HeLa cell invasion in the IFITM1 gene-transfected group was significantly lower than that in the pcDNA3.1-transfected group; the difference was statistically significant (P < 0.05)
After transfection by the IFITM1 gene construct and pcDNA3.1 vector, the invasion of HeLa cells was analyzed by a transwell matrix gel experiment. After 48 h, the number of HeLa cells at the bottom chamber was detected and images were taken. The numbers of HeLa cells of the IFITM1 recombinant construct group and pcDNA3.1 control group through the micropore were 64.33 ± 2.52 and 102.0 ± 11.53, respectively. The difference was statistically significant (P < 0.05). This result showed that overexpression of the IFITM1 gene inhibited the invasion of HeLa cells (Fig. 4b).
HeLa cell proliferation measured by cell counting kit-8 assay and cell cycle analysis by flow cytometry
After transfection by the IFITM1 gene construct and pcDNA 3.1 control vector, HeLa cell proliferation was measured by cell counting kit-8 assay. In terms of cell proliferation, the IFITM1 construct-transfected group was significantly lower than the pcDNA 3.1 vector control group at 48 and 72 h (P < 0.5, n = 5) (Fig. 5a). The effect of IFITM1 gene on the HeLa cell cycle was determined by PI staining, whereas FACS analysis was conducted to detect the DNA contents of cells. The number of cells in the G1/G0 phase, S phase, and G2/M phase can be used to determine the effect of IFITM1 gene on the cell cycle. After overexpression of IFITM1 gene, the number of HeLa cells obviously increased in the S phase of the cell cycle. Therefore, IFITM1 gene overexpression blocked HeLa cells in the S phase. The numbers of HeLa cells in the S phase of the IFITM1 construct-transfected group and pcDNA3.1-transfected group were 33.21% ± 6.22% and 21.38% ± 6.19%, respectively. The difference was statistically significant (P < 0.05; Fig. 5b, c). The IFITM1 construct-transfected group and pcDNA3.1 vector-transfected group showed no obvious difference in the G1 and G2/M phase of the HeLa cell cycle.

HeLa cell proliferation in the S phase of the cell cycle was inhibited by the IFITM1 gene. a HeLa cell proliferation in the IFITM1 gene-transfected group was significantly lower than that in the pcDNA 3.1 plasmid control group by cell counting kit-8 assay at 48 and 72 h; the difference was statistically significant (P < 0.05). b HeLa cell number of the S phase of the cell cycle in the IFITM1 gene recombinant construct group was greater than that in the pcDNA3.1 control group, as revealed by flow cytometry. c HeLa cell number in the S phase of the cell cycle in the IFITM1 gene-transfected group was significantly greater than that in the pcDNA3.1-transfected group; the difference was statistically significant (P < 0.05)
Effect of IFITM1 gene on HeLa cell apoptosis measured by Annexin V-FITC-PI assay
HeLa cell apoptosis was analyzed by Annexin V-FITC-PI assay. The apoptosis rate was 6.88 ± 1.52 in the IFITM1 construct-transfected group and 5.26 ± 0.83 in the pcDNA3.1 vector-transfected group. Therefore, overexpression of the IFITM1 gene increased HeLa cell apoptosis (P < 0.05; Fig. 6).

Effect of the IFITM1 gene on the HeLa cell apoptosis rate analyzed by Annexin V-FITC-PI assay. a The apoptosis rates of HeLa cells of the IFITM1 gene-transfected group and pcDNA3.1-transfected group were quantified by Annexin V/PI staining via flow cytometry. Results shown are the representative examples of flow cytometry. b The apoptosis rate of the IFITM1 gene-transfected group was obviously greater than that of the pcDNA3.1-transfected group; the difference was statistically significant (n = 3; P < 0.05)
Discussion
IFITM1 gene is an important factor that controls cell growth, and IFITM1 protein is initially proven to possess Leu-13 protein function. Leu-13 is a known leukocyte antigen, which forms a membrane complex involved in transduction, anti-proliferation, and homotypic adhesion signals in lymphocytes [19]. IFITM1 is an anti-HCV interferon-stimulated gene, and controls HCV infection through the interruption of viral coreceptor function [20]. IFITM1 serves as an important molecule to restrict HCV infection, and it may have implications in the development of therapeutic modalities [21]. IFITM1 restricts an early step in influenza A viral replication. Meanwhile, IFITM proteins confer basal resistance to influenza A virus but are also inducible by types I and II interferons and are critical for an interferon’s virustatic actions [22].
Previous studies found that IFITM1 gene overexpression can increase cell proliferation, migration, and invasion of esophageal SCC [23], head and neck cancer [24], and glioma [25]. However, IFITM1 gene expression in cervical cancer tissues was lower than that in normal cervical tissues. The reason why IFITM1 protein expression decreased in cervical cancer tissues is unclear. In the current study, we investigated the expression of IFITM1, Ki-67, and PCNA proteins in cervical cancer tissues and chronic cervicitis tissues. Our results showed that IFITM1 protein significantly decreased in cervical cancer tissues relative to that in chronic cervicitis tissues. However, the expression levels of Ki-67 and PCNA proteins in the corresponding cervical cancer tissues increased relative to those in chronic cervicitis tissues. We found that decreased IFITM1 protein expression resulted in cell proliferation in cervical tissues. Our results were similar to those of a previous study on hepatoma cells, which showed that the overexpression of IFITM1 inhibits cell proliferation [26].
Gene methylation regulates cell growth and is related to carcinogenesis [27,28,29]. Methylation in the IFITM1 gene promoter was analyzed in cervical cancer tissues and normal cervical tissues by MSP. Methylation in the IFITM1 gene promoter in cervical cancer tissues increased significantly compared with that in normal cervical tissues. However, the mRNA expression level in the corresponding cervical cancer tissues decreased. Reduction in IFITM1 mRNA expression in cervical tissues may lead to cervical carcinogenesis. In addition, we found that IFITM1 gene inhibited the invasion and migration of HeLa cells, block HeLa cells in the S phase, and enhance apoptosis. However, a previous study showed that IFITM1 plays an important role during invasion at the early stage of head and neck squamous cell carcinoma (HNSCC) progression, and IFITM1 can be a therapeutic target for HNSCC [30]. The reason why IFITM1 showed various effects in different types of cancer remains unknown, and further studies are needed.
Conclusion
The decrease in IFITM1 protein expression in cervical cancer may lead to cell proliferation. Therefore, the expression level of IFITM1 may be changed to improve the status of cervical cancer patients. In addition, reduced IFITM1 gene expression in cervical cancer tissues may be a result of methylation of the IFITM1 gene promoter, which is a potential target for cervical cancer treatment.
Abbreviations
- IFITM1:
-
interferon-induced transmembrane protein 1
- SCC:
-
squamous cell cancer
- GAPDH:
-
glyceraldehyde-3-phosphate dehydrogenase
- PI:
-
propidium iodide
- HNSCC:
-
head and neck squamous cell carcinoma
- PCR:
-
polymerase chain reaction
- HPV:
-
human papillomaviruses
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90.
Tian Y, Ding W, Wang Y, Ji T, Sun S, Mo Q, Chen P, Fang Y, Liu J, Wang B, Zhou J, Ma D, Wu P. Ubiquitin B in cervical cancer: critical for the maintenance of cancer stem-like cell characters. PLoS ONE. 2013;8:e84457.
Huang RL, Chang CC, Su PH, Chen YC, Liao YP, Wang HC, Yo YT, Chao TK, Huang HC, Lin CY, Chu TY, Lai HC. Methylomic analysis identifies frequent DNA methylation of zinc finger protein 582 (ZNF582) in cervical neoplasms. PLoS ONE. 2012;7:e41060.
Grigsby PW, Watson M, Powell MA, Zhang Z, Rader JS. Gene expression patterns in advanced human cervical cancer. Int J Gynecol Cancer. 2006;16:562–7.
Kim TJ, Choi JJ, Kim WY, Choi CH, Lee JW, Bae DS, Son DS, Kim J, Park BK, Ahn G, Cho EY, Kim BG. Gene expression profiling for the prediction of lymph node metastasis in patients with cervical cancer. Cancer Sci. 2008;99:31–8.
Kirkpatrick A, Bidwell J, van den Brule AJ, Meijer CJ, Pawade J, Glew S. TNFalpha polymorphism frequencies in HPV-associated cervical dysplasia. Gynecol Oncol. 2004;92:675–9.
Yugawa T, Narisawa-Saito M, Yoshimatsu Y, Haga K, Ohno S, Egawa N, Fujita M, Kiyono T. DeltaNp63alpha repression of the Notch1 gene supports the proliferative capacity of normal human keratinocytes and cervical cancer cells. Cancer Res. 2010;70:4034–44.
Pan Z, Zheng W, Zhang J, Gao R, Li D, Guo X, Han H, Li F, Qu S, Shao R. Down-regulation of the expression of CCAAT/enhancer binding protein alpha gene in cervical squamous cell carcinoma. BMC Cancer. 2014;14:417.
Sun Y, Liu JH, Jin L, Lin SM, Yang Y, Sui YX, Shi H. Over-expression of the Beclin1 gene upregulates chemosensitivity to anti-cancer drugs by enhancing therapy-induced apoptosis in cervix squamous carcinoma CaSki cells. Cancer Lett. 2010;294:204–10.
Pan Z, Li J, Pan X, Chen S, Wang Z, Li F, Qu S, Shao R. Methylation of the RASSF1A gene promoter in Uigur women with cervical squamous cell carcinoma. Tumori. 2009;95:76–80.
Lai HC, Lin YW, Huang TH, Yan P, Huang RL, Wang HC, Liu J, Chan MW, Chu TY, Sun CA, Chang CC, Yu MH. Identification of novel DNA methylation markers in cervical cancer. Int J Cancer. 2008;123:161–7.
Pan Z, Chen S, Pan X, Wang Z, Han H, Zheng W, Wang X, Li F, Qu S, Shao R. Differential gene expression identified in Uigur women cervical squamous cell carcinoma by suppression subtractive hybridization. Neoplasma. 2010;57:123–8.
Tanaka SS, Yamaguchi YL, Tsoi B, Lickert H, Tam PP. IFITM/Mil/fragilis family proteins IFITM1 and IFITM3 play distinct roles in mouse primordial germ cell homing and repulsion. Dev Cell. 2005;9:745–56.
Nibbe RK, Markowitz S, Myeroff L, Ewing R, Chance MR. Discovery and scoring of protein interaction subnetworks discriminative of late stage human colon cancer. Mol Cell Proteom. 2009;8:827–45.
Yu F, Xie D, Ng SS, Lum CT, Cai MY, Cheung WK, Kung HF, Lin G, Wang X, Lin MC. IFITM1 promotes the metastasis of human colorectal cancer via CAV-1. Cancer Lett. 2015;368:135–43.
Ogony J, Choi HJ, Lui A, Cristofanilli M, Lewis-Wambi J. Interferon-induced transmembrane protein 1 (IFITM1) overexpression enhances the aggressive phenotype of SUM149 inflammatory breast cancer cells in a signal transducer and activator of transcription 2 (STAT2)-dependent manner. Breast Cancer Res. 2016;18:25.
Yang G, Xu Y, Chen X, Hu G. IFITM1 plays an essential role in the antiproliferative action of interferon-gamma. Oncogene. 2007;26:594–603.
Akyerli CB, Beksac M, Holko M, Frevel M, Dalva K, Ozbek U, Soydan E, Ozcan M, Ozet G, Ilhan O, Gurman G, Akan H, Williams BR, Ozcelik T. Expression of IFITM1 in chronic myeloid leukemia patients. Leuk Res. 2005;29:283–6.
Bradbury LE, Kansas GS, Levy S, Evans RL, Tedder TF. The CD19/CD21 signal transducing complex of human B lymphocytes includes the target of antiproliferative antibody-1 and Leu-13 molecules. J Immunol. 1992;149:2841–50.
Wilkins C, Woodward J, Lau DT, Barnes A, Joyce M, McFarlane N, McKeating JA, Tyrrell DL, Gale M Jr. IFITM1 is a tight junction protein that inhibits hepatitis C virus entry. Hepatology. 2013;57:461–9.
Raychoudhuri A, Shrivastava S, Steele R, Kim H, Ray R, Ray RB. ISG56 and IFITM1 proteins inhibit hepatitis C virus replication. J Virol. 2011;85:12881–9.
Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan BJ, Weyer JL, van der Weyden L, Fikrig E, Adams DJ, Xavier RJ, Farzan M, Elledge SJ. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell. 2009;139:1243–54.
Ma Z, Guo W, Niu HJ, Yang F, Wang RW, Jiang YG, Zhao YP. Transcriptome network analysis reveals potential candidate genes for esophageal squamous cell carcinoma. Asian Pac J Cancer Prev. 2012;13:767–73.
Deraz EM, Kudo Y, Yoshida M, Obayashi M, Tsunematsu T, Tani H, Siriwardena SB, Keikhaee MR, Qi G, Iizuka S, Ogawa I, Campisi G, Lo Muzio L, Abiko Y, Kikuchi A, Takata T. MMP-10/stromelysin-2 promotes invasion of head and neck cancer. PLoS ONE. 2011;6:e25438.
Yu F, Ng SS, Chow BK, Sze J, Lu G, Poon WS, Kung HF, Lin MC. Knockdown of interferon-induced transmembrane protein 1 (IFITM1) inhibits proliferation, migration, and invasion of glioma cells. J Neuro oncol. 2011;103:187–95.
Yang G, Xu Y, Chen X, Hu G. IFITM1 plays an essential role in the antiproliferative action of interferon-gamma. Oncogene. 2007;26:594–603.
Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, Leonhardt H, Jaenisch R. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300:489–92.
Luczak MW, Jagodzinski PP. DNA methylation in cancer development. Folia Histochem Cytobiol. 2006;44:143–54.
Kurkjian C, Kummar S, Murgo AJ. DNA methylation: its role in cancer development and therapy. Curr Probl Cancer. 2008;32:187–235.
Hatano H, Kudo Y, Ogawa I, Tsunematsu T, Kikuchi A, Abiko Y, Takata T. IFN-induced transmembrane protein 1 promotes invasion at early stage of head and neck cancer progression. Clin Cancer Res. 2008;14:6097–105.
Authors’ contributions
WZ and ZP carried out the molecular genetic studies, participated in the gene analysis and wrote the manuscript. ZZ and XY carried out the immunohistochemistry Staining. PF and HL carried out the qRT-PCR. QZ and HH carried out gene methylation analysis. XG and DL carried out gene transfection. FL participated in the design of the study and performed the statistical analysis. YL and RS participated in experimental design and wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Nos. U1503125, 30860302, and 30660193), the International Science and Technology Collaboration Projector of Xinjiang Production and Construction Corps (Grant No. 2013BC003), the National Science and Technology Supporting Project (The Twelfth Five-Year-Researching Project) (Grant No. 2013BAI05B0503), the Importance of Scientific Research and Innovation Project of Shihezi University (Grant No. gxjs2013-zdgg05), the Ministry of Education of China in the Year 2011 for Promotion with the Region of Americas and Oceania Cooperation in Scientific Research and Cultivation of High Level Talent Project (Grant No. [2011] 1056 with International Relation Bureau of Ministry of Education), the Youth Scientific Innovation Special Grant of Xinjinag Production and Construction Corps (Grant No. 2012CB018), and Start-up Project Special Grant of Shihezi University (Grant No. RCZX201534). RS acknowledges the funding support from the Australia–China Science and Research Fund (ACSRF00980).
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Please contact the corresponding author for data requests.
Consent for publication
This manuscript is approved by all authors for publication in Cancer Cell International.
Ethics approval and consent to participate
Written informed consent was obtained from each patient; approval was obtained from the Ethics Committee of the Medical College of Shihezi University, China (2016-027-01).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional information
The original version of this article was revised to amend corresponding author details.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zheng, W., Zhao, Z., Yi, X. et al. Down-regulation of IFITM1 and its growth inhibitory role in cervical squamous cell carcinoma. Cancer Cell Int 17, 88 (2017). https://doi.org/10.1186/s12935-017-0456-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-017-0456-0