
Abstract
Background
The gut-lung axis, pivotal for respiratory health, is inadequately explored in pulmonary and critical care medicine (PCCM) inpatients.
Methods
Examining PCCM inpatients from three medical university-affiliated hospitals, we conducted 16S ribosomal RNA sequencing on stool samples (inpatients, n = 374; healthy controls, n = 105). We conducted statistical analyses to examine the gut microbiota composition in PCCM inpatients, comparing it to that of healthy controls. Additionally, we explored the associations between gut microbiota composition and various clinical factors, including age, white blood cell count, neutrophil count, platelet count, albumin level, hemoglobin level, length of hospital stay, and medical costs.
Results
PCCM inpatients exhibited lower gut microbiota diversity than healthy controls. Principal Coordinates Analysis revealed marked overall microbiota structure differences. Four enterotypes, including the exclusive Enterococcaceae enterotype in inpatients, were identified. Although no distinctions were found at the phylum level, 15 bacterial families exhibited varying abundances. Specifically, the inpatient population from PCCM showed a significantly higher abundance of Enterococcaceae, Lactobacillaceae, Erysipelatoclostridiaceae, Clostridiaceae, and Tannerellaceae. Using random forest analyses, we calculated the areas under the receiver operating characteristic curves (AUCs) to be 0.75 (95% CIs 0.69–0.80) for distinguishing healthy individuals from inpatients. The four most abundant genera retained in the classifier were Blautia, Subdoligranulum, Enterococcus, and Klebsiella.
Conclusions
Evidence of gut microbiota dysbiosis in PCCM inpatients underscores the gut-lung axis's significance, promising further avenues in respiratory health research.
Similar content being viewed by others


Background
The human gastrointestinal tract hosts a diverse consortium of microorganisms collectively referred to as the gut microbiota, playing a pivotal role in sustaining host well-being and regulating various physiological processes [1]. Recent advancements have underscored the crucial involvement of the gut microbiota in the etiology of diverse diseases, transcending the boundaries of the gastrointestinal system [2, 3]. The reciprocal interplay between the gut and distant organs, notably the lungs, has led to the identification of the “gut-lung axis,” a conceptual framework with profound implications for respiratory and critical care medicine [4,5,6].
Gut microbiota dysbiosis signifies an imbalance in the composition and function of the gut microbial community, marked by shifts in the relative abundance of specific bacterial taxa. This imbalance has been implicated in a spectrum of diseases, encompassing inflammatory bowel disorders, obesity, diabetes, and even neurological conditions [7,8,9]. Importantly, a growing body of evidence links gut microbiota dysbiosis to respiratory diseases [6, 10, 11]. The gut-lung axis constitutes a complex bidirectional communication system, wherein alterations in gut microbiota can impact lung health, and vice versa. Lung-derived inflammatory mediators and metabolites reciprocally influence the gut environment, instigating changes in gut microbial composition and function [5].
Despite limited research, studies suggest that the gut microbiome plays a significant role in respiratory health. Thibeault et al., Pérez-Cobas et al., and Dang et al. demonstrate that microbial dysbiosis and gut microbiota-produced metabolites can influence respiratory disease outcomes through immune modulation [12, 13]. These studies highlight the importance of considering the gut-lung axis in understanding and potentially treating respiratory diseases in critically ill patients. However, its significance among inpatients undergoing pulmonary and critical care medicine (PCCM) treatment remains relatively unexplored. This study endeavors to fill this knowledge gap by employing 16S ribosomal RNA sequencing of stool samples to delineate the features of gut microbiota in PCCM patients across three distinct medical university-affiliated hospitals: the First Affiliated Hospital of Guangzhou Medical University, the Second Affiliated Hospital of Guangzhou Medical University, and the Second Hospital, University of South China. The primary objective is to scrutinize the prevalence and clinical implications of gut microbiota dysbiosis in PCCM inpatients. Delving into the composition and diversity of the gut microbiota in this specific patient cohort aims to illuminate potential associations between gut microbiota dysbiosis and respiratory diseases.
Methods
Study design and participants
To explore the characteristics of the gut microbiota in inpatients from the Department of PCCM, we devised a systematic and replicable workflow. This study constitutes a hospital-based, population-centered, cross-sectional, multicenter survey of inpatients within the PCCM. Drawing from our previous chronic obstructive pulmonary disease (COPD) cohort investigations [14, 15], we secured a representative healthy sample from the same village or residential area. All participants, including both patients and healthy subjects, were residents of Guangzhou and Nanhua city, sharing comparable lifestyles and dietary practices.
Between January and April 2022, a total of 374 stool samples were collected randomly from inpatients at three hospitals: the First Affiliated Hospital of Guangzhou Medical University (Hospital 1, n = 91), the Second Affiliated Hospital of Guangzhou Medical University (Hospital 2, n = 133) and the Second Hospital, University of South China (Hospital 3, n = 150). These inpatient samples were paired with stool samples from 105 healthy individuals to form the healthy control group. Demographic and clinical data were extracted from electronic medical records. All clinical information was collected following standardized procedures by the State Key Laboratory of Respiratory Disease at Guangzhou Medical University. Prior to stool donation, all enrolled patients and healthy individuals provided written informed consent. The study received ethical approval from Guangzhou Medical University (Approval No. 2021-YJS-ks-14).
Inclusion and exclusion criteria
Inclusion criteria: 1. Inpatients from the PCCM at the three designated hospitals. 2. Age range from 18–80 years. 3. Patients with a confirmed diagnosis of the target condition or disease (e.g., COPD, respiratory-related disease). 4. Subjects who provide informed consent to participate in the study. Exclusion criteria: 1. Treatment with systemic (e.g., oral, intravenous, or intramuscular) corticosteroids within the preceding 4 weeks. 2. Patients with comorbidities or underlying conditions that may significantly influence the gut microbiota or confound the study results. 3. History of gastrointestinal disease diagnosis. 4. Treatment with antibiotics (including macrolide antibiotics) within the preceding 4 weeks. 5. Subjects who are unable to provide informed consent or unwilling to participate in the study. 6. Patients deemed unsuitable for inclusion in the study by the investigators.
Considering disease characteristics and for the sake of convenient analysis, the prevalent diseases observed among inpatients in the PCCM can be primarily classified into four major groups: 1. Airway diseases (which include asthma, COPD, and bronchiectasis) 2. Lung infections (covering pneumonia, tuberculosis, and pulmonary fungal infections) 3. Respiratory failure 4. Non-small cell lung cancer (NSCLC) 5. Other conditions (such as pneumothorax, pulmonary embolism, and interstitial pneumonia).
Collection of fecal samples and 16SrRNA sequencing
On the first day of hospital admission, fresh fecal samples were collected before any examinations or treatments began. Each patient was provided with sterile culture dishes, sterile forceps, and sterile gauze. The fecal collection method involved placing a sterile gauze on the toilet surface, and after defecation, approximately 10 g of fecal matter was collected using sterile forceps and transferred to the sterile culture dish, with relevant information recorded. After collection, the fecal samples were immediately frozen and stored in liquid nitrogen and then transferred to a − 80 °C freezer within 24 h for subsequent analysis.
Total bacterial DNA extraction from stool samples was performed using the QIAamp® DNA Stool Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions on 100 mg of each sample. The extracted DNA from each sample served as a template for amplifying the V3-V4 region of 16S rRNA genes using PCR. 16S Amplicon PCR Forward Primer 5′(TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG CCT ACG GGN GGC WGC AG) and 16S Amplicon PCR Reverse Primer 5′(GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA GGA CTA CHV GGG TAT CTA ATC C) [16]. The amplification, in vitro transcription and labeling, as well as hybridization, were carried out following the Illumina 16S metagenomic sequencing library preparation guide [17]. Subsequently, all libraries were sequenced using an Illumina MiSeq platform (San Diego, CA, USA) at the Majorbio Co. Ltd. (Shanghai, China).
Sequencing data processing
The data were analyzed on the online tool of Majorbio Cloud Platform (https://cloud.majorbio.com/page/tools/) [18]. In summary, the raw sequencing data underwent quality filtering (Q30) and were merged using FLASH (https://ccb.jhu.edu/software/FLASH/). The QIIME software pipeline (Quantitative Insights into Microbial Ecology, v1.9.1, http://qiime.org/install/index.html) was employed to cluster high-quality reads into operational taxonomic units (OTUs) at the 97% identity level and summarize the representation of taxonomic groups. Taxonomic information for each OTU was obtained using the RDP classifier Bayesian algorithm (http://sourceforge.net/projects/rdp-classifier/, v2.11) to analyze representative sequences at a 97% similarity level. This analysis covered various taxonomic ranks, including Phylum, Class, Order, Family, Genus, and Species, allowing assessment of the species composition of each sample's microbial community. The GreenGenes 13.8 database (http://greengenes.secondgenome.com/) was used for OTU search. α-diversity indices, including ACE index, Shannon index, Chao1 index and Simpson index, and β-diversity indices, including Bray–Curtis and weighted UniFrac metrics, were calculated using QIIME.
Statistical analysis
Appropriate basic statistical analyses, such as the chi-square test, ANOVA, and the Kruskal–Wallis H test, were conducted using SPSS version 27 (IBM SPSS, Armonk, NY, USA) to compare continuous and categorical variables. The P values were adjusted for multiple testing using the Bonferroni method, and the analyses were further adjusted for age and smoking history. Briefly, the significance of differences in alpha diversity was determined using ANOVA and the Kruskal–Wallis test. Principal Coordinates Analysis (PCoA) was conducted to visualize differences in microbial composition between inpatients from three hospitals and healthy controls. Non-Metric Multidimensional Scaling (NMDS) was performed at the OTU level to assess similarities and shifts in gut enterotypes. Comparisons of the relative proportions of major bacterial phyla and families between healthy and inpatient groups were also conducted using Wilcoxon rank-sum test. The Receiver Operating Characteristic (ROC) Curve was used to evaluate the performance of classifiers distinguishing between healthy controls and inpatients. ANOVA was used to compare bacterial phyla and families in different respiratory disease groups and healthy controls. P values of < 0.05 were considered statistically significant.
Enterotyping was conducted as described by Costea et al. [18]. The Jensen-Shannon Distance (JSD) and other distance metrics were calculated based on the relative abundance of the bacterial community at the chosen taxonomic classification level. Partitioning Around Medoids (PAM) clustering was then performed. The optimal number of clusters (K) was determined using the Calinski-Harabasz (CH) index. Visualization was achieved through Between-class Analysis (BCA) when K was greater than or equal to 3, or Principal Coordinates Analysis (PCoA) when K was 2 or greater.
Results
Characteristics of the study population
A total of 479 Chinese participants were included in the study and categorized into two groups: healthy controls (n = 105) and the inpatient group (n = 374). The characteristics of the study participants are presented in Table 1. In comparison to healthy controls, inpatients from PCCM exhibited significantly lower body mass index (BMI), as well as significantly higher smoking history and smoking index. There were 81 cases (21.7%) of airway disease, 104 cases (27.8%) of lung infection, 37 cases (9.9%) of respiratory failure, 128 cases (34.2%) of NSCLC, and 24 cases (6.4%) classified as “other.” The clinical factors (age, white blood cell counts, neutrophil counts, platelet counts, albumin level, hemoglobin level, duration of hospital stay, and medical costs) for all patients are also summarized in Table 1.
Changes of gut microbial diversity in inpatients from PCCM
We found a significant difference in the alpha diversity of the gut microbiota among the healthy group and inpatients group. The ACE index, Shannon index and Chao index of inpatients were significantly lower than those of healthy control, while the Simpson index was significantly higher than that of healthy control (P < 0.01 or P < 0.001, Fig. 1A–H). The microbiome of the healthy controls had higher alpha diversity than either group of inpatients from the three hospitals.

Gut microbial diversity differs between healthy controls and inpatients from PCCM. A, C, E and G The alpha diversity index of inpatients from PCCM were significantly lower than those of healthy control. B, D, F and H The ACE index, Shannon index, Chao index, and Simpson index of inpatients from PCCM were grouped based on the source hospital. Each dot corresponds to a sample. Significance was determined by ANOVA and by kruskal–wallis test, and P values were corrected using the Bonferroni method. **P < 0.01, ***P < 0.001. PCCM, pulmonary and critical care medicine
To compare species diversity among groups and investigate the potential similarities or differences in the overall gut microbiota community structure between inpatients and the healthy group, we conducted a β-diversity analysis on both sets. To visualize differences in microbial composition, we conducted both PCoA and NMDS. PCoA was chosen for its effectiveness in displaying linear relationships, while NMDS was selected for its robustness with non-linear ecological data, providing a comprehensive analysis of microbial community structures. The PCoA indicated statistically significant differences in the overall gut microbiota community structure between the healthy and inpatient groups, based on Bray–Curtis dissimilarities (R = 0.0371, P = 0.001, ANOSIM; Fig. 2A, B). This observation was accentuated by relatively scattered inpatient samples, suggesting heightened variability within the inpatient groups. Similarly, we observed notably different microbiota compositions among the inpatients and healthy group based on the NMDS method (Stress: 0.165, R = 0.0371, P = 0.001, ANOSIM) (Fig. 3A). In the ordination plot (Fig. 3B), the healthy group was distinctly separated from the other groups (Hospital 1, Hospital 2, or Hospital 3) (R2 = 0.035, P = 0.001, ANOSIM).

Principal coordinates analysis (PCoA) was conducted at the OTU level for both inpatients from three hospital and healthy controls. A PCoA box plot: various colors depict sample groups across distinct conditions, with box plots illustrating the distinct distribution of sample groups along the PC1 axis in the figure. B PCoA analysis (R = 0.0371, P = 0.001). Each dot corresponds to a sample

Analysis of similarities and shifts in gut enterotypes in fecal samples between inpatients and healthy controls. A NMDS on OTU level (Stress:0.165, R = 0.0371, P = 0.001). B Calculation of Distances on OTU Level for Each Sample Group (R2 = 0.035, P = 0.001). C Enterotypes in clinical groups. The study identified three distinct enterotypes based on dominant bacterial families from the Firmicutes and Proteobacteria phyla: Lachnospiraceae (Enterotype 1), Enterobacteriaceae (Enterotype 2) and Enterococcaceae (Enterotype 3). D Barplot of typing analysis. The Enterococcaceae enterotype was exclusively observed in inpatients from PCCM, with proportions of 12.8%, 15.4%, and 7.33% in Hospitals 1, 2, and 3, respectively. Each dot corresponds to a sample. NMDS, non-metric multidimensional scaling
Enterotypes were categorized within the extensive study population (n = 479) using previously established methods [19]. The microbial profiling revealed the presence of three distinct enterotypes at the family level (Fig. 3C), characterized by dominant bacterial families from the Firmicutes and Proteobacteria phyla. Specifically, these enterotypes were represented by Lachnospiraceae (Enterotype 1), Enterobacteriaceae (Enterotype 2) and Enterococcaceae (Enterotype 3). Remarkably, the Enterococcaceae enterotype was exclusively identified in inpatients from PCCM. The proportions of the Enterococcaceae enterotype from Hospital 1, Hospital 2, and Hospital 3 were recorded as 12.8%, 15.4%, and 7.33%, respectively (Fig. 3D). A permutational multivariate analysis of variance using bray–curtis dissimilarity unveiled statistically significant differences in gut microbiota composition between healthy controls and all inpatient subjects (P = 0.021).
Altered gut microbial composition in inpatients from PCCM
A total of 1172 OTUs were identified through the clustering of 479 samples. Among these, 1,073 OTUs were shared between the healthy and inpatient groups, while 3 OTUs were exclusive to the healthy group and 96 OTUs were unique to the inpatient group (Additional file 1: Supplementary Fig. 1A, B). At the phylum level, both the healthy group and inpatients from PCCM exhibited a consistent composition of five major bacterial phyla: Bacteroidetes, Firmicutes, Proteobacteria, Actinobacteria, and Verrucomicrobiota. The first four phyla collectively accounted for over 97% of the total sequences in both groups (Fig. 4A, B). Notably, there was no significant distinction in phylum-level distribution between the healthy group and inpatients from PCCM.

Comparisons were made regarding the relative proportions of bacterial phyla and families between the healthy and inpatient groups. Specifically, panels (A) and (B) illustrate the comparisons of major bacterial phylum abundance, while panels (C) and (D) depict the comparisons of major bacterial family abundance between the healthy and inpatient groups. Significance was determined by ANOVA and by Wilcoxon rank-sum test, and P values were corrected using the Bonferroni method
Among the sequences categorized within these four phyla, they were primarily distributed across 18 bacterial families, which collectively represented over 95% of the total sequences across all four groups (Fig. 4C, D). While the distribution of these phyla was generally similar, fifteen of bacterial families exhibited varying relative abundances between the two groups (P < 0.05, P < 0.01, or P < 0.001; Fig. 5A). Specifically, a significantly higher abundance of Enterococcaceae, Lactobacillaceae, Erysipelatoclostridiaceae, Clostridiaceae, and Tannerellaceae was observed within the inpatient population from PCCM (P < 0.05, P < 0.01, or P < 0.001; Fig. 5A). On the contrary, ten gut microbiota such as Lachnospiraceae, Ruminococcaceae and Prevotellaceae were significantly increased in healthy group. The differential abundance analysis was conducted using the Wilcoxon rank-sum test at the family level to ensure robust identification of differentially abundant taxa.

The correlation between gut microbiota and clinical factors in inpatients, as well as identifies gut microbial markers distinguishing inpatients from healthy controls. A Fifteen bacterial families differed in their relative abundance in samples from the healthy and inpatient groups. B Relationship between gut microbiota and clinical factors in inpatients. Distance-based redundancy analysis of clinical factors and bacterial community. Wilcoxon rank-sum test bar plot on family level. C The receiver operating characteristic curve (ROC) illustrates the performance of the classifiers for distinguishing healthy status and inpatients, with a display of the 95% confidence interval. D Notch plots visualize the relative abundances of the microbial species retained in the classifier. Each dot corresponds to a sample. *P < 0.05, **P < 0.01 and ***P < 0.001. Neu, neutrophil; WBC, white blood cell
Relationship between gut microbiota and clinical factors in inpatients
As shown in Fig. 5B, the relationship between clinical factors (age, white blood cell counts, neutrophil counts, platelet counts, albumin level, hemoglobin level, days in hospital and medical costs) and microbial communities at genus level was investigated by distance-based redundancy analysis (db-RD). The factor most strongly correlated with bacterial community and species distribution was age (R2 = 0.0735, P = 0.001), followed by medical costs (R2 = 0.0518, P = 0.001), days in hospital (R2 = 0.0378, P = 0.002), hemoglobin (R2 = 0.0305, P = 0.005) and white blood cell counts (R2 = 0.0265, P = 0.01).
Specific gut microbial markers differentiated inpatients and healthy controls
With the confirmation of gut microbial dysbiosis among the inpatients, our subsequent goal was to investigate the potential of gut microbiota as non-invasive indicators for this group. Employing random forest analyses, we sought to identify taxonomic markers within the gut microbiome that could predict the distinction between the two groups (Additional file 2: Supplementary Fig. 2A, B). Utilizing taxonomic profiles at the genus level, the areas under the receiver operating characteristic curves (AUCs) were calculated to be 0.75 (95% CIs 0.69 − 0.80) for distinguishing healthy individuals from inpatients, underscoring the predictive capability of gut microbiota in classifying these groups (Fig. 5C). Following feature selection based on variable importance, a set of 15 genus-level taxa were retained within the classifier for both groups (Fig. 5D). To distinguish inpatients from healthy controls, Blautia, Subdoligranulum, Enterococcus and Klebsiella were the four most abundant genus retained in the classifier.
Specific gut microbiota signature in patients with different respiratory diseases
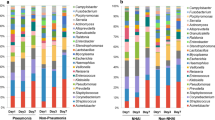
To explore whether gut microbiota dysbiosis is associated with different types of respiratory diseases, we categorized patients into four distinct disease groups: airway diseases, lung infections, respiratory failure, NSCLC, and others. In comparison to healthy individuals, the diversity index (P < 0.01 or P < 0.001, Fig. 6A) exhibited a significant reduction in the airway diseases group, lung infections group, respiratory failure group, and NSCLC group. Based on NMDS analysis (stress: 0.167, R = 0.0324, P = 0.002, ANOSIM, Fig. 6B), we observed substantial differences in microbiota compositions between patients with different diseases and the healthy groups. Similarly, the Enterococcaceae enterotype was exclusively observed in the patient group (Fig. 6C, D). The recorded proportions of the Enterococcaceae enterotype were 13.0% in the airway diseases group, 13.5% in the lung infections group, 8.1% in the respiratory failure group, 8.6% in the NSCLC group, and 17.4% in the others group. There were no significant distinctions in phylum-level distribution between the healthy group and the four different disease groups (Fig. 7A). When compared to the healthy control group, the airway diseases, lung infections, respiratory failure, and NSCLC groups all exhibited a significant increase in the abundance of Enterococcaceae (P < 0.001), along with a significant decrease in the abundance of Prevotellaceae and Lachnospiraceae (P < 0.001, Fig. 7B, Additional file 3: Supplementary Table 1).

Gut microbial diversity and enterotypes in patients with different respiratory diseases. A The Shannon index of the airway diseases group, lung infections group, respiratory failure group, and NSCLC group were significantly lower than that of the healthy control. B Remarkably distinct microbiota compositions were observed among the different respiratory diseases and the healthy group. C Distribution of samples across different enterotypes. D Distribution of enterotypes among the different respiratory diseases and the healthy group. Each dot corresponds to a sample. **P < 0.01 and ***P < 0.001

Gut microbial composition in patients with different respiratory diseases. A Relative proportions of bacterial phyla in the different respiratory diseases group and the healthy group. B Comparison of the abundance of major bacterial families in the different respiratory diseases group and the healthy group. Microbiota data were tested by ANOVA and the P values were corrected for multiple testing using the Bonferroni method
Discussion
The gut microbiota has gained significant attention due to its crucial role in maintaining host health and influencing various physiological processes. In this study, we aimed to explore the characteristics of the gut microbiota in inpatients admitted to the PCCM departments of three different medical university-affiliated hospitals. Our findings provide evidence of dysbiosis in the gut microbiota of inpatients from the PCCM.
The role of the gut microbiota in influencing overall systemic health is gaining increasing recognition. The concepts of the “gut-lung” axis and the “lung-gut” axis highlight the intricate communication between the gut and distant organs, notably the lungs [20,21,22]. Our results indicate that inpatients from PCCM exhibit gut microbiota dysbiosis, as evidenced by altered microbial diversity and composition. The lower alpha diversity indices (ACE, Shannon, Chao, and Simpson) observed in inpatients compared to healthy controls align with previous studies linking reduced diversity to various disease states, including chronic obstructive pulmonary disease [15, 23], asthma [24, 25], NSCLC [26] and pneumonia [27]. Our study has revealed several intriguing insights into the relationship between gut microbiota and inpatients from PCCM, highlighting the potential association between respiratory disease status and gut microbiota diversity, and vice versa.
The identification of four distinct enterotypes among the study population adds a novel dimension to the investigation of gut microbiota in PCCM inpatients. Notably, the exclusive presence of the Enterococcaceae enterotype in inpatients from PCCM points towards potential disease-specific associations. Consistently, a significantly higher abundance of the Enterococcus genus was observed within the inpatient population from the PCCM. Enterococci are gram-positive, facultative anaerobic bacteria that are typically present in low abundance within the healthy human gut microbiota. However, certain species of Enterococci, such as Enterococcus faecalis and E. faecium, can act as opportunistic pathogens, often leading to severe bloodstream infections in patients [28]. These bacteria have also been implicated in the progression of liver cancer and cholangitis [29, 30]. Previous research has demonstrated that a state known as Enterococcus intestinal domination (EID), where enterococci constitute ≥ 30% of the microbiota, is linked to conditions like Enterococcus bacteremia, graft-versus-host disease, and mortality in individuals undergoing allogeneic hematopoietic stem cell transplantation [31]. The clinical implications of this finding warrant further investigation to determine whether the Enterococcaceae enterotype could serve as a biomarker or potential therapeutic target for respiratory-related diseases.
Such differences may arise due to factors such as disease-related inflammation, treatment regimens, and altered immune responses. Furthermore, patients with respiratory disease are also more likely to suffer from comorbidities than individuals without respiratory disease. Compared with individuals without respiratory diseases, patients with respiratory diseases exhibit a higher prevalence of conditions such as hypertension, diabetes, gastroesophageal reflux disease, congestive heart failure, chronic kidney disease, psychiatric disorders, and osteoporosis [32,33,34,35]. Additionally, they tend to use more glucocorticoid-related drugs. Many of these diseases have the potential to influence the gut microbiome.
Our study's integration of clinical factors with microbial community data highlights the complex interplay between the gut microbiota and host health. Age, emerged as a significant factor influencing microbial diversity, consistent with previous reports [36]. The relationship between gut microbiota and medical costs, duration of hospital stay and other clinical factors underscores the potential of microbiota-based approaches for prognostication or treatment stratification in critical care settings. The random forest analysis's identification of taxonomic markers with discriminatory potential further supports the notion of gut microbiota as a non-invasive indicator of disease states. The identification of specific genera, such as Blautia, Subdoligranulum, Enterococcus, and Klebsiella, opens avenues for further research into their functional roles and potential links to respiratory diseases' severity or prognosis. Prospective studies are needed to validate these markers and their clinical significance.
Study limitations and future directions. Several limitations must be acknowledged in this study. The cross-sectional design precludes establishing causality, and longitudinal studies are needed to elucidate the temporal dynamics of the gut-lung axis in PCCM. Moreover, the study's focus on a Chinese population raises questions about the generalizability of the findings to other ethnicities or geographic regions. Future research should delve into the mechanistic underpinnings of the observed gut microbiota alterations in PCCM inpatients. Animal models and in vitro experiments could provide insights into the functional implications of specific taxa and their interactions with the immune system.
Conclusions
In conclusion, our study provides valuable insights into the gut microbiota composition of inpatients from PCCM. The observed dysbiosis and distinct microbial profiles highlight the potential involvement of gut microbiota in the pathogenesis of respiratory diseases.
Availability of data and materials
The raw sequencing read data for all the samples have been deposited in the Genome Sequence Archive of the BIG Data Center (https://bigd.big.ac.cn; Beijing Institute of Genomics (BIG), Chinese Academy of Sciences) under the accession number CRA012416. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AUC:
-
Area under the curve
- BMI:
-
Body mass index
- COPD:
-
Chronic obstructive pulmonary disease
- EID:
-
Enterococcus intestinal domination
- NMDS:
-
Non-metric multidimensional scaling
- NSCLC:
-
Non-small cell lung cancer
- OTU:
-
Operational taxonomic unit
- PCCM:
-
Pulmonary and critical care medicine
- PCoA:
-
Principal coordinates analysis
References
Nie P, Li Z, Wang Y, Zhang Y, Zhao M, Luo J, Du S, Deng Z, Chen J, Wang Y, Chen S, Wang L. Gut microbiome interventions in human health and diseases. Med Res Rev. 2019;39(6):2286–313.
Gallacher D, Mitchell E, Alber D, Wach R, Klein N, Marchesi JR, Kotecha S. Dissimilarity of the gut-lung axis and dysbiosis of the lower airways in ventilated preterm infants. Eur Respir J. 2020;55(5):1901909.
Saint-Criq V, Lugo-Villarino G, Thomas M. Dysbiosis, malnutrition and enhanced gut-lung axis contribute to age-related respiratory diseases. Age Res Rev. 2021;66: 101235.
Narayana JK, Aliberti S, Mac Aogáin M, Jaggi TK, Ali NABM, Ivan FX, Cheng HS, Yip YS, Vos MIG, Low ZS, Lee JXT, Amati F, Gramegna A, Wong SH, Sung JJY, Tan NS, Tsaneva-Atanasova K, Blasi F, Chotirmall SH. Microbial dysregulation of the gut-lung axis in bronchiectasis. Am J Respir Crit Care Med. 2023;207(7):908–20.
Wang L, Cai Y, Garssen J, Henricks PAJ, Folkerts G, Braber S. The bidirectional gut-lung axis in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2023;207(9):1145–60.
Wang Z, Liu J, Li F, Luo Y, Ge P, Zhang Y, Wen H, Yang Q, Ma S, Chen H. The gut-lung axis in severe acute pancreatitis-associated lung injury: the protection by the gut microbiota through short-chain fatty acids. Pharmacol Res. 2022;182: 106321.
Kim YJ, Womble JT, Gunsch CK, Ingram JL. The gut/lung microbiome axis in obesity, asthma, and bariatric surgery: a literature review. Obesity. 2021;29(4):636–44.
Dohnalová L, Lundgren P, Carty JRE, Goldstein N, Wenski SL, Nanudorn P, Thiengmag S, Huang KP, Litichevskiy L, Descamps HC, Chellappa K, Glassman A, Kessler S, Kim J, Cox TO, Dmitrieva-Posocco O, Wong AC, Allman EL, Ghosh S, Sharma N, Sengupta K, Cornes B, Dean N, Churchill GA, Khurana TS, Sellmyer MA, FitzGerald GA, Patterson AD, Baur JA, Alhadeff AL, Helfrich EJN, Levy M, Betley JN, Thaiss CA. A microbiome-dependent gut-brain pathway regulates motivation for exercise. Nature. 2022;612(7941):739–47.
Martínez-López YE, Esquivel-Hernández DA, Sánchez-Castañeda JP, Neri-Rosario D, Guardado-Mendoza R, Resendis-Antonio O. Type 2 diabetes, gut microbiome, and systems biology: a novel perspective for a new era. Gut Microbes. 2022;14(1):2111952.
Wang YH, Yan ZZ, Luo SD, Hu JJ, Wu M, Zhao J, Liu WF, Li C, Liu KX. Gut microbiota-derived succinate aggravates acute lung injury after intestinal ischaemia/reperfusion in mice. Eur Respir J. 2023;61(2):2200840.
Garcia ER, Vergara A, Aziz F, Narváez S, Cuesta G, Hernández M, Toapanta D, Marco F, Fernández J, Soriano A, Vila J, Casals-Pascual C. Changes in the gut microbiota and risk of colonization by multidrug-resistant bacteria, infection, and death in critical care patients. Clin Microbiol Infect. 2022;28(7):975–82.
Dang AT, Marsland BJ. Microbes, metabolites, and the gut-lung axis. Mucosal Immunol. 2019;12(4):843–50.
Pérez-Cobas AE, Baquero F, de Pablo R, Soriano MC, Coque TM. Altered ecology of the respiratory tract microbiome and nosocomial pneumonia. Front Microbiol. 2022;12: 709421.
Zhou Y, Zhong NS, Li X, Chen S, Zheng J, Zhao D, Yao W, Zhi R, Wei L, He B, Zhang X, Yang C, Li Y, Li F, Du J, Gui J, Hu B, Bai C, Huang P, Chen G, Xu Y, Wang C, Liang B, Li Y, Hu G, Tan H, Ye X, Ma X, Chen Y, Hu X, Tian J, Zhu X, Shi Z, Du X, Li M, Liu S, Yu R, Zhao J, Ma Q, Xie C, Li X, Chen T, Lin Y, Zeng L, Ye C, Ye W, Luo X, Zeng L, Yu S, Guan WJ, Ran P. Tiotropium in early-stage chronic obstructive pulmonary disease. N Engl J Med. 2017;377(10):923–35.
Li N, Dai Z, Wang Z, Deng Z, Zhang J, Pu J, Cao W, Pan T, Zhou Y, Yang Z, Li J, Li B, Ran P. Gut microbiota dysbiosis contributes to the development of chronic obstructive pulmonary disease. Respir Res. 2021;22(1):274.
Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glöckner FO. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucl Acid Res. 2013;41(1): e1.
16S Metagenomic Sequencing Library Preparation. San Diego, CA: Illumina. Updated 2014. https://www.illumina.com/. 15 July 2016
Ren Y, Yu G, Shi C, Liu L, Guo Q, Han C, Huang H. Majorbio cloud: a one-stop, comprehensive bioinformatic platform for multiomics analyses. iMeta. 2022;1:e12.
Costea PI, Hildebrand F, Arumugam M, Bäckhed F, Blaser MJ, Bushman FD, de Vos WM, Ehrlich SD, Fraser CM, Hattori M, Huttenhower C, Jeffery IB, Knights D, Lewis JD, Ley RE, Ochman H, O’Toole PW, Quince C, Relman DA, Shanahan F, Sunagawa S, Wang J, Weinstock GM, Wu GD, Zeller G, Zhao L, Raes J, Knight R, Bork P. Enterotypes in the landscape of gut microbial community composition. Nat Microbiol. 2018;3(1):8–16.
Barcik W, Boutin RCT, Sokolowska M, Finlay BB. The role of lung and gut microbiota in the pathology of asthma. Immunity. 2020;52(2):241–55.
Price CE, O’Toole GA. The gut-lung axis in cystic fibrosis. J Bacteriol. 2021;203(20): e0031121.
Mazumder MHH, Gandhi J, Majumder N, Wang L, Cumming RI, Stradtman S, Velayutham M, Hathaway QA, Shannahan J, Hu G, Nurkiewicz TR, Tighe RM, Kelley EE, Hussain S. Lung-gut axis of microbiome alterations following co-exposure to ultrafine carbon black and ozone. Part Fibre Toxicol. 2023;20(1):15.
Li N, Yi X, Chen C, Dai Z, Deng Z, Pu J, Zhou Y, Li B, Wang Z, Ran P. The gut microbiome as a potential source of non-invasive biomarkers of chronic obstructive pulmonary disease. Front Microbiol. 2023;14:1173614.
Yang Z, Chen Z, Lin X, Yao S, Xian M, Ning X, Fu W, Jiang M, Li N, Xiao X, Feng M, Lian Z, Yang W, Ren X, Zheng Z, Zhao J, Wei N, Lu W, Roponen M, Schaub B, Wong GWK, Su Z, Wang C, Li J. Rural environment reduces allergic inflammation by modulating the gut microbiota. Gut Microbes. 2022;14(1):2125733.
Lee-Sarwar K, Dedrick S, Momeni B, Kelly RS, Zeiger RS, O’Connor GT, Sandel MT, Bacharier LB, Beigelman A, Laranjo N, Gold DR, Lasky-Su J, Litonjua AA, Liu YY, Weiss ST. Association of the gut microbiome and metabolome with wheeze frequency in childhood asthma. J Allergy Clin Immunol. 2022;150(2):325–36.
Saifon W, Sensorn I, Trachu N, Oranratnachai S, Charoenyingwattana A, Runcharoen C, Monnamo N, Sukkasem W, Inchareon P, Suwatanapongched T, Chansriwong P, Ativitavas T, Panvichian R, Chantratita W, Reungwetwattana T. Gastrointestinal microbiota profile and clinical correlations in advanced EGFR-WT and EGFR-mutant non-small cell lung cancer. BMC Cancer. 2022;22(1):963.
Xia GH, Zhang MS, Wu QH, Wang HD, Zhou HW, He Y, Yin J. Dysbiosis of gut microbiota is an independent risk factor of stroke-associated pneumonia: a chinese pilot study. Front Cell Infect Microbiol. 2021;11: 715475.
Brinkwirth S, Ayobami O, Eckmanns T, Markwart R. Hospital-acquired infections caused by enterococci: a systematic review and meta-analysis, WHO European region, 1 January 2010 to 4 february 2020. Euro Surveill. 2021;26(45):2001628.
Iida N, Mizukoshi E, Yamashita T, Yutani M, Seishima J, Wang Z, Arai K, Okada H, Yamashita T, Sakai Y, Masuo Y, Agustina R, Kato Y, Fujinaga Y, Oshima M, Honda M, Lebreton F, Gilmore MS, Kaneko S. Chronic liver disease enables gut Enterococcus faecalis colonization to promote liver carcinogenesis. Nat Cancer. 2021;2(10):1039–54.
Zigmond E, Zecher BF, Bartels AL, Ziv-Baran T, Rösch T, Schachschal G, Lohse AW, Ehlken H, Schramm C. Bile duct colonization with Enterococcus sp. associates with disease progression in primary sclerosing cholangitis. Clin Gastroenterol Hepatol. 2023;21(5):1223-1232e3.
Messina JA, Tan CY, Ren Y, Hill L, Bush A, Lew M, Andermann T, Peled JU, Gomes A, van den Brink MRM, Chao NJ, Surana NK, Sung AD. Enterococcus intestinal domination is associated with increased mortality in the acute leukemia chemotherapy population. Clin Infect Dis. 2021. https://doi.org/10.1093/cid/ciab1043.
Chen W, FitzGerald JM, Sin DD, Sadatsafavi M. Canadian respiratory research network. Excess economic burden of comorbidities in COPD: a 15 year population-based study. Eur Respir J. 2017;50(1):1700393.
Metwally EM, Rivera MP, Durham DD, Lane L, Perera P, Lamb D, Henderson LM. Lung cancer screening in individuals with and without lung-related comorbidities. JAMA Netw Open. 2022;5(9): e2230146.
Cardet JC, Bulkhi AA, Lockey RF. Nonrespiratory comorbidities in asthma. J Allerg Clin Immunol Pract. 2021;9(11):3887–97.
Greulich T, Weist BJD, Koczulla AR, Janciauskiene S, Klemmer A, Lux W, Alter P, Vogelmeier CF. Prevalence of comorbidities in COPD patients by disease severity in a German population. Respir Med. 2017;132:132–8.
Miyaho K, Sanada K, Kurokawa S, Tanaka A, Tachibana T, Ishii C, Noda Y, Nakajima S, Fukuda S, Mimura M, Kishimoto T, Iwanami A. The potential impact of age on gut microbiota in patients with major depressive disorder: a secondary analysis of the prospective observational study. J Pers Med. 2022;12(11):1827.
Funding
Funding for this work was provided by the Foundation of the National Key Laboratory of Respiratory Diseases (grant number SKLRD-Z-202308), the Guangdong University Featured Innovation Program Project 2024 (grant number naijianli) and the General Financial Support Project of Hunan Provincial Health Commission (grant number 20231107).
Author information
Authors and Affiliations
Contributions
NJL, SL, ZW and MZH were responsible for the study concept, project direction, and experiment design. NJL, GYT, ZLX, ZWY and WXC acquired the samples and clinical information, analyzed the results, and drafted the manuscript. NJL and ZLX conducted the microbial sequencing analysis. All authors reviewed and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Medical Ethics Committee of Guangzhou Medical University (Ethics No. 2021-YJS-ks-14). Prior to enrollment, all subjects were informed about the study's content, and they provided their consent to participate by signing the informed consent form.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, N., Tan, G., Xie, Z. et al. Distinct enterotypes and dysbiosis: unraveling gut microbiota in pulmonary and critical care medicine inpatients. Respir Res 25, 304 (2024). https://doi.org/10.1186/s12931-024-02943-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-024-02943-7