
Abstract
Background
Numerous bacterial human growth hormone (hGH) expression methods under conventional fermentation and induction conditions have been described. Despite significant progress made in this area over the past several years, production of recombinant hGH by using cellular expression systems still requires further optimization. Fusion of the ubiquitin (Ub) tag to the hGH protein allowed to increase of the overall efficiency of the biosynthesis and improve the protein stability. Ub is a protein composed of 76 amino acid residues with a molecular mass of 8.6 kDa, expressed in all eukaryotes. This protein is an element of the universal protein modification system, which does not occur in bacteria, and is a useful carrier for heterologous proteins obtained through expression in Escherichia coli. Purification of Ub-fusion proteins is easier than that of unconjugated recombinant proteins, and Ub can be removed by deubiquitinating proteases (DUBs or UBPs).
Results and Conclusion
In the present study the UBPD2C protease, a stable UBP1 analog, was produced as a recombinant protein in E. coli and used for production of recombinant human growth hormone (rhGH). hGH was expressed as a fusion protein with Ub as a tag. Our findings show that the UBPD2C protease is very effective in removing the Ub moiety from recombinant Ub-fused hGH. The described approach enables obtaining a considerable yield of rhGH in a purity required for pharmaceutical products.
Similar content being viewed by others

Background
UBP1 protease is a yeast cysteine protease, 809 amino acids long, which cleaves ubiquitin from proteins fused to its C-terminus [1], and binds to Ub through an ester bond during the reaction. The protease activity depends on its ability to cleave the Ub peptide fused via its C-terminus to other polypeptides, regardless of the amino acid sequence of the fused moiety [2].
In some expression systems, proteins of interest are first expressed as fusions with Ub [3] or its derivatives [4]-[10], and then recovered using an Ub-removing enzyme (e.g. UBP1) [11],[12]. This approach has many advantages, including improved quality and efficiency of protein expression as well simplified purification process, which are of great importance in the industrial production of recombinant proteins [13]-[18]. We previously described [19] UBP1 protease deletion and point mutations, which improved the expression level of the protease in a microbiological expression system [20].
In technological processes, large quantities of deubiquitylating enzyme (DUB) are required for maximum possible catalytic activity. However, the majority of the currently available expression protocols does not lead to efficient expression of DUB, which greatly limits their applicability, especially in industrial processes. In the production of a specific protein for therapeutic application, particularly human growth hormone (hGH), purity and activity of the enzymes used are important factors. Accordingly, we have presented various solutions to meet these requirements [19],[20]. Nevertheless, there is still a need for a method to obtain highly active DUB enzyme in a gram-scale quantity for the production of bioactive peptides [4],[5],[21].
In the present study, we describe the development of an efficient method for obtaining hGH from bacterial cells, particularly from Escherichia coli. This method could yield gram-scale quantities of the protein with appropriate activity for application in the production of pharmaceutical preparations.
Human growth hormone
22 kDa hGH, also known as somatotropin, is a single chain polypeptide consisting of total 191 amino acids [22],[23]. It is synthesized and secreted by cells known as somatotrophs in the anterior pituitary. In the lifetime of an individual, the highest quantities of growth hormone are produced during puberty in the anterior lobe or glandular portion of the pituitary [24]-[28]. hGH is synthesized as a precursor and released into the blood following post-translational modifications. The hormone is used in the treatment of certain forms of dwarfism caused by hGH deficiencies, in obesity therapy, and in wound and burn treatment. Clinical trials indicate that hGH helps diminish the devastating effects of the hypermetabolic response to burn injury especially among children. These are the reasons why the demand for this hormone is very high [29]-[33].
Production of recombinant hGH requires optimization of vectors used to express the protein in those heterologous hosts. A series of methods have been designed [8],[34]-[39]. A generic expression vector features promoter sequences facilitating transcription and translation as well as sequences ensuring the stability of the synthesized protein. The vector described in this study contains strong promoters that enable efficient synthesis and accumulation of 30% or more of the targeted recombinant proteins among the total cellular proteins. In this study, we have reported the cloning, expression, refolding, and purification of hGH.
Results and discussion
The Ub tag was used to obtain a mature form of hGH
Ub, the best-known short protein, was used as the tag in this study. The use of Ub to enhance the levels of protein expression has been reported previously [4],[13],[14],[40]-[43]. In the present study, efficient expression of the Ub::hGH fusion gene in E. coli strains was achieved, and the UBPD2C analog of the UBP1 protease was used to cleave the Ub moiety from the N-terminus of the hGH. The hybrid polypeptide gene was cloned into several expression vectors such as pIGAL1, pIGDM1, pIGRKAN, and pIGMS31KAN. The resulting plasmids pIGDMKUH, pIGALUH, pIGALUHM, pIGRKKhGH, and pIGMS31PRH were used to transform E. coli strains DH5α and NM522. The E. coli DH5α cells were found to be most suitable with regard to the expression level of this fusion gene and the protein purification process. We used several vectors to determine the most suitable one, and the differences between them have been described in the Materials and methods section. The above-mentioned expression vectors with constitutive promoters pms or P1, P2 and terminators T1, T2 and pms (in appropriate combinations) were used. All of the constructed E. coli strains carried one of the following plasmids: pIGDMKUH, pIGALUH, pIGALUHM, pIGRKKhGH, or pIGMS31PRH, and were tested according to the procedure briefly presented in Figure 1.

Schematic showing the steps involved in the purification of the growth hormone. The values obtained for selected parameters of the process as well as the protein yields are indicated.
The scheme presented in Figure 1 summarizes the workflow after harvest of the inclusion bodies: purification of Ub::hGH by chromatography on SP Sepharose FF column, renaturation, protein precipitation and cleavage of Ub from Ub::hGH fusion protein. Purification of hGH was performed on Phenyl Sepharose FF column and then the protein was concentrated on Q Sepharose FF column. The purity of the preparations at each stage of purification was monitored via SDS-PAGE (Figures 2 and 3).

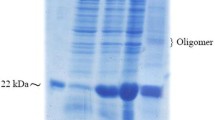
SDS-PAGE of the growth hormone samples from successive purification stages. (A) M, low molecular weight (LMW) protein marker (94, 67, 43, 30, 20.1, and 14.4 kDa); 1, the protein dissolved in DRCI buffer; 2, peak I obtained after purification on DEAE Sepharose FF column; 3, peak I obtained after purification on SP Sepharose FF column; 4, protein after renaturation; 5, sample after ammonium sulfate precipitation. (B) M, low molecular weight (LMW) protein marker (94, 67, 43, 30, 20.1, and 14.4 kDa); 1, growth hormone European standard; 2, Phenyl Sepharose FF column entry; 3, fraction I obtained after purification on Phenyl Sepharose FF column; 4, fraction II obtained after purification on Phenyl Sepharose FF column; 5, fraction III obtained after purification on Phenyl Sepharose FF column.

SDS-PAGE of Ub::hGH fusion protein cleaved by UBPD2C protease: M, low molecular weight (LMW) protein marker (94, 67, 43, 30, 20.1, and 14.4 kDa); 1, 10-min digestion; 2, 15-min digestion; 3 and 7, 30-min digestion; 4, 40-min digestion; 5, 50-min digestion; 6, 60-min digestion; 8, standard Ub::hGH.
Ub::hGH cleavage by UBPD2C-the UBP1 analog
The time course of Ub::hGH fusion protein digestion by the UBP1 protease analog was analyzed. Aliquots of the reaction mixture were collected at different time points and resolved by SDS-PAGE (Figure 3). It can be clearly seen from the figure that most of the Ub:hGH was converted to hGH already after 10 min. The reaction proceeds up to 40 min. From that point the intensity of the Ub::hGH band does not change indicating termination of the cleavage.
The stability of the UBPD2C enzyme prepared in an earlier study [19] was further investigated. The UBP1 protease analog was incubated for 22 h at temperatures of 4°C, 20°C, 25°C and 37°C, respectively. Subsequently, each of these test preparations was used for digesting the Ub::hGH fusion protein under the conditions described in the Materials and methods section. Aliquots of the samples collected at different time points were analyzed by SDS-PAGE. As shown in Figure 4, the protease was stable at all the different temperatures tested (Figure 4).

SDS-PAGE analysis of the UBPD2C activity after incubation for 22 h at temperatures of 4°C, 20°C, 25°C and 37°C. 1, 6 Ub::hGH undigested; 2, incubated at 4°C; 3, incubated at 20°C; 4, incubated at 25°C; 5, incubated at 37°C; M, low molecular weight standard (kDa).
Furthermore, the stability of the purified UBP1 analog protease was also tested by storing aliquots of the samples at 20°C, 4°C, -4°C and -20°C for 25 weeks. Subsequently, standard digestion procedure using the Ub::hGH purified substrate was carried out and the extent of cleavage was assessed by SDS-PAGE, followed by densitometric quantification of the reaction products (GelScan v. 1.45). The results of this analysis, shown in Figure 5, clearly demonstrated that UBPD2C enzyme can be stored for 25 weeks at -20°C. If stored at 4°C it is completely inactivated after 2 weeks and at -4°C after 3 weeks. Incubation of the protease at 20°C for three days allowed to retain 50% of its activity. After 1 week in this temperature it became completely inactivated. Protease stored at -70°C remained active for 25 weeks (data not shown). The enzyme is capable working in a wide range of pH (from pH 10.0 to pH 6.0) and temperature (from 15°C to 50°C) (data not shown).

Stability of UBPD2C protease. Stability of UBP1 protease measured every week after incubation of this enzyme at +20°C, +4°C, -4°C or -20°C. The first measurement of UBP1 protease activity incubated at +20°C followed after 3 days.
Final preparations of the hGH purified from the five tested E. coli strains were analyzed by high-performance liquid chromatography (HPLC) using the method described in the European Pharmacopoeia (5th Edition, 2005) [44]. Accordingly, E. coli DH5α cells carrying the pIGDMKUH plasmid were found to be the best construct for obtaining hGH. The hGH preparation obtained from this strain and purified according to the method described earlier exhibited approximately 92.2% purity, thus fulfilling the specifications/conditions required by United States Pharmacopeial Convention [45] and European Pharmacopoeia (7th Edition, 2010) [44] (Table 1). Accordingly, in order to monitor the size of molecules present in the Ub::hGH UBPD2C digest and hGH after Phenlyl Sepharose FF Column, size exclusion chromatography (SEC) was performed (data not shown, please refer to the Figure 1 in the manuscript, showing the steps involved in the purification of the growth hormone). The protein purity analysis allowed to observe a significant improvement in hGH purity by removal of high and low molecular weight impurities present in the digest.
Analysis of the stability of various hGH expression constructs in E. coli DH5α cultures grown in an antibiotic-free LB medium
Stability experiments were conducted according to the above-described method using the E. coli DH5α host strain with various expression vectors: pIGMS31PRH, pIGRKKhGH, pIGDMKUH, pIGALUH, pIGALUHM.
SDS-PAGE and phase contrast microscopy revealed that the most stable plasmids used for the transformation of E. coli DH5α cells was pIGDMKUH. The plasmid was found to remain stable through 80 generations (four passages) in antibiotic-free cultures (Figure 6). This finding is important for the use of the protein in the pharmaceutical industry.

SDS-PAGE assessment of the stability of the pIGDMKUH plasmid in E. coli DH5α cells. Every passage was cultured for 18 h. M, low molecular weight standard (94, 67, 43, 30, 20.1, and 14.4 kDa); 1, 1st passage of culture with antibiotic pressure; 2, 2nd passage of culture with antibiotic pressure; 3, 3rd passage of culture with antibiotic pressure; 4, 4th passage of culture with antibiotic pressure; 5, purified Ub::hGH as control; 6, 1st passage of culture without antibiotic pressure; 7, 2nd passage of culture without antibiotic pressure; 8, 3rd passage of culture without antibiotic pressure; 9, 4th passage of culture without antibiotic pressure.
Production of hGH through fermentation using organisms transformed by recombinant hGH expression constructs requires optimization of vectors used to express the proteins of interest in those heterologous hosts. A generic expression vector features promoter sequences facilitating transcription and translation as well as sequences ensuring the stability of the synthesized protein. The vectors described in this study contained strong promoters that could direct efficient synthesis and accumulation of 30% or more of recombinant proteins among the total cellular proteins. Each construct was different with respect to the kind of promoter used or its orientation or transcription terminator or resistance to antibiotics. These were the main variations leading to selection of one final vector construct enabling most efficient recombinant hGH expression.
In many cases the overall yield of biologically active protein is very low, and the methods of purification hGH are often not efficient or time-consuming. An alternative approach that has been developed involve complete solubilization and purification of rhGH produced in E. coli. [34]. Kim et al described obtaining of hGH and His-hGH expression by inducing IPTG and purifying them on the Ni-NTA column. The other work [46] described seven N-terminal fusion partners His6, Trx, GST, b’ and a’ domain of PDIb’ a’, NusA, PDI for soluble overexpression of hGH in E. coli. To cleave the tag proteins from fused hGH, the TEVrs protease was used. The tobacco etch virus recognition site ENLYFQ/G was placed at the N-terminus of hGH. In this case, only the DNA sequence was changed rather than amino acids. We tried to obtain hGH with a native growth hormone sequence for medical/pharmaceutical application.
In addition to the above-mentioned analyses, HPLC purification, sequencing of the N-terminus (15 amino acid residues; performed at Biocenter Kraków), and mass spectrometry of the purified preparation were performed to confirm that the protein is hGH.
Biological activity of rhGH
To determine the biological activity of the obtained rhGH, the protein was allowed to stimulate mitosis, and then tetrazolium salt (MTT) was added to determine the number of live cells. In this method, active mitochondrial dehydrogenases in the live cells changed the yellow color of the MTT to a purple product of formazan with a concentration proportional to the number of live cells.
This method allowed the examination of the biological activity of the rhGH preparation, and confirmed that proper tertiary structure of the protein had been obtained. The calculated biological activity of the preparation was 4.4 IU/mg and that of the somatotropin standard was 2.0 IU/mg. The rhGH protein purified under our conditions retained its biological activity at the comparable level to that of control hGH. In contrast, no growth stimulation was observed with BSA, which was used as a negative control (Figure 7).

Results of the measured biological activity of the prepared rhGH obtained with the use of KC Junior program in ELISA μQuant Bio-Tek Instruments. (■) control hGH (somatotropin) curve; (♦) curve for absorbance vs. rhGH protein concentration in the preparation and (▲) BSA at the indicated concentration.
Once satisfactory yield of purified hGH was obtained from 1 l of bacterial culture, the process was further scaled-up and the growth hormone was produced on a large scale (data not shown). Usage of 150 liter fermentor enabled to achieve the production capacity of 250 g per year.
Conclusions
The purpose of this study was to develop an efficient method for obtaining recombinant hGH from E. coli. We showed that the strains of E. coli, transformed with expression plasmids carrying the Ub-growth hormone gene, yielded satisfactory expression of the Ub-growth hormone protein in LB medium. In each case, most of the Ub-growth hormone fusion protein was produced in the inclusion bodies. The efficiency of protein expression was found to be sufficiently high, enabling the developed strains of E. coli to be used for industrial-scale production of hGH (220 mg of inclusion bodies/1 l of culture). This method could facilitate the production of relatively large quantities of the protein with appropriate quality (purity: 92.2%) and activity (4.4 IU/mg while the standard reached 2.0 IU/mg) and application in pharmaceutical industry.
Furthermore, another purpose of this study was to establish a protocol for growth hormone purification, in particular, the renaturation conditions. Accordingly, it was found that the UBP1 protease analog is a flexible tool that may be successfully used in experimental research as well as in the manufacture of recombinant proteins.
To summarize, the use of the above-mentioned system (expression of the final substrate in the form of Ub::hGH fusion protein and the use of UBP1 analog protease for deubiquitination), exploiting a highly specific and stable protease allows retrieving the required protein with high yield. 20 mg of the purified hGH was obtained from 1 l of bacterial culture. These properties make it suitable for a large-scale production in the pharmaceutical industry. Moreover, it must be noted that the E. coli DH5α strain with pIGDMKUH plasmid was the most efficient construct, which remained stable and facilitated bacterial growth in the absence of any antibiotic.
Materials and methods
Primers, plasmids, and bacterial strains
The UBPD2C protease gene was obtained as described previously [19],[20], [47]. The Ub::hGH fusion gene was cloned into plasmid pIGCMST derivative [48] using restriction enzymes, NdeI and SalI. The recombinant plasmid was named pIGCMUHGH and was deposited in GenBank (Accession No. HQ845201) [49].
The pIGCMUHGH plasmid was used as a template for PCR. The hGHubF and hGHubF primers were used (Table 2) to amplify the Ub::hGH sequence. The amplified 450-bp DNA fragment was purified using a DNA gel extraction kit (A&A Biotechnology, Gdansk, Poland), and was digested with NdeI and SalI and subcloned into pIGMS31KAN, pIGRKKAN, pIGDMKUH, pIGALUH, and pIGALUHM expression vectors. Each of the obtained plasmids was sequenced to confirm the identity of the DNA sequence. Subsequently, these plasmids were transformed into E. coli strain NM522. The genotypes of the bacterial strains used in this study are listed in Table 3. The strains were grown in Luria-Bertani (LB) medium as described previously [50].
Construction of expression vectors
pms promoter and deo P1 P2 promoter
The pms promoter was isolated from a plasmid present in a clinical strain of Klebsiella pneumonia (GenBank Accession No. AY543071) [49],[50]. It is a strong, constitutive promoter (Patent PL 213561). The sequence of the deo P1P2 promoter/operator region is known and the entire deo P1P2 promoter region is approximately 760 bp long, with the two promoters being separated by a distance of about 600 bp. Expression of a gene product under deo P2-driven transcription is very low in the presence of glucose and very high in the presence of other energy producing sources. Initiations from P1 are negatively controlled by the deoR repressor, the inducer being deoxyribose-5-phosphate (deoxyribose-5-P). Initiations from P2 depend on the cyclic AMP/cyclic AMP receptor protein complex (cAMP/CRP) and are negatively controlled by the cytR repressor, the inducer being cytidine [51].
pIGMS31KAN plasmid
The promoter sequence of retron Ec86 and its following transcription terminator sequence [52] were inserted into pIGMS31KAN plasmid. The resulting pIGMS31PR plasmid was used to clone the Ub::hGH fusion gene (Figure 8).

Schematic representation of the expression vectors A-pIGRKKAN, B-pIGMS31KAN, C-pIGALUHM, D-pIGALUH and E-pIGDMKUH. hGH was expressed as a fusion protein with the Ub tag. The restriction enzyme sites used to clone the Ub::hGH gene are indicated.
pIGRKKAN plasmid
P1 and P2 deo operon of E. coli strain K-12 promoters, and the gene encoding the fusion protein composed of synthetic Ub and the gene growth hormone in R.NdeI and R.SalI restriction sites were cloned into pIGRKKAN plasmid, which was isolated from K. pneumonia strain 287-w. The restriction map of the recombinant plasmid is presented in Figure 8.
pIGDMKUH plasmid
The pIGDMKUH plasmid was derived from pIGDM1 plasmid (GenBank Accession No. AF014880) [49]. The length of the pIGDMKUH plasmid was 3887 bp and contained the pms promoter and T1, T2 transcription terminators [53]. The synthetic Ub::hGH fusion gene was cloned into R.NdeI and R.SalI sites (Figure 8).
pIGALUH and pIGALUHM plasmids
The pIGALUH and pIGALUHM plasmids were derived from pIGAL1 plasmid (GenBank Accession No. AY424310). The synthetic Ub::hGH fusion genes were cloned into the pIGALUH and pIGALUHM plasmids. The pIGALUH plasmid contained the ampicillin resistance gene, pms promoter, and terminator sequences, while the pIGALUHM plasmid contained the ampicillin resistance gene, pms promoter in reverse orientation, as well as an additional transcription terminator located upstream of the pms promoter (Figure 8).
Creating bacterial glycerol stocks for long-term storage of plasmids
The method of preparing bacterial stocks is critical for maintaining efficient expression and plasmid stability in host cells. One bacterial colony was inoculated into 3 ml of LB medium with an appropriate antibiotic. The culture was incubated under shaking for 4 h at 37°C, and then transferred into 20 ml of LB medium with antibiotic and again incubated under shaking at 37°C until the optical density measured at 600 nm (OD600) reached ≈ 1. The culture was subsequently aliquoted into sterile Eppendorf tubes (0.5 ml/tube) and 0.5 ml of 40% glycerol was added to each tube. The bacterial stocks were stored at −70°C. Later, 0.5 ml of the stock was added to 1 l of LB medium, and 200 ml of the LB medium supplemented with 200 μl of ampicillin (100 μg/ml) or 200 μl of kanamycin (50 μg/ml) and 100 μl of the inoculate was added to a 500-ml flask. The culture was incubated in a rotary shaker at 160 rpm and 37°C for 18 h, and then centrifuged at 6000 rpm for 5 min at 4°C. The obtained bacterial cell pellet was processed as described in the subsequent section.
Purification of Ub::hGH and cleavage of the fusion protein by deubiquitinating UBPD2C
Isolation of inclusion bodies
Wet cell biomass of 5–7 g, obtained from 1 l of culture, was suspended in 100 ml of Buffer C (50 mM Tris HCl, 0.5 M NaCl, and 5 mM β-mercaptoethanol; pH 7.5), and lysozyme was added to a final concentration of 0.43 mg/ml. The suspension was incubated for 35 min at 20°C. Then, Triton X-100 was added to a final concentration of 1% and the mixture was sonicated. Subsequently, phenylmethylsulfonyl fluoride (PMSF) was added to a final concentration of 1 mM and then glycerol followed in a quantity corresponding to one-third of the mass of the cell suspension. The mixture was centrifuged at 8000 rpm for 20 min at 20°C. The precipitate containing inclusion bodies was suspended in 100 ml of CT buffer (50 mM Tris HCl, 0.5 M NaCl, 5 mM β-mercaptoethanol, and 1% Triton X-100; pH 7.5) and sonicated for 10 min on ice, as previously described [50]. After centrifugation at 8000 rpm for 20 min, the precipitate was suspended in 100 ml of phosphate buffered saline (PBS) with 1% Triton X-100 and sonicated [50]. The precipitate was centrifuged again and suspended in 100 ml of PBS with 2 M urea, sonicated as reported [50], and centrifuged for another time to obtain the inclusion bodies. The yield of inclusion bodies was between 0.7 and 1.2 g/l of culture.
The inclusion bodies were dissolved in DRCI buffer (6–8 M urea, 50 mM phosphate buffer, and 5 mM β-mercaptoethanol; pH 12). The solution was cleared by centrifugation, its pH was adjusted to 7.0 with concentrated phosphoric acid, and was subsequently loaded onto DEAE Sepharose fast flow (FF) column (approximately 100 ml; GE Healthcare Life Sciences) equilibrated with 6–8 M urea and 20 mM phosphate buffer at pH 7.0. The flow through material, containing the Ub::hGH protein, was collected and the fusion protein was loaded onto SP Sepharose FF column.
SP Sepharose FF column
The pooled Ub::hGH peak fractions from the DEAE Sepharose FF column were directly loaded onto a 30-ml column filled with SP Sepharose FF (Amersham Pharmacia Biotech AB) equilibrated with 6–8 M urea and 20 mM phosphate buffer at pH 7.0. The flow through material, containing the Ub::hGH protein, was collected and renatured. Separation on this column was not necessary if the Ub::hGH protein yield from the DEAE column was about 70%.
Renaturation
Ub::hGH in fractions from the SP Sepharose FF column was renatured by approximately 10-fold dilution in BR buffer (20 mM phosphate buffer pH 7.0, 50 mM NaCl) to a concentration of 0.09–0.15 mg/ml protein and then incubated for 1 h at room temperature.
Ammonium sulfate precipitation
After renaturation, the fusion protein was precipitated by adding ammonium sulfate to 80% saturation at 4°C under constant stirring. The samples were centrifuged at 12000 rpm for 15 min at 4°C. The pellet was suspended in 1/40th of the pre-centrifugation sample volume (approximately 50 ml) of 20 mM phosphate buffer (pH 7.5) and 0.5 M NaCl, and then centrifuged at 12000 rpm for 15 min at 4°C.
Cleavage of Ub::hGH fusion protein using UBPDC, UBPD2C protease
The supernatant (protein in 20 mM phosphate buffer (pH 7.5) + 0.5 M NaCl) was incubated with appropriate DUB preparations to cleave the Ub moiety from the growth hormone polypeptide. The reaction was performed at 37°C for 1 h (1 μl of UBPD2C 60 μg of protein; 1 μl of UBPDC – 14 μg of protein). Then, the sample was centrifuged at 12000 rpm for 15 min at 4°C. The supernatant was fractionated by chromatography on Phenyl Sepharose FF resin. The purification process of UBPD2C and UBPDC proteases has been described elsewhere [19],[47].
Phenyl Sepharose FF column
Approximately 20 ml of Phenyl Sepharose FF column (GE Healthcare Life Sciences) was equilibrated with 0.5 M NaCl and 20 mM phosphate buffer (pH 7.0). The hGH protein was eluted with a 3 mM phosphate buffer at pH 7.0–9.0. The fractions containing the growth hormone were collected, and the protein solution was adjusted to pH 7.0 using concentrated phosphoric acid and stored at 4°C.
Q Sepharose FF column
The growth hormone fractions from Phenyl Sepharose FF column were concentrated on a 10-ml Q Sepharose FF column (Amersham Pharmacia Biotech AB). The carrier was equilibrated with 20 mM phosphate buffer at pH 7.5, transferred to the column, and eluted with a linear gradient of 0.5 M NaCl in 20 mM phosphate buffer (growth hormone containing fractions were eluted at 0.25 M NaCl).
Storage
The purified growth hormone was transferred into 3.1 mM Na2HPO4 (pH 7.0; by hydrophobic interaction chromatography, carrier: Phenyl Sepharose FF, isocratic distribution) for storage at 4°C.
Verification of plasmid stability in cultures grown in LB medium without antibiotic
One stock was inoculated into 50 ml of antibiotic-free LB medium. The culture was incubated under shaking at 37°C until an OD600 of ~1 was reached. Subsequently, 1 ml of the culture was used for successive passages. Four passages were carried out for each culture for 18 h, and 1-ml samples were collected to obtain bacterial lysates. The Ub::hGH gene expression was verified in the host cells. The experiment was repeated four times independent of the transformation phase, and the accuracy of the results was confirmed.
Densitometric analysis using Scangel v. 1.45 program
Quantitative analyses of the proteins Ub::hGH, Ub, and hGH were performed by staining with 0.05% Coomassie Brilliant Blue R250 and obtaining electropherograms using the software package Scangel v. 1.45 program (Kucharczyk T.E., Warsaw, Poland). For destaining, the gels were incubated in the same solution without the dye.
Biological activity assay method
The hGH biological activity assay in the Nb-2 rat cell line was carried out as described previously [51],[54]-[56]. Nb-2 is a prolactin-related cell line sensitive to hGH (somatotropin; European Collection of Cell Cultures (ECACC)). The Nb-2 cells were prepared for the hGH assay as described by Tanaka et al. [57]. After a 24-h incubation period, the cells were suspended at a concentration of 1 × 105 cell/ml in RPMI-1640 medium containing 10% horse serum, glutamine, antibiotic, and 2-mercaptoethanol. Subsequently, 100 μl of the suspension were added to each well of a 96-well cell culture plate, flat bottom (Nunc).
hGH standard solution
Somatotropin (2.0 IU/mg protein specific activity (SIGMA S-4776)) was used as the standard for hGH biological activity assay. The somatotropin solution was distributed onto a plate with Nb-2 cells so that its concentrations on the plate were 0.004, 0.008, 0.015, 0.03, 0.06, 0.12, 0.265, 0.53, 0.8, 1.07, and 1.33 ng/ml. Six replicates were assayed for each somatotropin concentration.
Preparation of dilutions of the test hGH preparation
The growth hormone protein concentration in the test preparation was assayed at a wavelength of 280 nm, considering the amount of tyrosine, tryptophan, and cysteine in the hGH. Solutions of the growth hormone preparation were distributed onto a plate with Nb-2 cells in six replicates. The scheme of solutions distribution and their concentrations were similar to those employed for somatotropin, the commercial marker of hGH.
Absorbance reading
The plates were incubated for 72 h (37°C, 5% CO2). Subsequently, 15 μl of MTT solution (5 mg MTT/1 ml of PBS) were added to each well, and the plates were incubated for 3 h at 37°C. Then, 60 μl of lysis buffer (10% Triton X-100, 0.2 N HCl, and 20% isopropanol) were added to each well and gently stirred. After that, 50 μl of DMSO were added and the plates were incubated for 0.5 h at 37°C. The absorbance was read with an ELISA plate reader (μQuant Bio-Tek Instruments) at two wavelengths: 585 and 655 nm. The somatotropin standard curves and hGH activity were determined using the KC Junior software program.
Authors’ contributions
AP, GP and PB conceived the project and designed research. AWK, IS, MS, DM, LChP, NL, AMR, ABK, AM, AJP, LG, JKS and RW performed the experiments. AP, GP, AWK and IS analyzed the data. ABK performed MALDI-TOF mass spectra of hGH (data not shown) and revised the manuscript. GP and PB contributed reagents/materials/analysis tools. AWK wrote the first draft of the manuscript. IS, AJP contributed to the writing of the manuscript. All authors read and approved the final manuscript.
Abbreviations
- aa:
-
Amino-acid residue
- bp:
-
Base pair(s)
- DUB:
-
Deubiquitinating protease
- IPTG:
-
Isopropyl-β-D-galactoside
- hGH:
-
Human growth hormone
- kDa:
-
Kilodalton
- LB:
-
Luria-Bertani
- PCR:
-
Polymerase chain reaction
- IU:
-
International unit
- UB:
-
Ubiquitin
- UBP:
-
Ubiquitin-specific processing protease
- SDS:
-
Sodium dodecyl sulfate
- SDS-PAGE:
-
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
References
Tobias JW, Varshavsky A: Cloning and functional analysis of the ubiquitin-specific protease gene UBP1 of Saccharomyces cerevisiae. J Biol Chem. 1991, 266: 12021-12028.
Yu HA, Kim SG, Kim EJ, Lee WJ, Kim DO, Park K, Park YC, Seo JH: Characterization of ubiquitin C-terminal hydrolase 1 (YUH1) from Saccharomyces cerevisiae expressed in recombinant Escherichia coli. Protein Expr Purif. 2007, 56 (1): 20-26. 10.1016/j.pep.2007.07.005.
Waugh DS: Making the most of affinity tags. Trends Biotechnol. 2005, 23 (6): 316-320. 10.1016/j.tibtech.2005.03.012.
Malakhov MP, Mattern MR, Malakhova OA, Drinker M, Weeks SD, Butt TR: SUMO fusions and SUMO-specific protease for efficient expression and purification of proteins. J Struct Funct Genomics. 2004, 5 (1–2): 75-86. 10.1023/B:JSFG.0000029237.70316.52.
Butt TR, Edavettal SC, Hall JP, Mattern MR: SUMO fusion technology for difficult-to-express proteins. Protein Expr Purif. 2005, 43 (1): 1-9. 10.1016/j.pep.2005.03.016.
Fong BA, Wu WY, Wood DW: The potential role of self-cleaving purification tags in commercial-scale processes. Trends Biotechnol. 2010, 28 (5): 272-279. 10.1016/j.tibtech.2010.02.003.
Lu Q: Seamless cloning and gene fusion. Trends Biotechnol. 2005, 23 (4): 199-207. 10.1016/j.tibtech.2005.02.008.
Hannig G, Makrides SC: Strategies for optimizing heterologous protein expression in Escherichia coli. Trends Biotechnol. 1998, 16 (2): 54-60. 10.1016/S0167-7799(97)01155-4.
Wang T, Evdokimov E, Yiadom K, Yan Z, Chock PB, Yang DC: Biotin-ubiquitin tagging of mammalian proteins in Escherichia coli. Protein Expr Purif. 2003, 30 (1): 140-149. 10.1016/S1046-5928(03)00098-6.
Costa SJ, Almeida A, Castro A, Domingues L, Besir H: The novel Fh8 and H fusion partners for soluble protein expression in Escherichia coli: A comparison with the traditional gene fusion technology. Appl Microbiol Biotechnol. 2013, 97 (15): 6779-6791. 10.1007/s00253-012-4559-1.
Baker RT, Varshavsky A: Inhibition of the N-end rule pathway in living cells. Proc Natl Acad Sci U S A. 1991, 88 (4): 1090-1094. 10.1073/pnas.88.4.1090.
Baker RT, Catanzariti AM, Karunasekara Y, Soboleva TA, Sharwood R, Whitney S, Board PG: Using deubiquitylating enzymes as research tools. Methods Enzymol. 2005, 398: 540-554. 10.1016/S0076-6879(05)98044-0.
Xu X, Jin F, Yu X, Ren S, Hu J, Zhang W: High-level expression of the recombinant hybrid peptide cecropinA(1-8)-magainin2(1-12) with an ubiquitin fusion partner in Escherichia coli. Protein Expr Purif. 2007, 55 (1): 175-182. 10.1016/j.pep.2007.04.018.
Baker RT, Smith SA, Marano R, McKee J, Board PG: Protein expression using cotranslational fusion and cleavage of ubiquitin. Mutagenesis of the glutathione-binding site of human Pi class glutathione S-transferase. J Biol Chem. 1994, 269 (41): 25381-25386.
Baker RT: Protein expression using ubiquitin fusion and cleavage. Curr Opin Biotechnol. 1996, 7 (5): 541-546. 10.1016/S0958-1669(96)80059-0.
Pilon AL, Yost P, Chase TE, Lohnas GL, Bentley WE: High-level expression and efficient recovery of ubiquitin fusion proteins from Escherichia coli. Biotechnol Prog. 1996, 12 (3): 331-337. 10.1021/bp9600187.
Wang A, Clapper J, Guderian JA, Foy TM, Fanger GR, Retter MW, Skeiky YA: A novel method for increasing the expression level of recombinant proteins. Protein Expr Purif. 2003, 30 (1): 124-133. 10.1016/S1046-5928(03)00075-5.
Catanzariti AM, Soboleva TA, Jans DA, Board PG, Baker RT: An efficient system for high-level expression and easy purification of authentic recombinant proteins. Protein Sci. 2004, 13 (5): 1331-1339. 10.1110/ps.04618904.
Wojtowicz A, Mazurkiewicz-Pisarek A, Plucienniczak G, Mikiewicz-Sygula D, Chojnacka L, Lukasiewicz N, Plucienniczak A: Expression of yeast deubiquitination enzyme UBP1 analogues in E. coli. Microb Cell Fact. 2005, 4 (1): 17-10.1186/1475-2859-4-17.
Wójtowicz A, Mikiewicz Syguła D, Płucienniczak G, Płucienniczak A: A UBP1 protease mutant, and its coding sequence, their application and methods of production. WO2004/097011. 2004
Zuo X, Li S, Hall J, Mattern MR, Tran H, Shoo J, Tan R, Weiss SR, Butt TR: Enhanced expression and purification of membrane proteins by SUMO fusion in Escherichia coli. J Struct Funct Genomics. 2005, 6 (2–3): 103-111. 10.1007/s10969-005-2664-4.
Lewis UJ, Sinha YN, Haro LS: Variant forms and fragments of human growth hormone in serum. Acta Paediatr Suppl. 1994, 399: 29-31. 10.1111/j.1651-2227.1994.tb13282.x.
Ikehara M, Ohtsuka E, Tokunaga T, Taniyama Y, Iwai S, Kitano K, Miyamoto S, Ohgi T, Sakuragawa Y, Fujiyama K, Ikari T, Kobayashi M, Miyake T, Shibahara S, Onot A, Uedat T, Tanakat T, Babat H, Mikit T, Sakurait A, Oishi T, Chisaka O, Matsubara K: Synthesis of a gene for human growth hormone and its expression in Escherichia coli. Proc Natl Acad Sci U S A. 1984, 81: 5956-5960. 10.1073/pnas.81.19.5956.
Isaksson OG, Eden S, Jansson JO: Mode of action of pituitary growth hormone on target cells. Annu Rev Physiol. 1985, 47: 483-499. 10.1146/annurev.ph.47.030185.002411.
Garcia-Barros M, Costoya JA, Rios R, Arce V, Devesa J: N-glycosylated variants of growth hormone in human pituitary extracts. Horm Res. 2000, 53: 40-45. 10.1159/000023512.
Olson KC, Fenno J, Lin N, Harkins RN, Snider C, Kohr WH, Ross MJ, Fodge D, Prender G, Stebbing N: Purified human growth hormone from E. coli is biologically active. Nature. 1981, 293 (5831): 408-411. 10.1038/293408a0.
Persson J, Andersen DC, Lester PM: Evaluation of different primary recovery methods for E. coli-derived recombinant human growth hormone and compatibility with further down-stream purification. Biotechnol Bioeng. 2005, 90 (4): 442-451. 10.1002/bit.20434.
Dagnaes-Hansen F, Holst HU, S Ndergaard M, Vorup-Jensen T, Flyvbjerg A, Jensen UB, Jensen TG: Physiological effects of human growth hormone produced after hydrodynamic gene transfer of a plasmid vector containing the human ubiquitin promotor. J Mol Med (Berl). 2002, 80 (10): 665-670. 10.1007/s00109-002-0371-1. Epub 2002
Mehta A, Hindmarsh PC: The use of somatropin (recombinant growth hormone) in children of short stature. Paediatr Drugs. 2002, 4 (1): 37-47. 10.2165/00128072-200204010-00005. Review
Climent V, Mar N F, Picó A: Pharmacologic therapy in growth hormone disorders and the heart. Curr Med Chem. 2007, 14 (13): 1399-1407. 10.2174/092986707780831195.
Breederveld RS, Tuinebreijer WE: Recombinant human growth hormone for treating burns and donor sites. Cochrane Database Syst Rev. 2012, 12: CD008990-Review
Herndon DN, Pierre EJ, Stokes KN, Barrow RE: Growth hormone treatment for burned children. Horm Res. 1996, 45 (Suppl 1): 29-31. 10.1159/000184825. Review
Rojas Y, Finnerty CC, Radhakrishnan RS, Herndon DN: Burns: an update on current pharmacotherapy. Expert Opin Pharmacother. 2012, 13 (17): 2485-2494. 10.1517/14656566.2012.738195.
Kim MJ, Park HS, Seo KH, Yang HJ, Kim SK, Choi JH: Complete solubilization and purification of recombinant human growth hormone produced in Escherichia coli. PLoS One. 2013, 8 (2): e56168-10.1371/journal.pone.0056168.
Salamone D, Barañao L, Santos C, Bussmann L, Artuso J, Werning C, Prync A, Carbonetto C, Dabsys S, Munar C, Salaberry R, Berra G, Berra I, Fern Ndez N, Papouchado M, Foti M, Judewicz N, Mujica I, Muñoz L, Alvarez SF, Gonz lez E, Zimmermann J, Criscuolo M, Melo C: High-level expression of bioactive recombinant human growth hormone in the milk of a cloned transgenic cow. J Biotechnol. 2006, 124 (2): 469-472. 10.1016/j.jbiotec.2006.01.005.
Patra AK, Mukhopadhyay R, Mukhija R, Krishnan A, Garg LC, Panda AK: Optimization of inclusion body solubilization and renaturation of recombinant human growth hormone from Escherichia coli. Protein Expr Purif. 2000, 18 (2): 182-192. 10.1006/prep.1999.1179.
Liu X, Chen Y, Wu X, Li H, Jiang C, Tian H, Tang L, Wang D, Yu T, Li X: SUMO fusion system facilitates soluble expression and high production of bioactive human fibroblast growth factor 23 (FGF23). Appl Microbiol Biotechnol. 2012, 96 (1): 103-111. 10.1007/s00253-011-3864-4.
Khan MA, Sadaf S, Sajjad M, Waheed Akhtar M: Production enhancement and refolding of caprine growth hormone expressed in Escherichia coli. Protein Expr Purif. 2009, 68 (1): 85-89. 10.1016/j.pep.2009.05.011.
Koo TY, Park TH: Expression of recombinant human growth hormone in a soluble form in Escherichia coli by slowing down the protein synthesis rate. J Microbiol Biotechnol. 2007, 17 (4): 579-585.
Hondred D, Walker JM, Mathews DE, Vierstra RD: Use of ubiquitin fusions to augment protein expression in transgenic plants. Plant Physiol. 1999, 119 (2): 713-724. 10.1104/pp.119.2.713.
Butt TR, Khan MI, Marsh J, Ecker DJ, Crooke ST: Ubiquitin-metallothione in fusion protein expression in yeast. A genetic approach for analysis of ubiquitin functions. J Biol Chem. 1988, 263 (31): 16364-16371.
Ecker DJ, Stadel JM, Butt TR, Marsh JA, Monia BP, Powers DA, Gorman JA, Clark PE, Warren F, Shatzman A, Crooke ST: Increasing gene expression in yeast by fusion to ubiquitin. J Biol Chem. 1989, 264 (13): 7715-7719.
Butt TR, Jonnalagadda S, Monia BP, Sternberg EJ, Marsh JA, Stadel JM, Ecker DJ, Crooke ST: Ubiquitin fusion augments the yield of cloned gene products in Escherichia coli. Proc Natl Acad Sci U S A. 1989, 86 (8): 2540-2544. 10.1073/pnas.86.8.2540.
European Pharmacopoeia 7th edition. Strasbourg: European Directorate for the Quality of Medicines and Healthcare, Council of Europe; 2011.
The United States Pharmacopeia, USP 35, NF 30, United States Pharmacopeial Convention, Rockville, MD 20852. 2011:1991.
Nguyen MT, Koo BK, Thi Vu TT, Song JA, Chong SH, Jeong B, Ryu HB, Moh SH, Choe H: Prokaryotic soluble overexpression and purification of bioactive human growth hormone by fusion to thioredoxin, maltose binding protein, and protein disulfide isomerase. PLoS One. 2014, 9 (3): e89038-10.1371/journal.pone.0089038.
Wojtowicz A, Mazurkiewicz-Pisarek A, Chojnacka L, Płucienniczak A: A UBP1 protease mutant, and its coding sequence, their applications and heterogonous protein expression system. Patent WO 2006/073320 A1. 2006
Mikiewicz D, Wróbel B, Wegrzyn G, Płucienniczak A: Isolation and characterization of a ColE1-like plasmid from Enterobacter agglomerans with a novel variant of rom gene. Plasmid. 1997, 38 (3): 210-219. 10.1006/plas.1997.1312.
Zaleski P, Wawrzyniak P, Sobolewska A, Mikiewicz D, Wojtowicz-Krawiec A, Chojnacka-Puchta L, Zielinski M, Plucienniczak G, Plucienniczak A: New cloning and expression vector derived from Escherichia coli plasmid pIGWZ12: A potential vector for a two-plasmid expression system. Plasmid. 2012, 67 (3): 264-271. 10.1016/j.plasmid.2011.12.011.
Plucienniczak A, Sokołowska I, Marciniak-Rusek A, Lukasiewicz N, Smorawińska M, Chojnacka L, Płucienniczak G, Mikiewicz-Syguła D, Kuthan J, Kruszyńska A, Wolinowska R, Strzeżek K, Wójtowicz A, Mazurkiewicz-Pisarek A, Wójcik E: Method for production of recombinant growth hormone in form of hybrid protein. US Patent. 2011, 892787-
Valentin-Hansen P, Aiba H, Schumperli D: The structure of tandem regulatory regions in the deo operon of Escherichia coli K12. EMBO J. 1982, 1: 317-322.
Lim D, Maas WK: Reverse transcriptase-dependent synthesis of a covalently linked, branched DNA-RNA compound in E. coli B. Cell. 1989, 56 (5): 891-904. 10.1016/0092-8674(89)90693-4.
Brosius J, Holy A: Regulation of ribosomal RNA promoters with a synthetic lac operator. Proc Natl Acad Sci U S A. 1984, 81 (22): 6929-6933. 10.1073/pnas.81.22.6929.
Denizot F, Lang R: Rapid colometric assay for growth and survival. Modification to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods. 1986, 89 (2): 271-277. 10.1016/0022-1759(86)90368-6.
Goodwin CJ, Holt SJ, Downes S, Marshal NJ: The use of intermediate electron acceptors to enhance MTT bioreduction in microculture tetrazolium assay for human growth hormone. Life Sci. 1996, 59 (20): 1745-1753. 10.1016/S0024-3205(96)00512-7.
Goodwin CJ, Holt SJ, Riley PA, Downes S, Marshal NJ: Growth hormone-responsive DT-diadhorase-mediated bioreduction of tetrazolium salts. Biochem Biophys Res Commun. 1996, 226 (3): 935-941. 10.1006/bbrc.1996.1453.
Tanaka T, Shiu RP, Gout PW, Beer CT, Noble RL, Friesen HG: A new sensitive and specific bioassay for lactogenic hormones: measurement of prolactin and growth hormone in human serum. J Clin Endocrinol Metab. 1980, 51 (5): 1058-1063. 10.1210/jcem-51-5-1058.
Acknowledgments
We thank Jacek Skowronski, Departments of Molecular Biology and Microbiology and Pharmacology, Case Western Reserve School of Medicine, Cleveland, Ohio, for critical reading of the manuscript and editorial help.
Funding
This work was supported by the European Union Project Innovative Economy POIG 01.01.02-00-007/08-00.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Wojtowicz-Krawiec, A., Sokolowska, I., Smorawinska, M. et al. Use of Ubp1 protease analog to produce recombinant human growth hormone in Escherichia coli . Microb Cell Fact 13, 113 (2014). https://doi.org/10.1186/s12934-014-0113-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-014-0113-4