
Abstract
Background
Although EGFR-TKI resistance mechanisms in non-small cell lung cancer (NSCLC) have been extensively studied, certain patient subgroups remain with unclear mechanisms. This retrospective study analysed mutation data of NSCLC patients with EGFR-sensitive mutations and high programmed death-ligand 1 (PD-L1) expression or high TMB to identify primary resistance mechanisms.
Methods
Hybrid capture-based next-generation sequencing (NGS) was used to analyse mutations in 639 genes in tumor tissues and blood samples from 339 NSCLC patients. PD-L1 immunohistochemical staining was also performed on the same cell blocks. Molecular and pathway profiles were compared among patient subgroups.
Results
TMB was significantly higher in lung cancer patients with EGFR-sensitive mutations and high PD-L1 expression. Compared with the high-expression PD-L1 or high TMB and low-expression or TMB groups, the top 10 genes exhibited differences in both gene types and mutation rates. Pathway analysis revealed a significant mutations of the PI3K signaling pathway in the EGFR-sensitive mutation group with high PD-L1 expression (38% versus 12%, p < 0.001) and high TMB group (31% versus 13%, p < 0.05). Notably, PIK3CA and PTEN mutations emerged as the most important differentially mutated genes within the PI3K signaling pathway.
Conclusions
Our findings reveal that the presence of PI3K signaling pathway mutations may be responsible for inducing primary resistance to EGFR-TKIs in NSCLC patients with EGFR-sensitive mutations along with high PD-L1 expression or high TMB. This finding is of great significance in guiding subsequent precision treatments in NSCLC.
Similar content being viewed by others

Background
Approximately 20% of individuals diagnosed with non-small cell lung cancer (NSCLC) harbor a distinct genetic mutation in the epidermal growth factor receptor (EGFR) gene, referred to as an activating somatic mutation [1]. The most frequently observed mutations within this category include the exon 19 deletion (E19del) and the exon 21 substitution at position 858 (L858R), in which the amino acid arginine is substituted with leucine [2, 3]. Small molecule tyrosine kinase inhibitors (TKIs) targeting EGFR, including first/second-generation TKIs (gefitinib, erlotinib, and afatanib) and third-generation TKIs (osimertinib), have been extensively employed in the treatment of NSCLC patients with EGFR-sensitive mutations and have exhibited favorable outcomes [4,5,6,7]. However, there was significant variability in response duration and survival among these patients.
Previous studies have revealed a spectrum of resistance mechanisms to EGFR inhibitors, including EGFR-dependent resistance, such as the C797X mutation, which confers resistance to osimertinib [8], and non-EGFR-dependent resistance caused by the activation of bypass or downstream signaling pathways [9, 10], as well as histological or phenotypic transformation [11]. Additionally, the resistance mechanisms remain unidentified in some patients. Studies have indicated that NSCLC patients with EGFR mutations (E19del/L858R) accompanied by a higher tumor mutational burden (TMB) tend to have a less favorable prognosis when treated with EGFR-TKIs than those with low TMB [12]. Furthermore, another study has suggested that in NSCLC patients with EGFR mutations (E19del/L858R) and high programmed death-ligand 1 (PD-L1) expression, the prognosis with EGFR-TKI treatment is less favorable than that with low PD-L1 expression [13]. Nevertheless, the underlying mechanisms responsible for the poorer prognosis in these subgroups of EGFR mutations have not yet been thoroughly investigated.
To explore the potential mechanisms underlying these subgroups, in this study, we conducted a retrospective analysis of the genetic mutation data of patients in these NSCLC subgroups and analysed the differences in pathway mutations. Our research aimed to offer new treatment opportunities for patients with these specific types of NSCLC.
Materials and methods
Patients and sample characteristics
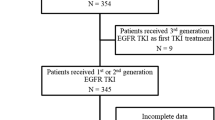
From November 2022 to August 2023, a total of 339 patients with pathologically diagnosed NSCLC who had not received EGFR-TKI treatment were enrolled in the present study at the Affiliated Cancer Hospital of Nanjing Medical University and Deyang Pelple’s Hospital. Each patient underwent a pathological diagnosis and was needed to provide both tumor tissue and paired blood samples. Cancer diagnosis was initially established through clinical and X-ray findings and later confirmed via histological analysis of tumor biopsies. Exclusion criteria for the study included cases where NSCLC was not pathologically confirmed, cases where tissue or blood samples were not provided, and cases where the cell blocks of the samples contained tumor cells in quantities less than 20%. Clinical data, including information on age and gender, were retrieved from the medical records. Written informed consent was obtained from all participants, and this study was approved by the institutional review board of our hospital.
DNA extraction and library construction
According to the manufacturer’s protocols, tumor DNA and blood genomic DNA were extracted using a human tissue DNA extraction kit (Shanghai YunYing) and a human blood genomic DNA extraction kit (Shanghai YunYing), respectively. DNA was eluted in an elution buffer, and its concentration and purity were evaluated using a NanoDrop spectrophotometer. DNA was stored at -20 °C until use. Library preparation was performed using the VAHTS Universal DNA Library Prep Kit for Illumina. Target enrichment was performed using Shanghai YunYing’s optimized probes, which target the exons and some introns of 639 cancer-related genes. Sequencing was performed on an Illumina NextSeq500 platform using the manufacturer’s protocols.
Next-generation sequencing (NGS)-based assay and bioinformatics analysis
FastQC software (version 0.11.2) and customized Python script were used to screen FASTQ files, with the adaptor sequences and sequences with Q below 30 removed. Clean reads were mapped to the reference human genome GRCh37/hg19 using BWA (Burrows Wheeler Aligner version 0.7.7). BAM files were then realigned and recalled using GATK3.5, which was also used to detect mutations. Duplicate sequences were removed using Picard MarkDuplicates (version 1.35) to reduce any potential polymerase chain reaction bias. VarScan (version 2.3.2) was used to select single nucleotide variations (SNVs) satisfying the following criteria: depth ≥ 100, reads ≥ 10, and allele frequency ≥ 5% (if hotspot, ≥ 1%). Pindel (version 0.2.5b8) was used for insertion or deletion (indel) detection using default parameters, with at least 5 unique reads.
Compared with matched normal samples, somatic SNVs and InDels of tumors were named and functionally annotated using MuTect v. 1.1.4 and Varscan2 v. 2.3.9 software. Mutations with a variant allele frequency of ≥ 5% were defined as high-confidence mutations (≥ 1% for hotspots). Tumor mutational burden (TMB) was calculated using the number of all somatic, coding, base substitution, and indel mutations per megabase including synonymous mutations. The total number of mutations counted was divided by the size of the coding region of the targeted territory (1.36 Mb of the coding genome) to calculate the TMB per megabase. Microsatellite instability (MSI) scores of all samples were calculated using MSIsensor [14] with default parameters, a software tool for quantifying MSI in genome sequencing data using tumor-only or paired tumor-normal samples. We used 29 microsatellite sites as input files for MSI detection of tumor-only patterns. The MSI score was defined as the percentage of unstable microsatellites among all microsatellites used. Each microsatellite site had at least 20 spanning reads and single-nucleotide mutations.
PD-L1 expression test
The PD-L1 expression level for each patient was determined using the Dako 22C3 pharmDx system (Agilent Technologies Inc., Santa Clara, CA, USA) assay, and the results are presented as a tumor proportion score (TPS) [15].
Statistical analysis
The prevalence and distribution of genomic alterations were visualized using the R package “maftools” [16]. The R package “ggplot2” was used to draw the boxplots. The nonparametric Wilcox test was subsequently used to test for the significance of the difference in means between the two populations.
Results
Patient characteristics
A total of 339 patients, consisting of 295 with lung adenocarcinoma (LUAD) and 44 with lung squamous cell carcinoma (LUSC), who were newly diagnosed with NSCLC cancer, were included in the present study. The mean age at diagnosis of the study participants was 61.9 years (range, 25–86 years; median, 64 years), and a significant difference was observed between PD-L1 expression level groups (Table 1). Additional characteristics of the patient cohort are summarized in Table 1, and more detailed information can be found in Table S1. All participants successfully completed the targeted sequencing, which included all exons and partial introns of the 639 genes listed in Table S2.
TMB exhibits significant differences among various clinical indicators and molecular features
As shown in Fig. 1A, LUAD exhibited significantly lower TMB values than LUSC (average: 3.4 muts/Mb versus 6.8 muts/Mb, p < 0.0001). Patients aged > 60 years had higher TMB values than those aged < 60 years old (average: 4.5 muts/Mb versus 2.8 muts/Mb, p < 0.0001) (Fig. 1B). In terms of gender differences, males display significantly higher TMB values than females (average: 4.7 muts/Mb versus 3.0 muts/Mb, p < 0.0001) (Fig. 1C). Regarding molecular features, no significant differences in TMB values were observed between EGFR E19del and L858R mutations. Similarly, no notable distinctions were found between the other EGFR mutations and the wild-type (as shown in Fig. 1D). However, when looking at specific EGFR mutations, TMB values were notably lower for EGFR E19del (average: 2.5 muts/Mb versus 3.8 muts/Mb, p < 0.01; 2.5 muts/Mb versus 5.3 muts/Mb, p < 0.0001) or L858R (average: 2.6 muts/Mb versus 3.8 muts/Mb, p < 0.05; 2.6 muts/Mb versus 5.3 muts/Mb, p < 0.0001) in comparison to other EGFR mutations or wild-type. Likewise, no significant differences were observed in TMB between the PD-L1 negative and PD-L1 moderate expression groups. However, the high PD-L1 expression group exhibited significantly higher TMB values when compared to the PD-L1 negative or moderate expression groups (average: 4.9 muts/Mb versus 3.4 muts/Mb, p < 0.001; 4.9 muts/Mb versus 3.6 muts/Mb, p < 0.05) (Fig. 1E).

The relationship between TMB and clinical indicators. (A) Lung cancer pathological type; (B) Age at diagnosis in years; (C) Gender; (D) EGFR mutation status; (E) PD-L1 expression level; and (F) EGFR mutation status and PD-L1 expression level. “ns”, “*”, “**”, “***” and “****” indicate P > 0.05, P < 0.05, P < 0.01, P < 0.001 and P < 0.0001, Wilcoxon test. LUAD for adenocarcinoma; LUSC for squamous cell carcinoma
According to the results of Fig. 1D and E, we divided the patients into PD-L1 high or nonhigh-expression groups (TPS ≥ 50% or TPS < 50%), EGFR-sensitive or non-EGFR-sensitive groups (with or without EGFR E19del/L858 mutation), and TMB high (TMB-H) or low (TMB-L) groups (TMB ≥ 4.4 muts/Mb or TMB < 4.4 muts/Mb, greater than or less than the third quartile). High PD-L1 expression was associated with a significantly higher TMB than nonhigh PD-L1 expression (average: 3.2 muts/Mb versus 2.4 muts/Mb, p < 0.05) in patients harboring EGFR-sensitive mutations (Fig. 1F). According to the data analysis, MSI did not affect the TMB value (Fig. S1). More information is displayed in Table S3.
Mutation overview and analysis with PD-L1 status
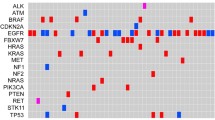
Among the observed mutation types, missense mutations were the most common, followed by frameshift deletions and nonsense mutations (see Fig. S2A). In terms of variant types, single nucleotide polymorphisms (SNPs) constituted a larger portion than insertions or deletions (as illustrated in Fig. S2B). Notably, the C > T transition was the dominant single nucleotide variant (SNV) observed in NSCLCs (Fig. S2C). The number of altered bases in each sample and a summary of the variant classifications were counted, as depicted in Fig. S2D and Fig. S2E, respectively. In NSCLCs, the top 10 mutated genes were EGFR (57%), TP53 (46%), LRP1B (10%), PIK3CA (8%), KRAS (8%), FAT1 (6%), KEAP1 (6%), ATM (5%), CDKN2A (5%), and NF1 (4%) (Fig. S2F) which might play an important role in the biological processes of NSCLC. According to the waterfall plot of the top 20 mutated genes, where the mutation type is denoted by various colors with annotations, nonsense and missense mutations were mostly observed (Fig. S2G).
Figure 2 A presents the PD-L1 staining results of four representative NSCLC patients, corresponding to high and low expression levels. To understand the mutation difference in distinct PD-L1 expression groups, we plotted the mutation profiles of the top 10 mutated genes. As depicted in Fig. 2B, the top 10 genes in the PD-L1 high-expression group were TP53, EGFR, LRP1B, PK3CA, KRAS, ALK FAT1, BRAF, CDKN2A, and KMT2D. Conversely, in the nonhigh-expression group, the top 10 genes were EGFR, TP53, LRP1B, KRAS, PK3CA, ATM, KEAP1, ERBB2, FAT1, and NF1 (Fig. 2C). These profiles also revealed differences in the gene mutation rates between the two groups. In the high PD-L1 expression group, several significant associations and mutual exclusions among the gene mutations were observed. Specifically, TP53 mutations were significantly associated with CDKN2A mutations, and KRAS mutations were significantly associated with FAT1 mutations. Conversely, BRAF and ALK mutations were mutually exclusive with EGFR mutations and mutually exclusive with BRAF and KRAS mutations (Fig. 2D). In the nonhigh PD-L1 expression group, comutated genes included FAT1, LRP1B, and PIK3CA, as well as NF1 and KEAP1, and ATM and PIK3CA. EGFR mutations were significantly mutually exclusive with ERBB2, KRAS, KEAP1, and LRP1B mutations (see Fig. 2E). These associations and exclusions shed light on the complex genetic relationships among different PD-L1 expression groups.

Mutation analysis in the high and nonhigh PD-L1 expression groups. (A) Immunohistochemical image of lung cancer patients with high expression of PD-L1 (TPS ≥ 50%) and nonhigh expression (TPS < 50%); (B) Overview of mutation profiles in patients with high expression of PD-L1; (C) Overview of mutation profiles in patients with nonhigh expression of PD-L1; (D) Comutation analysis in patients with high expression of PD-L1; (E) Comutation analysis in patients with nonhigh expression of PD-L1; (F) Forest plot of comparing mutations between patients with high expression and nonhigh expression of PD-L1; (G) Co-bar plot of differentially mutated genes in patients with high expression and nonhigh expression of PD-L1. “*”, “**”, and “***” indicate P < 0.05, P < 0.01, and P < 0.001
Through a comparative analysis of mutations between the high and nonhigh PD-L1 expression groups, we identified significant differences in the mutation frequency of several genes: BRAF (8% versus 0%), ALK (9% versus 1%), EGFR (42% versus 58%), PIK3CA (15% versus 6%), DICER1 (7% versus 2%) and EPHA5 (7% versus 2%) (Fig. 2F and G) (P < 0.05).
Mutation analysis with TMB status
Similarly, we generated mutation profiles for the top 10 mutated genes based on TMB status. In the TMB-H group, the top 10 genes were TP53, EGFR, LRP1B, FAT1, KEAP1, KRAS, CDK2NA, PIK3CA, ATM, and BRCA2, whereas in the low TMB group, they were EGFR, TP53, PK3CA, KRAS, ERBB2, CTNNB1, LRP1B, APC, ATM and RB1, and there were also differences in gene mutation rates (Fig. 3A and B). In the TMB-H group, TP53 mutation and KEAP1 were significantly associated with CDKN2A mutation and BRCA2 mutations, respectively (Fig. 3C). EGFR mutations were significantly mutually exclusive to LRP1B. In the TMB-L group, EGFR mutations were significantly mutually exclusive to ERBB2 and KRAS (Fig. 3D). Comparative mutation analysis of TMB-H and TMB-L groups showed that multiple genes are related to TMB: SPIA1 (7% versus 1%), ARID1B (6% versus 0%), SLIT2 (6% versus 0%), PRKDC (9% versus 0%), KMT2D (9% versus 0%), CDKN2A (12% versus 1%), EPHA5 (8% versus 0%), EPHA3 (8% versus 0%), KEAP1 (14% versus 1%), FAT1 (14% versus 1%), LRPIB (24% versus 3%), ATR (6% versus 0%), BRCA2 (10% versus 0%), ATM (10% versus 3%), TP53 (66% versus 33%), PDGFRA (7% versus 0%), NF1 (10% versus 1%), FLT3 (5% versus 0%), BRAF (6% versus 0%), KRAS (14% versus 5%), and EGFR (33% versus 55%)(Fig. 3E and F) (P < 0.01).

Mutation analysis in groups with high or low TMB values. (A) Overview of mutation profiles in patients with high tumor mutational burden (TMB-H, TMB ≥ 4.4 muts/Mb); (B) Overview of mutation profiles in patients with low tumor mutational burden (TMB-L, TMB < 4.4 muts/Mb); (C) Comutation analysis in patients with high tumor mutational burden; (D) Comutation analysis in patients with low tumor mutational burden; (E) Phylogenetic tree comparing mutations between patients with high and low tumor mutational burden; (F) Cobar plot of differentially mutated genes in patients with high and low tumor mutational burden. “**” and “***” indicate P < 0.01 and P < 0.001
Tumor signaling pathway analysis
To further investigate the potential impact of high PD-L1 expression or high TMB on the tumor signaling pathways in NSCLC patients with EGFR mutations, we conducted a tumor signaling pathway mutation analysis (pathways and related genes referenced in Table S2 in the previous study [17]). As shown in Table S4, among the EGFR mutated group with PD-L1 expression, there were differences in the mutation rates of the following pathways: chromatin, histone modifiers, genome integrity, histone modification, mitogen-activated protein kinase (MAPK) signaling, other signaling, PI3K (phosphoInositide 3-kinase) signaling, RNA abundance, receptor tyrosine kinase (RTK) signaling, splicing and target of rapamycin (TOR) signaling. Similarly, among the EGFR mutated group with TMB (Table S5), there were differences in the mutation rates of the following pathways: cell cycle, chromatin histone modifiers, chromatin SWI/SNF (SWItch/Sucrose Non-Fermentable) complex, genome integrity, MAPK signaling, other, other signaling, PI3K signaling, RTK signaling, transforming growth factor beta (TGFB) signaling, and transcription factor.
These pathways were selected for further differential analysis. In the context of EGFR-sensitive mutations, accompanied by high PD-L1 expression compared to low expression, we identified a substantial difference in the mutation rates of the PI3K signaling pathway (38% versus 12%, p < 0.001) pathways (Fig. 4). In EGFR-sensitive mutations accompanied by TMB-H compared to TMB-L, there were significant differences in the mutation rates of the cell cycle (14% versus 3%, p < 0.05), chromatin SWI/SNF (14% versus 4%, p < 0.05), Genome. integrity (69% versus 36%, p < 0.01), Other (10% versus 2%, p < 0.01), Other signaling (17% versus 2%, p < 0.001), PI3K signaling (31% versus 13%, p < 0.05), and Transcription factor (17% versus 6%, p < 0.05) pathways (Fig. 5). The analysis of mutation differences in other selected pathways between groups is displayed in Figures S3 and S4, and no significant difference was observed in the group of EGFR-sensitive mutations with high versus low PD-L1 expression or high versus low TMB values. Specific mutations of the key genes PIK3CA and PTEN in the PI3K pathway are shown in the lollipop plot (Figure S5). PIK3CA primarily has activating mutations such as H1407, E545, or E542 (Figures S5A and S5C), while PTEN mainly has loss-of-function mutations (Figures S5B and S5D).

Pathway mutation differential and profile analysis in groups with high or nonhigh PD-L1 expression. Differential analysis of the PI3K signaling pathway (A); The mutation profile of the PI3K signaling pathway in the group with EGFR E19del/L858R mutation and high ( TPS ≥ 50%) or nonhigh (TPS < 50%) PD-L1 expression levels (B). “*” and “***” indicate P < 0.05 and P < 0.001, respectively. Wilcox test

Pathway mutation differential and profile analysis in the high or low TMB value group. Differential analysis of signaling pathways: cell cycle (A), chromatin SWI/SNF complex (B), genome integrity (C), other (D), other signaling (E), PI3K signaling (F), and transcription factor (G); the mutation profile of signaling pathways in the group with EGFR E19del/L858R and high (H, TMB ≥ 4.4 muts/Mb) or low (I, TMB < 4.4 muts/Mb) TMB values. “ns”, “*”, “**”, “***” and “****” indicate P > 0.05, P < 0.05, P < 0.01, P < 0.001 and P < 0.0001, Wilcoxon test
Discussion
EGFR-TKI is a crucial therapy for NSCLC patients with EGFR mutations, but there are significant variations in their prognosis 4–7. Prior research has highlighted factors such as primary and acquired [18,19,20], tumor histology, and phenotype transformation [11] as potential reasons for these differences. Notably, recent studies have indicated that high TMB or high PD-L1 expression plays a significant role as a primary resistance mechanism to EGFR-TKIs in EGFR-mutated NSCLC. However, the specific resistance mechanisms of these distinct EGFR mutations remain poorly understood. In our study, we further categorized patients with NSCLC based on their PD-L1 expression levels and TMB values, in addition to their EGFR mutation status. We conducted a detailed analysis of mutation characteristics within these subgroups.
To the best of our knowledge, our study represents the first evidence suggesting that NSCLC patients with EGFR-sensitive mutations, coupled with high PD-L1 expression, exhibit elevated TMB. While PD-L1 and TMB are typically regarded as two distinct immune markers, our findings suggest a potential correlation between PD-L1 and TMB in the context of EGFR-sensitive mutations. TMB is generally defined as the number of somatic mutations per megabase in the analysed genomic sequence. This implies that NSCLC patients with EGFR-sensitive mutations and high PD-L1 expression may harbor a higher burden of genetic mutations, potentially influencing tumor-related pathways. This correlation may also contribute to the relatively unfavorable prognosis observed in patients with EGFR-sensitive mutations and high TMB [12], as well as in those with EGFR-sensitive mutations accompanied by high PD-L1 expression 13. Our mutation profile analysis demonstrated notable differences in the top 10 mutated genes and their mutation rates between the high and low PD-L1 expression or TMB-H and TMB-L groups. Comparative analysis of mutations further supports these distinctions, suggesting potential variations in tumor signaling pathways between these groups. A more in-depth pathway mutation analysis revealed that EGFR-sensitive mutations, whether accompanied by high TMB or high PD-L1 expression, exhibit a higher mutation rate in the PI3K signaling pathway.
The PI3K pathway plays an important role in tumor development and progression. It is a signaling pathway involved in the regulation of multiple biological processes such as cell growth, survival, proliferation, and metabolism. Aberrant activation of the PI3K pathway is closely associated with the occurrence and progression of various types of cancer. Studies have shown that aberrant activation of the PI3K pathway can lead to increased tumor cell growth, inhibition of apoptosis, promotion of angiogenesis, and enhanced metastasis and invasion capabilities [21,22,23]. This is because the activation of the PI3K pathway can promote cell cycle progression, enhance signaling for cell proliferation and growth, inhibit programmed cell death, and facilitate tumor cell invasion and metastasis by regulating the cell cytoskeleton and matrix metalloproteinase expression. Our results confirm that the PI3K pathway is often activated through PIK3CA mutations/amplifications and PTEN loss, which aligns with the established knowledge. Contrary to the mutual exclusivity observed in most oncogenic driver gene mutations, PIK3CA mutations often cooccur with other oncogenic driver gene mutations in NSCLC. In the AURA3 study [24], the incidence of PIK3CA amplification/mutation in patients with acquired resistance to second-line treatment with osimertinib was 5%, with two patients having concurrent PIK3CA amplification and HER2 amplification. Among patients who developed resistance to first-line treatment with osimertinib [9], 7% were found to have PIK3CA mutations, with the most common being the E545K mutation (4%), followed by E453K and H1047R. These studies suggest that activation of the PI3K pathway may be the reason for the poor response to EGFR-TKIs in patients with EGFR-sensitive mutations accompanied by high TMB or high PD-L1 expression.
It should be noted that our study has some limitations. First, the number of patients with EGFR-sensitive mutations accompanied by high PD-L1 expression or high TMB in our study was relatively small, which may introduce bias, and further expansion of the sample size is needed. Second, we only analysed pathway-related gene mutations, and further validation is needed to determine whether the mutations truly affect pathway expression. Most importantly, we lacked relevant follow-up data and EGFR-TKI medication data, and further validation of our conclusions is needed by considering the patient’s prognosis.
In summary, our research found that NSCLC patients with EGFR-sensitive mutations accompanied by high expression of PD-L1 or high TMB values may have a higher frequency of abnormal activation in the PI3K pathway. This, in turn, may lead to a poorer response to EGFR-TKI treatment compared with other types of patients. Our findings provide an understanding of the resistance mechanisms in these patients and offer new insights and directions for precise treatment.
Data availability
The data supporting this study’s findings are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Abbreviations
- E19del:
-
exon 19 deletion
- InDel:
-
insertion or deletion
- LUAD:
-
lung adenocarcinoma
- LUSC:
-
lung squamous cell carcinoma
- MSI:
-
Microsatellite instability
- NGS:
-
next-generation sequencing
- NSCLC:
-
non-small cell lung cancer
- PD-L1:
-
programmed death-ligand 1
- PI3K:
-
phosphoInositide 3-kinase
- SNP:
-
single nucleotide polymorphism
- SNV:
-
single nucleotide variation
- TKI:
-
tyrosine kinase inhibitor
- TMB:
-
tumor mutational burden
- TPS:
-
tumor proportion score
References
Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell Lung cancer. Nat Rev Cancer. 2010;10:760–74.
Jordan EJ, Kim HR, Arcila ME, Barron D, Chakravarty D, Gao J, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov. 2017;7:596–609.
Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, Mitsudomi T. Mutations of the epidermal growth factor receptor gene in Lung cancer: biological and clinical implications. Cancer Res. 2004;64:8919–23.
Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell Lung cancer. N Engl J Med. 2018;378:113–25.
Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell Lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–46.
Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell Lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121–8.
Sequist LV, Yang JC, Yamamoto N, O’Byrne K, Hirsh V, Mok T, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31:3327–34.
Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell Lung cancer. Br J Cancer. 2019;121:725–37.
Ramalingam SS, Cheng Y, Zhou C, Ohe Y, Imamura F, Cho BC, et al. Mechanisms of acquired resistance to first-line osimertinib: preliminary data from the phase III FLAURA study. Ann Oncol. 2018;29:viii740.
Chmielecki J, Mok T, Wu YL, Han JY, Ahn MJ, Ramalingam SS, et al. Analysis of acquired resistance mechanisms to osimertinib in patients with EGFR-mutated advanced non-small cell Lung cancer from the AURA3 trial. Nat Commun. 2023;14:1071.
Schoenfeld AJ, Chan JM, Kubota D, Sato H, Rizvi H, Daneshbod Y, et al. Tumor analyses reveal squamous transformation and off-target alterations as early resistance mechanisms to first-line osimertinib in EGFR-mutant Lung cancer. Clin Cancer Res. 2020;26:2654–63.
Offin M, Rizvi H, Tenet M, Ni A, Sanchez-Vega F, Li BT, et al. Tumor mutation burden and efficacy of EGFR-tyrosine kinase inhibitors in patients with EGFR-mutant Lung Cancers. Clin Cancer Res. 2019;25:1063–9.
Hsu KH, Huang YH, Tseng JS, Chen KC, Ku WH, Su KY, et al. High PD-L1 expression correlates with primary resistance to EGFR-TKIs in treatment naïve advanced EGFR-mutant lung adenocarcinoma patients. Lung Cancer. 2019;127:37–43.
Han X, Zhang S, Zhou DC, Wang D, He X, Yuan D et al. MSIsensor-ct: microsatellite instability detection using cfDNA sequencing data. Brief Bioinform. 2021;22.
Roach C, Zhang N, Corigliano E, Jansson M, Toland G, Ponto G, et al. Development of a companion diagnostic PD-L1 immunohistochemistry assay for pembrolizumab therapy in non-small-cell Lung cancer. Appl Immunohistochem Mol Morphol. 2016;24:392–7.
Mayakonda A, Lin DC, Assenov Y, Plass C, Koeffler HP. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018;28:1747–56.
Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, et al. Comprehensive characterization of cancer driver genes and mutations. Cell. 2018;173:371–85e18.
Chabon JJ, Simmons AD, Lovejoy AF, Esfahani MS, Newman AM, Haringsma HJ, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in Lung cancer patients. Nat Commun. 2016;7:11815.
Garraway LA, Jänne PA. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012;2:214–26.
Jackman D, Pao W, Riely GJ, Engelman JA, Kris MG, Jänne PA, et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell Lung cancer. J Clin Oncol. 2010;28:357–60.
Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–19.
Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554.
Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Reviews Drug Discov. 2014;13:140–56.
Papadimitrakopoulou VA, Wu Y-L, Han J-Y, Ahn M-J, Ramalingam SS, John T, et al. Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann Oncol. 2018;29:viii741.
Acknowledgements
We thank the patients who agreed to participate to this study. We thank the clinical teams who cared for the patients and the yunying laboratory teams who realized NGS assay and bioinformatics analysis.
Funding
This work was supported by the Key Research and Development Program of Zhejiang province [2023C03057]; Zhejiang Leading Talent Entrepreneurship Project [2021R02019]; and the Jiaxing Leading Talent Entrepreneurship Project.
Author information
Authors and Affiliations
Contributions
Wuwu Ding: Conceptualization, Validation, Investigation, Writing - review & editing; Pengmin Yang: Formal analysis, Visualization, Writing - original draft; Xiaokai Zhao: Methodology, Formal analysis, Visualization, Writing - review & editing; Xiaozhi Wang: Conceptualization, Data curation, Supervision; Huaqing Liu: Performed and analyzed experiments; Qing Su:: Performed and analyzed experiments; Xintao Wang: Data curation. Jieyi Li: Writing - review & editing, Visualization; Ziying Gong: Methodology, Formal analysis, Supervision, Funding acquisition; Daoyun Zhang: Conceptualization, Data curation, Supervision, Project administration, Funding acquisition; Xinwei Wang: Conceptualization, Data curation, Supervision, Project administration, Funding acquisition. All authors contributed to the manuscript revision, and read, and approved the submitted version.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Approval of the research protocol by an Institutional Reviewer Board: Ethical approval to report this study was obtained from the institutional review board of the Affiliated Cancer Hospital of Nanjing Medical University. All research activities were conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from patients for their anonymized information to be published in this article.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 1:
Figure S1: The relationship between TMB and MSI. Figure S2: Mutation overview of collected samples. Figure S3: Pathway mutation differential analysis in high or nonhigh PD-L1 expression group. Figure S4: Pathway mutation differential analysis in high or low TMB value group. Figure S5: Distribution of mutations in the PIK3CA and PTEN genes. Table S1: Detailed information for each patient. Table S2: The gene list of AllNGS-Panel 639. Table S3: Association between TMB status and clinical features. Table S4: Differential analysis of mutations in signaling pathways related to EGFR-sensitive mutations or high PD-L1 expression. Table S5: Differential analysis of mutations in signaling pathways related to EGFR-sensitive mutations or TMB-H
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ding, W., Yang, P., Zhao, X. et al. Unraveling EGFR-TKI resistance in lung cancer with high PD-L1 or TMB in EGFR-sensitive mutations. Respir Res 25, 40 (2024). https://doi.org/10.1186/s12931-023-02656-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-023-02656-3