
Abstract
Background
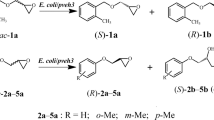
Epoxide hydrolase can regioselectively catalyze the oxirane ring-opening hydrolysis of rac-epoxides producing the corresponding chiral diols. In our laboratory, a gene named pveh1 encoding an EH from Phaseolus vulgaris was cloned. Although the directed modification of PvEH1 was carried out, the mutant PvEH1Y3 showed a limited degree of enantioconvergence towards racemic (rac-) m-chlorostyrene oxide (mCSO).
Results
PvEH1 and PvEH1Y3 were combinatively subjected to laboratory evolution to further enhance the enantioconvergence of PvEH1Y3 towards rac-mCSO. Firstly, the substrate-binding pocket of PvEH1 was identified using a CAVER 3.0 software, and divided into three zones. After all residues in zones 1 and 3 were subjected to leucine scanning, two E. coli transformants, E. coli/pveh1Y149L and /pveh1P184L, were selected, by which rac-mCSO was transformed into (R)-m-chlorophenyl-1,2-ethanediol (mCPED) having 55.1% and 27.2% eep. Secondly, two saturation mutagenesis libraries, E. coli/pveh1Y149X and /pveh1P184X (X: any one of 20 residues) were created at sites Y149 and P184 of PvEH1. Among all transformants, both E. coli/pveh1Y149L (65.8% αS and 55.1% eep) and /pveh1P184W (66.6% αS and 59.8% eep) possessed the highest enantioconvergences. Finally, the combinatorial mutagenesis was conducted by replacements of both Y149L and P184W in PvEH1Y3, constructing E. coli/pveh1Y3Z2, whose αS reached 97.5%, higher than that (75.3%) of E. coli/pveh1Y3. In addition, the enantioconvergent hydrolysis of 20 mM rac-mCSO was performed by E. coli/pveh1Y3Z2, giving (R)-mCPED with 95.2% eep and 97.2% yield.
Conclusions
In summary, the enantioconvergence of PvEH1Y3Z2 was successfully improved by laboratory evolution, which was based on the study of substrate-binding pocket by leucine scanning. Our present work introduced an effective strategy for the directed modification of enantioconvergence of PvEH1.
Similar content being viewed by others

Background
Optically pure epoxides and vicinal diols, the highly value-added and versatile building blocks, were widely applied in pharmaceutical, fine chemical and agrochemical industries [1,2,3]. For example, (R)-pCPED was used for the synthesis of (R)-Eliprodil, a neuroprotective agent for the treatment of ischemic stroke, while (R)-mCPED for β3-adrenergic receptor agonists, such as SR 58611 and AJ-9677 [4]. With the ever-increasing environmental consciousness, the biocatalysis mediated by whole resting cells or enzymes was considered to be an alternative to chemocatalysis that required expensive chiral ligands and hazardous metals, such as Jacobsen’s asymmetric ring-opening hydrolysis and epoxidation [5, 6].
Epoxide hydrolases (EHs; 3.3.2.-), existing widely in nature, can enantioselectively and/or regioselectively catalyze the oxirane ring-opening hydrolysis of rac-epoxides, retaining epoxide enantiomers and/or producing the corresponding chiral diols. Based on the catalytic mechanisms of given EH-substrate pairs, the asymmetric hydrolysis of rac-epoxides was divided into two pathways: kinetic resolution and enantioconvergent hydrolysis [7]. The former can retain single epoxide enantiomers with an intrinsic limitation of 50% maximum yield, while the latter can produce optically pure vicinal diols with 100% theoretically yield [8].
The mono-enzymatic catalysis was an ideal bioprocess for preparing chiral diols via convergent hydrolysis of epoxides, but few naturally existing EHs had high and opposite regioselectivities towards (R)- and (S)-epoxides [9]. To completely and quickly hydrolyze rac-epoxide, EH applied in enantioconvergent hydrolysis also have to possess a low enantioselectivity (i.e., enantiomeric ratio, E). In view of the merits of mono-enzymatic catalysis and the shortage of highly enantioconvergent EHs, it is necessary to excavate novel EHs or to modify specific local configurations of the existing EHs by protein engineering [10]. Among the different gene mutagenesis techniques, saturation mutagenesis (SM) at sites lining the enzyme’s binding pocket has emerged as a particularly viable approach to improve selectivity [11, 12]. For example, through five rounds of iterative saturation mutagenesis of nine residues at sites lining in the substrate-binding pocket (SBP) of Aspergillus niger M200 EH (AnEH), its best mutant, named H:12-A1, was selected, whose regioselectivity coefficients (αS values) towards (S)-SO and -pCSO were higher than those of AnEH, and by which rac-SO and -pCSO were transformed into (R)-PED and -pCPED, respectively, with over 70% enantiomeric excess (eep) [13]. Several other research groups also reported the laboratory evolution of residues located in the SBP of EHs [14, 15].
Previously, to improve the enantioconvergence of PvEH1 towards styrene epoxides, its laboratory evolution was carried out based on the computer-aided design. Among all tested mutants of PvEH1, PvEH1L105I/M160A/M175I (renamed PvEH1Y3), was selected (Additional file 1: Table S1). Its enantioconvergence towards rac-mCSO increased to 69.7% from 1.0% eep of PvEH1 [10]. In our present work, the SBP of PvEH1, identified and analyzed using a CAVER 3.0 software (http://www.caver.cz/), was divided into three zones. To substantially improve the enantioconvergence of PvEH1, the zones 1 and 3 were subjected, in which all residues were subjected to leucine scanning [16], namely, substituting target residues with Leu, to identify the sites where Leu mutants had best EH enantioconvergence. Two saturation mutagenesis libraries, E. coli/pveh1Y149X and /pveh1P184X, were constructed and screened to select the best substituted residues at their respective sites. Then, the combinatorial mutagenesis of PvEH1Y3 was conducted by replacing its Y149 and P184 with the selected best substituted residues, thereby creating one five-site mutant of PvEH1, named PvEH1Y3/Y149L/P184W or PvEH1Y3Z2. Lastly, the enantioconvergent hydrolysis of rac-mCSO was carried out using resting cells of E. coli/pveh1Y3Z2.
Materials and methods
Strains, plasmids and chemicals
E. coli BL21(DE3) and pET-28a(+) (Novagen, Madison, WI) were used for the construction of recombinant plasmid and expression of pveh1 or its variant, while PrimeSTAR HS DNA polymerase and DpnI endonuclease (TaKaRa, Dalian, China) were used for the leucine scanning and site-saturation mutagenesis of pveh1 as well as the combinatorial mutagenesis of pveh1Y3. pET-28a-pveh1 and -pveh1Y3 and their corresponding E. coli/pveh1 and /pveh1Y3 were constructed and stored in our lab. Rac-mCSO, (R)- and (S)-mCPED were purchased from Energy (Shanghai, China). All other chemicals were of analytical purity.
Homology modeling of PvEH1 and identification of its SBP
Using the known crystal structure of a Vigna radiata EH at 2.0 Å resolution (VrEH1, PDB: 5XMD), sharing 87.4% identity with PvEH1, as the template, the three-dimensional (3-D) structure of PvEH1 was modeled using the MODELLER 9.11 program (http://salilab.org/modeller/) (Additional file 1: Figure S2), and subjected to molecular mechanics optimization by CHARMM27 force field in the GROMACS 4.5 package (http://www.gromacs.org/) (Additional file 1: Figure S3). The 3-D structure with the best geometry quality, which was validated by Structure Assessment in SWISS-MODEL (https://swissmodel.expasy.org/assess), was obtained, and visualized by a PyMOL software (http://pymol.org/) (Additional file 1: Figures S4 and S5). The SBP of a modeled PvEH1’s 3-D structure was identified and analyzed using a CAVER 3.0 software, and artificially divided into three zones 1–3 (Fig. 1).

The SBP of PvEH1 (a) and its three zones (b)
Leucine scanning and site-saturation mutagenesis of PvEH1
The zones 1 and 3 of PvEH1’s SBP were subjected, in which all residues were subjected to leucine scanning. The variants of pveh1 were designed by substituting the target residues-encoding codons with Leu-encoding codon, synthesized by Genewiz (Suzhou, China), and transformed into E. coli BL21, respectively, thereby constructing the corresponding E. coli transformants, such as E. coli/pveh1Y149L and /pveh1P184L. Through screening, the specific residue sites where Leu mutants catalyzed the enantioconvergent hydrolysis of rac-mCSO with the highest eep values of (R)-mCPED were identified for the further studies.
Based on the results of leucine scanning, the saturation mutagenesis of a Y149- or P184-encoding codon in pveh1 was carried out using a one-step whole-plasmid PCR method [17]. The primers of saturation mutagenesis were designed as reported previously [18], and synthesized by Sangon (Shanghai, China) as listed in Additional file 1: Table S2. Using pET-28a-pveh1 as a template, the mutagenesis PCR was performed by PrimeSTAR HS DNA polymerase using a pair of primers, Y149X-F/Y149X-R or P184X-F/P184X-R, as following conditions: a denaturation at 95 °C for 4 min, 30 cycles of at 98 °C for 10 s, 55 °C for 15 s and 72 °C for 6 min, and an extra elongation at 72 °C for 10 min. The target PCR products, pET-28a-pveh1Y149X or -pveh1P184X (X: any one of 20 residues), were digested by DpnI at 37 °C for 6 h to decompose the methylated template DNA, and transformed into E. coli BL21(DE3), respectively, thereby constructing the mutagenesis library, E. coli/pveh1Y149X or /pveh1P184X. Using both the eep of (R)-mCPED and c of rac-mCSO as indexes, E. coli/pveh1Y149X and /pveh1P184X were screened, respectively. The best E. coli transformant of each library at residue site Y149 or P184 of PvEH1 was selected, expressing the EH mutant with the highest enantioconvergence towards rac-mCSO.
Combinatorial site-directed mutagenesis of PvEH1Y3
The combinatorial site-directed mutagenesis was designed by residue replacements of Y149L and P184W in PvEH1Y3, and also carried out by one-step whole-plasmid PCR. The PCR primers were designed according to the nucleotide sequence of pveh1Y3 and codons encoding the selected mutation residues, and synthesized by Sangon (Additional file 1: Table S3). Using pET-28a-pveh1Y3 as the template, the first round of PCR was conducted using a pair of primers, Y149L-F/Y149L-R, under the same PCR conditions as described above. Thereafter, using the first-round PCR product as the template, the second round of PCR was carried out using another pair of primers, P184W-F/P184W-R. The target PCR product, pET-28a-pveh1Y3/Y149L/P184W or -pveh1Y3Z2, was digested by DpnI, and transformed into E. coli BL21(DE3), thereby constructing one E. coli transformant, named E. coli/pveh1Y3Z2, harboring a five-site variant of pveh1 or a two-site one of pveh1Y3.
Analytic methods of HPLC and GC chromatographies
The activity of PvEH1 or its mutant as well as the conversion ratio (c) of rac-mCSO defined as the ratio of its depleted concentration to initial one was assayed by high-performance liquid chromatography (HPLC), using an e2695 apparatus with an XBridge BEH C18 column (Waters, Milford, MA). The mobile phase of methanol/H2O (7:3, v/v) was used at 0.8 mL/min, and continuously monitored using a Waters 2489 UV–Vis detector at 220 nm. The generated diols (R)- and (S)-mCPED were analyzed by HPLC with a Chiralcel OD-H column (Daicel, Osaka, Japan). The n-hexane/isopropanol (9:1, v/v) was used as a mobile phase. Because (R)- and (S)-mCSO can not be separated by OD-H, they were assayed by chiral gas chromatography (GC), using a GC-2010 system (Shimadzu, Tokyo, Japan) with a CP-Chirasil-DEX CB column (Agilent, Santa Clara, CA) and a flame ionization detector. The injector and detector temperatures were 220 °C, while the column temperature was programmed from 110 to 190 °C at 10 °C/min. The nitrogen gas carrier was used at 3.0 mL/min.
EH expression of E. coli transformant and EH activity assay
E. coli transformant harboring pveh1 or its variant, such as E. coli/pveh1 or /pveh1Y3Z2, was inoculated into LB medium supplemented with 100 μg/mL kanamycin, and cultured at 37 °C overnight as the seed culture. Then, the same fresh medium was inoculated with 2% (v/v) seed culture, and grown until OD600 reached 0.6–0.8. The expression of PvEH1 or its mutant was induced by 0.5 mM IPTG at 20 °C for 10 h. The induced transformant cells were collected, and resuspended in 100 mM Na2HPO4–NaH2PO4 buffer (pH 7.0) to 100 mg wet cells/mL. E. coli BL21(DE3) transformed with pET-28a(+), named E. coli/pET-28a, was used as the negative control.
The hydrolytic conditions for the PvEH1 or its mutant activity assay were as follows: 475 μL cell suspension of E. coli transformant, suitably diluted with 100 mM phosphate buffer (pH 7.0), was mixed with 25 μL 200 mM rac-mCSO, incubated at 25 °C for 10 min, and terminated by adding 2 mL methanol. The sample was assayed by HPLC with a C18 column. One EH activity unit (U) was defined as the amount of whole wet cells catalyzing the hydrolysis of 1 μmol rac-mCSO per minute under the given assay conditions.
EH enantioconvergence assay
EH enantioconvergence assay was carried out as follows: 1.8 mL suitably diluted cell suspension was mixed with 200 μL 200 mM rac-mCSO and incubated at 25 °C. Aliquots of 100 μL sample were periodically drawn out, and extracted with 900 μL ethyl acetate for chiral HPLC analysis (or containing 1 mM n-hexanol as the internal standard for GC analysis). (R)- and (S)-mCPED were analyzed by chiral HPLC, while (R)- and (S)-CSO by GC. Both the eep of (R)-mCPED and ees of (R)-CSO were calculated with the equations: eep = [(Rp − Sp)/(Rp + Sp)] × 100% and ees = [(Rs − Ss)/(Rs + Ss)] × 100%, in which Rp and Sp were the concentrations of (R)- and (S)-product, while Rs and Ss were the concentrations of (R)- and (S)-substrate. The EH regioselectivity coefficients (βR and αS) were applied to quantitatively evaluate the preference attacking on Cβ (a less hindered terminal carbon in the oxirane ring) of (R)-epoxide and on Cα (a more hindered carbon) of (S)-epoxide, respectively [17]. Based on the above parameters, βR and αS were deduced by linear regression: eep = (αS + βR − 1) + [(βR − αS) × ees × (1 − c)]/c [19].
Enantioconvergent hydrolysis of rac-mCSO by E. coli/pveh1 Y3Z2
As we using the whole cell as the biocatalyst, the regioselective hydrolysis of rac-mCSO, in a 5 mL 100 mM phosphate buffer (pH 7.0) system containing 20 mM rac-mCSO and 100 mg wet cells/mL of E. coli/pveh1Y3Z2, was carried out at 25 °C until the c reached over 99%. During the hydrolytic course, aliquots of 100 μL reaction sample were drawn out, extracted with 900 μL ethyl acetate, and analyzed by chiral HPLC to calculate the c of rac-mCSO and eep of (R)-mCPED.
Results and discussion
3-D structural analysis of PvEH1 and its SBP identification
Most EHs belonged to α/β hydrolase superfamily, which consisted of a core domain, an α/β domain and a lid domain (Fig. 2a) [20]. Like 3-D structures of other EHs, that of PvEH1 also contained a core domain, a β-sheet packed between two layers of 9 α-helices and a lid domain. Its active center was made up of two polarizing tyrosine residues: Y150 and Y234 and catalytic triad D101-H299-D264 (Fig. 2b). To avoid mutants losing catalytic activities, these five sites cannot be replaced by other residues. After being homologically modeled and optimized, 3-D structure of PvEH1was analyzed to identify the SBP of PvEH1. The SBP was located in the core domain and the lid domain mainly (Fig. 1a). It was reported that SBP can be divided according to the zone where the substrate binding with the enzyme [16]. Herein, as shown in Fig. 1b, the SBP of PvEH1 was divided into three zones subjectively: zone 1 (residues A120-V126, M129, V135-G142 and Y149), zone 2 (residues D101, Y150, Y234, D264 and H299) and zone 3 (residues M160-T162, A171-M175 and R180-L186). Five residue sites in active center which displayed important roles in the catalytic reaction were in zone 2. Compared with zone 3, zone 1 involved in more residues (17 vs. 15). Similar with the SBP of LEH [15], that of PvEH1 just looked like a dumbbell, which contained two big cavities and a tunnel connecting them.

3-D structure of PvEH1 (a) and its active center (b). The active center of was PvEH1 made up by two polarizing tyrosine residues: Y150 and Y234 and catalytic triad D101-H299-D264
Leucine scanning of the SBP
Amino acid substitutions at sites lining the SBP of EH may evolve its stereoselectivity [20]. As lacking the basic understanding of the prerequisites for regioselectivity in nucleophilic attack, each residue in zone 1 and zone 3 was replaced by leucine respectively to construct E. coli transformant expressing single-site mutant, except that the residue was leucine originally, for finding out the key sites influencing the enantioconvergence. The eep values and activities of all the mutants towards rac-mCSO were measured respectively. The results were outlined in Additional file 1: Table S4. Among all the mutants, PvEH1Y149L displayed a marked increase in enantioconvergence, and eep reached to 55.1%. On other side, residue replacement like M129L in zone 1 reversed the configuration of the main diol product partly (Fig. 3). The similar phenomenon was also observed in the reshaping of LEH SBP, in that case, a one-site mutant LEHL114F favored the formation of (R,R)-diol, while LEH the formation of (S,S)-diol [15]. Compared with residue replacements in zone 1, those in zone 3 had smaller effects on the catalytic activities (Fig. 3). For example, different from mutant PvEH1Y149L, PvEH1P184L had an increased enantioconvergence and its activity was not decreased. By the way, mutant PvEH1M129L displayed the highest catalytic activity, while mutant PvEH1Y149L the lowest except PvEH1P137L (no activity). There was an increase in the enantioconvergence of mutant PvEH1Y149L, but under the comparison with other studies which have been reported, it is still not ideal enough [21, 22].

The enantioconvergence and relative activity part mutants from leucine scanning of zone 1 (contain PvEH1Y149L, PvEH1M129L and PvEH1V126L) and zone 3 (contain PvEH1P184L, PvEH1T162L and PvEH1M160L), where catalytic activity of PvEH1 was used as a control
Saturation mutagenesis of Y149 or P184 in PvEH1
To figure out which residues at sites 149 and 184 are most beneficial for improving the enantioconvergence of PvEH1, saturation mutagenesis libraries E. coli/pveh1Y149X and E. coli/pveh1P184X were constructed and screened. According to the “22c-trick” method reported by Kille et al. [18] a special mixture of three primers was employed to create a degeneracy of 22 unique codons coding for the 20 amino acids. To ensure a full coverage of potential mutants, well above a theoretical coverage of > 95%, all libraries were oversampled at least threefold. That is, 66 E. coli transformants from each library were needed. All transformants were screened by HPLC to select the highest mutant in eep value, then confirmed by DNA sequencing.
As a result, in saturation mutagenesis library E. coli/pveh1P184X, four E. coli transformants with an over 35% in eep value were obtained, that is, E. coli/pveh1P184E, E. coli/pveh1P184D, E. coli/pveh1P184M and E. coli/pveh1P184W, whose P184-encoding codon (CCT) was verified to be mutated to E-, D-, M- and W-encoding codons (GAG, GAT, ATG and TGG). As shown in Fig. 4, E. coli/pveh1P184E and E. coli/pveh1P184D could not transform 10 mM rac-mCSO completely, while E. coli/pveh1P184M and E. coli/pveh1P184W could. E. coli/pveh1P184W displayed the highest enantioconvergence in saturation mutagenesis library E. coli/pveh1P184X. The eep of (R)-mCPED catalyzed by E. coli/pveh1P184W was 59.8%, which was nearly 58-fold higher than that by PvEH1. Unfortunately, most E. coli transformants in saturation mutagenesis library E. coli/pveh1Y149X displayed no catalytic activity towards mCSO except E. coli/pveh1Y149L. Taking into consideration of studies on active areas in VrEH2 and StEH1 [21, 23], it is likely that amino acid sites around site 149 (including 149) may display important roles in the catalytic activity. On the other hand, the exchanges (Y149L and P184W) were crucial for the regioselectivity regulation [13].

The screening of PvEH1P184X in for the c of rac-mCSO and eep of (R)-mCPED. After DNA sequencing, four best mutants (PvEH1P184M, PvEH1P184E, PvEH1P184D and PvEH1P184W) were confirmed. The eep and c by these four were measured
The regioselectivity coefficients (βR and αS) were applied to quantitatively evaluate the preference attacking on Cβ of (R)-epoxide which could afford the diol of unchanged (R)-configuration, and on Cα of (S)-epoxide which could lead to the (R)-diol by inversion of configuration. It was necessary to determine regioselectivity coefficients which were helpful to understand the mechanism of enantioconvergence [8]. The αS values of mutants PvEH1Y149L and PvEH1P184W increased from 10.3% to 65.8% and 66.6%, which straight contributed to the high enantioconvergence. But they were still lower than that of PvEH1Y3 (Table 1).
Residue replacements of both Y149L and P184 W in PvEH1Y3
According to the research of Ye et al. [10] mutant PvEH1Y3 displayed a limited enantioconvergence towards mCSO (eep = 69.7%). But it still did not meet the requirement of production of (R)-mCPED. For further enhancing the enantioconvergence, we took the advantage of a cooperative mutational effect from replacements Y149L and P184W [24]. Mutant PvEH1Y3Z2 (containing residues replacements: L105I, M160A, M175I, Y149L and P184W) was engineered, using PvEH1Y3 as the template. The enantioconvergence of mutant PvEH1Y3Z2 was 94-fold higher than that of PvEH1 (eep 1.0%). Compared regioselectivity coefficients of PvEH1Y3Z2 with those of PvEH1, it was observed that almost all the improvement in enantioconvergence was contributed by the increase of αS and there was no obviously change in the βR. The αS of PvEH1Y3Z2 (97.5%) was higher than that of StEH1 (97%) and mbEH A (79%) [4, 25]. Combining with the research by Kotik et al. [13] we surmised that with the beneficial amino acid exchanging, the position of nucleophilic attack from PvEH1Y3Z2 is switched from Cβ to Cα of the oxirane ring of (S)-mCSO. On the other hand, nucleophilic attack of the oxirane ring of (R)-mCSO remained largely unaltered in the PvEH1Y3Z2, which creates an enantioconvergence towards rac-mCSO.
Enantioconvergent hydrolysis of rac-mCSO by E. coli/pveh1 Y3Z2
To confirm the details in the improvement in enantioconvergence of PvEH1Y3Z2, the eep, c, (R)- and (S)-mCPED concentrations in the enantioconvergent hydrolysis of rac-mCSO catalyzed by E. coli/pveh1Y3Z2 were monitored by chiral HPLC. With 20 mM rac-mCSO and 100 mg/mL E. coli/pveh1Y3Z2 adding into a volume of 5.0 mL, hydrolysis of rac-mCSO was carried out by resting cells at 25 °C, until c reached over 99%. With a large amount of (R)-mCPED produced, few (S)-mCPED was formed, which decreased eep value. Among the hydrolysis, the eep was all over 93%. The rac-mCSO was transformed into mCPED within 6 h completely (c = 99.5%). According to the Fig. 5, at the end of the hydrolysis, the eep and yield were 95.2% and 97.2%, respectively, and the chiral HPLC spectra of production was seen in Additional file 1: Figure S1b. By now, among all the reported EHs which the author has ever known, PvEH1Y3Z2 owned the highest enantioconvergence towards rac-mCSO, even higher than that of StEH1 (eep 91%) and Kau2 (eep 92%) [4, 22] (Table 2). Compared with enantioconvergences of other EHs towards other substrates, that of PvEH1Y3Z2 also displayed an excellent enantioconvergence. On the other hand, compared with the laboratory evolution of AnEH from Aspergillus niger M200 by Kotik et al. [13] less screening effort was taken and more improvement in enantioconvergence was achieved in this work (eep from 1.0 to 95.2% vs. from 3 to 70.5%).

The reaction course of enantioconvergent hydrolysis of rac-mCSO by whole cells which expressed PvEH1Y3Z2. The hydrolysis of mCSO was carried out at 25 °C, 2 mL volume containing 20 mM rac-mCSO, 100 mg/mL E. coli/pveh1Y3Z2 wet cells until the conversion reached over 99%. The reaction course of aliquots of sample were drawn out periodically, extracted with ethyl acetate, and analyzed by chiral HPLC and GC
Conclusions
In this work, the enantioconvergence towards mCSO was conferred on PvEH1 by a laboratory evolution. Firstly, after identified and analyzed using a CAVER 3.0 software, the SBP of PvEH1 was studied by leucine scanning. The result shows that PvEH1Y149L and PvEH1P184L have a marked increase in enantioconvergence, which means that sites 149 and 184 played important roles in the enantioconvergent hydrolysis of rac-mCSO. Secondly, to confirm the best residue at each site, saturation mutagenesis libraries E. coli/pveh1Y149X and E. coli/pveh1P184X were constructed and screened. Mutants PvEH1Y149L and PvEH1P184W have the highest enantioconvergence in each saturation mutagenesis library. Thirdly, mutant PvEH1Y3Z2 containing five residue replacements was constructed using PvEH1Y3 as the template. The enantioconvergence of PvEH1Y3Z2 was 94-fold higher than that of PvEH1. The analysis of regioselectivity coefficients indicated that the position of nucleophilic attack from mutant PvEH1Y3Z2 switched from Cβ to Cα of the oxirane ring of (S)-mCSO, which led to the improvement in enantioconvergence. At last, enantioconvergent hydrolysis by E. coli/pveh1Y3Z2 was monitored. The result showed that the eep and yield of (R)-mCPED were 95.2% and 97.2%, when the rac-mCSO was transformed into mCPED completely (c = 99.5%).
Availability of data and materials
All data generated and/or analysed during this study are included in this article.
References
Xu LN, Fang GY, Yu YH, Ma YF, Ye ZH, Li ZY. Molecular mechanism of heterogeneous supramolecular catalysis of metal-free cucurbituril solid for epoxide alcoholysis. Mol Catal. 2018;467:1–8.
Kamble MP, Yadav GD. Biocatalytic resolution of (R,S)-styrene oxide using a novel epoxide hydrolase from red mung beans. Catal Today. 2018;309:236–41.
Tan CL, Zhang X, Zhu ZJ, Xu MJ, Yang TW, Osire T, Yang ST, Rao ZM. Asp305Gly mutation improved the activity and stability of the styrene monooxygenase for efficient epoxide production in Pseudomonas putida KT2440. Microb Cell Fact. 2019;18:12.
Monterde MI, Lombard M, Archelas A, Cronin A, Arand M, Furstoss R. Enzymatic transformations. Part 58: enantioconvergent biohydrolysis of styrene oxide derivatives catalysed by the Solanum tuberosum epoxide hydrolase. Tetrahedron Asymmetry. 2004;15:2801–5.
Kotik M, Archelas A, Wohlgemuth R. Epoxide hydrolases and their application in organic synthesis. Curr Org Chem. 2012;16:451–82.
Kamble MP, Yadav GD. Kinetic resolution of (R,S) phenyl glycidyl ether by red mung beans (Vigna angularis) epoxide hydrolases. Biocatal Agric Biotechnol. 2017;12:260–5.
Solares LF, Mateo C. Improvement of the epoxide hydrolase properties for the enantioselective hydrolysis of epoxides. Curr Org Chem. 2013;17:744–55.
Wu YW, Kong XD, Zhu QQ, Fan LQ, Xu JH. Chemoenzymatic enantioconvergent hydrolysis of p-nitrostyrene oxide into (R)-p-nitrophenyl glycol by a newly cloned epoxide hydrolase VrEH2 from Vigna radiate. Catal Commun. 2015;58:16–20.
Li C, Zhao J, Hu D, Hu BC, Wang R, Zang J, Wu MC. Multiple site-directed mutagenesis of a Phaseolus vulgaris epoxide hydrolase to improve its catalytic performance towards p-chlorostyrene oxide based on the computer-aided re-design. Int J Biol Macromol. 2019;121:326–32.
Ye HH, Hu D, Shi XL, Wu MC, Deng C, Li JF. Directed modification of a novel epoxide hydrolase from Phaseolus vulgaris to improve its enantioconvergence towards styrene epoxides. Catal Commun. 2016;87:32–5.
Li AT, Qu G, Sun ZT, Reetz MT. Statistical analysis of the benefits of focused saturation mutagenesis in directed evolution based on reduced amino acid alphabets. ACS Catal. 2019;9:7769–78.
Hibbert EG, Dalby PA. Directed evolution strategies for improved enzymatic performance. Microb Cell Fact. 2005;4:29.
Kotik M, Archelas A, Faměrová V, Oubrechtová P, Křen V. Laboratory evolution of an epoxide hydrolase—towards an enantioconvergent biocatalyst. J Biotechnol. 2011;156:1–10.
Zheng HB, Kahakeaw D, Acevedo JP, Reetz MT. Directed evolution of enantioconvergency: the case of an epoxide hydrolase-catalyzed reaction of a racemic epoxide. ChemCatChem. 2010;2:958–61.
Sun ZT, Lonsdale R, Kong XD, Xu JH, Zhou JH, Reetz MT. Reshaping an enzyme binding pocket for enhanced and inverted stereoselectivity: use of smallest amino acid alphabets in directed evolution. Angew Chem Int Ed. 2015;54:12410–5.
Kong XD, Yuan S, Li L, Chen S, Xu JH, Zhou J. Engineering of an epoxide hydrolase for efficient bioresolution of bulky pharmaco substrates. Proc Natl Acad Sci. 2014;111:15717–22.
Zou SP, Zheng YG, Wu Q, Wang ZC, Xue YP, Liu ZQ. Enhanced catalytic efficiency and enantioselectivity of epoxide hydrolase from Agrobacterium radiobacter AD1 by iterative saturation mutagenesis for (R)-epichlorohydrin synthesis. Appl Microbiol Biotechnol. 2018;102:733–42.
Kille S, Acevedo-Rocha CG, Parra LP, Zhang ZG, Opperman DJ, Reetz MT, Acevedo JP. Reducing codon redundancy and screening effort of combinatorial protein libraries created by saturation mutagenesis. ACS Synth Biol. 2013;2:83–92.
Li C, Hu D, Zong XC, Deng C, Feng L, Wu MC, Li JF. Asymmetric hydrolysis of styrene oxide by PvEH2, a novel Phaseolus vulgaris epoxide hydrolase with extremely high enantioselectivity and regioselectivity. Catal Commun. 2017;102:57–61.
Barth S, Fischer M, Schmid RD, Pleiss J. Sequence and structure of epoxide hydrolases: a systematic analysis. Proteins. 2004;55:846–55.
Li FL, Kong XD, Chen Q, Zheng YC, Xu Q, Chen FF, Fan LQ, Lin GQ, Zhou JH, Yu HL, Xu JH. Regioselectivity engineering of epoxide hydrolase: near-perfect enantioconvergence through a single site mutation. ACS Catal. 2018;8:8314–7.
Kotik M, Stepánek V, Grulich M, Kyslík P, Archelas A. Access to enantiopure aromatic epoxides and diols using epoxide hydrolases derived from total biofilter DNA. J Mol Catal B Enzym. 2010;65:41–8.
Amrein BA, Bauer P, Duarte F, Carlsson ÅJ, Naworyta A, Mowbray SL, Widersten M, Kamerlin SCL. Expanding the catalytic triad in epoxide hydrolases and related enzymes. ACS Catal. 2015;5:5702–13.
Reetz MT. The importance of additive and non-additive mutational effects in protein engineering. Angew Chem Int Ed. 2013;52:2658–66.
Xu W, Xu JH, Pan J, Gu Q, Wu XY. Enantioconvergent hydrolysis of styrene epoxides by newly discovered epoxide hydrolases in mung bean. Org Lett. 2006;8:737–1740.
Kotik M, Zhao W, Iacazio G, Archelas A. Directed evolution of metagenome-derived epoxide hydrolase for improved enantioselectivity and enantioconvergence. J Mol Catal B Enzym. 2013;91:44–51.
Hu BC, Li C, Wang R, Zong XC, Li JP, Li JF, Wu MC. Improvement in the activity and enantioconvergency of PvEH3, an epoxide hydrolase from Phaseolus vulgaris, for p-chlorostyrene oxide by site-saturation mutagenesis. Catal Commun. 2018;117:9–13.
Acknowledgements
We are grateful to Prof. Xianzhang Wu (School of Biotechnology, Jiangnan University) and Mr. Xiang Lv for providing the technical assistance.
Funding
This work was financially supported by the National Natural Science Foundation of China (No. 21676117) and Postgraduate Research and Practice Innovation Program of Jiangsu Provence (SJX17_0503).
Author information
Authors and Affiliations
Contributions
XZ and CL designed this study. XZ performed most experiments. All the authors analyzed the data. XZ and MW mainly wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Chiral HPLC spectra for enantioconvergence hydrolytic of rac-mCSO by PvEH1Y3Z2 including analysis of rac-mCSO and rac-mCPED (a), and analysis of (R)-mCPED (b). Figure S2. The comparison of the homology model (red) with the template model (green). Figure S3. The change in RMSD values of the whole model. The RMSD value of the whole model tends to 0.18 nm. Figure S4. The Ramachandran plots of the model. The Ramachandran favored residue sites was 96.07%, which means that the most distribution of residues is good and the model could be believed. Figure S5. The local quality estimate of every residue. Compared with the template model, the identity of amino acids lining the substrate-binding pocket was 93.94%. The local similarity value of most residue in the substrate-binding pocket was over 0.8, which means the residue was highly similar to the template model. Table S1. Enantioconvergences and activities of PvEH1 and its three-site mutant towards rac-mCSO. Table S2. PCR primers used for the site-saturation saturation mutagenesis of pveh1. Table S3. PCR primers used for combinatorial site-directed mutagenesis of pveh1Y3. Table S4. Catalytic characteristics of representative mutants from leucine scanning mutagenesis.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zong, XC., Li, C., Xu, YH. et al. Substantially improving the enantioconvergence of PvEH1, a Phaseolus vulgaris epoxide hydrolase, towards m-chlorostyrene oxide by laboratory evolution. Microb Cell Fact 18, 202 (2019). https://doi.org/10.1186/s12934-019-1252-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-019-1252-4