
Abstract
Background
Hospital-acquired and ventilator-associated-pneumonia (HAP/VAP) are one of the most prevalent health-care associated infections in the intensive care unit (ICU). Culture-independent methods were therefore developed to provide faster route to diagnosis and treatment. Among these, metagenomic next-generation sequencing (mNGS) has shown considerable promise.
Methods
This proof-of-concept study describes the technical feasibility and evaluates the clinical validity of the mNGS for the detection and characterization of the etiologic agents causing hospital-acquired and ventilator-associated pneumonia. We performed a prospective study of all patients with HAP/VAP hospitalized in our intensive care unit for whom a bronchoalveolar lavage (BAL) was performed between July 2017 and November 2018. We compared BAL fluid culture and mNGS results of these patients.
Results
A total of 32 BAL fluids were fully analyzed. Of these, 22 (69%) were positive by culture and all pathogens identified were also reported by mNGS. Among the culture-positive BAL samples, additional bacterial species were revealed by mNGS for 12 patients, raising the issue of their pathogenic role (colonization versus coinfection). Among BALF with culture-negative test, 5 were positive in mNGS test.
Conclusions
This study revealed concordant results for pneumonia panel pathogens between mNGS and culture-positive tests and identified additional pathogens potentially implicated in pneumonia without etiologic diagnosis by culture. mNGS has emerged as a promising methodology for infectious disease diagnoses to support conventional methods. Prospective studies with real-time mNGS are warranted to examine the impact on antimicrobial decision-making and clinical outcome.
Similar content being viewed by others

Background
Hospital-acquired and ventilator-associated-pneumonia (HAP/VAP) are one of the most prevalent health-care associated infections in the intensive care unit (ICU). HAP and, most prominently, VAP are associated with poor outcome and the overall attributable mortality of HAP/VAP is around 13% [1]. Critical care physicians are expected to provide a treatment adapted to causative agents and their antibiotic susceptibility profiles as early as possible in the course of the infection [2,3,4]. These data have been confirmed by the recent COVID-19 pandemic, many patients experienced bacterial pulmonary infection during the ICU stay [5]. International guidelines for the HAP/VAP management published in 2017 [6] pointed to potential benefits of narrow-spectrum antimicrobials in reducing the risk of toxicity and later emergence of microbial multiresistance. The HAP/VAP diagnosis is established when pneumonia occurs after ≥ 48 h of hospitalization and not incubating at the time of ICU admission. The time of onset of nosocomial pneumonia affects the etiology, and late onset is strongly associated with infection by multi-drugs-resistants pathogens. In this study, early- and late-onset VAP are defined as occurring within the first 5 and > 5 days of hospitalization, respectively [6, 7].
Conventional culture of a respiratory sample enables bacterial identification, quantification and antimicrobial susceptibility testing, which remains the gold standard to diagnose and treat lung infections. However, culture-based tests are time-consuming (48–72 h) and could lack sensitivity, particularly for slow-growing bacteria or when respiratory samples are collected after administration of an antibiotic therapy. Culture-independent tests for respiratory infections were developed to provide results within a few hours. These methods are usually based on PCR panels that detect only nucleic acids from common respiratory pathogens as well as selected antibiotic resistance genes (ARG) [8,9,10,11]. Metagenomic next-generation sequencing (mNGS) is a more recent methodology that refers to the concept of sequencing all the DNA of a sample to identify microorganisms but also their genomic traits like ARG, virulence factors or typing markers. Clinical applications of mNGS are currently drawing the attention of infectious diseases specialists and guidelines for mNGS-based diagnostics have emerged [12, 13]. Before launching clinical impact studies in the context of nosocomial pneumonia, it is of importance to evaluate the performance of mNGS on respiratory samples. We performed a proof-of-concept study to describe the technical feasibility of mNGS on respiratory samples and to evaluate its clinical relevance in detecting and characterizing causative agents of HAP/VAP.
Methods
We conducted a monocentric prospective study that enrolled consecutive ICU patients of Annecy-Genevois Hospital (Metz-Tessy, France) who developed a HAP/VAP between July 2017 and November 2018. Pneumonia was diagnosed based on clinical features and compatible radiological imaging. An adjudication committee composed of independent experts reviewed all clinical, radiological and microbiological documents, to retrospectively confirm or deny the diagnosis of HAP/VAP suspected at the time of inclusion.
Each patient could be included in the study several times during ICU stay, whenever a new HAP/VAP episode was suspected. Patients aged under 18, immunosuppressed, with cystic fibrosis, under guardianship or pregnant were excluded. Patient information, including comorbidities, diagnosis at admission, current or recent antimicrobial therapy were collected from electronic charts.
The study protocol was approved by the French institutional ethics committee (CPP Sud-Méditerranée, Marseille, 2017-A00253-50, FRANCE). All patients or their relatives received an information letter and a non-objection form for participation was signed.
Whenever a HAP/VAP diagnosis was suspected, experienced physicians collected a BAL sample with a single-use flexible bronchoscope. Collected fluids were immediately sent to the local microbiology laboratory to perform cultures for diagnostic purpose. Whenever available, 1.2 mL of the BAL leftover was sent to BIOASTER institute (Lyon, France) for metagenomic sequencing within 48 h, while maintaining the cold chain (maximum 4 °C). mNGS data are then analyzed at Christophe Mérieux Center (bioMérieux-Grenoble, France).
Conventional culture
BAL fluids were routinely cultured quantitatively on liquid and solid media and were examined daily for 72 h. Bacterial colonies were identified using matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS, MALDI Biotyper®; reference library 2016 and 2017) [14]. Significance threshold was defined as ≥ 104 colony forming units (CFU) per mL, counts below this cutoff were considered negative.
Metagenomic next-generation sequencing (mNGS): sample processing and bioinformatics pipeline
The complete clinical metagenomics workflow is described in Hauser et al. [15] and shown schematically in Fig. 1.

Complete workflow for clinical metagenomic analysis of BAL samples. A is the experimental workflow in which two independent samples are analyzed in the same sequencing run. B represents the rule of interpretation applied independently to each SOI to determine whether it is involved in patient infection or presence at normal concentration or absence in the sample or the inability to interpret the result. SOI species of interest, SPC sample processing control, MT metagenomic threshold
Briefly, to control the whole mNGS process, and to calculate the absolute concentration of the detected pathogen(s), a sample processing control (SPC) made with a defined amount of Bacillus subtilis (ATCC 19659 BioBall MultiShot 10E8 bioMérieux) was added to each BAL sample, before further processing. The final concentration of added B. subtilis within each sample was 1.7 × 104 CFU/mL, close to the clinical significance threshold (104 CFU/mL).
Bacterial DNA enrichment was performed using selective lysis of human cells with saponin and DNase I treatment to degrade the DNA released. The DNase I treatment also eliminates the extracellular DNAs and the DNAs of dead bacterial cells [16], thus favoring the detection of DNA originating from viable bacterial cells. After DNase inactivation, bacteria were disrupted by bead beating. Nucleic acids were extracted on an EasyMag® platform (bioMérieux) using the generic protocol (V2.0.1) and stored at − 20 °C.
Sequencing libraries were prepared with an optimized protocol of the Nextera® XT DNA Library Preparation Kit (Illumina).
The sequence data of each sample were analyzed with the bioinformatics pipeline described by Tournoud et al. [17] to quantify bacterial species. Briefly, reads were trimmed and filtered based on quality. Taxonomic read binning was performed with Kraken [18] using an internal reference database including sequences from (a) more than 10,000 genomes from the 19 species of interest (SOI)(common VAP causing pathogens listed in Table 1), (b) B. subtilis (used as SPC), (c) bacteria typically found in the human lung and oral cavity and (d) the human genome (Homo sapiens genome assembly GRCh37). Bacterial genome sequences were from both public (e.g. PATRIC [19], RefSeq [20], FDA ARGOS [21] and private (strains sequenced from BAL samples at bioMérieux) databases. [22]. Reads associated to a SOI with an average genome coverage depth > 1 × were assembled using the IDBA-UD 1.1.1 assembler.
To avoid spurious pathogen detection due to erroneous taxonomic assignments by Kraken, the assemblies were aligned with BLAST [23] against the pneumonia panel marker database built by selecting clade-specific MetaPhlAn2 [24] and 16S rDNA markers (for Hafnia alvei, Proteus vulgaris and Morganella morganii, for which no MetaPhlAn2 markers were available). The following rules were applied: (i) When at least one marker of the tested SOI is detected, taxonomic assignment of the sequences to the SOI was confirmed; (ii) When a marker from another species was detected and not the one of the tested SOI, the tested SOI was invalidated to avoid false positive detections induced by miss-classification of the reads. When no pathogen marker was detected, mainly because of low-coverage assemblies, no confirmation or invalidation of taxonomic classification to SOI were reported.
The metagenomics analysis report contained two sections. The first is focused on the 19 pneumonia panel pathogens. The results of each of these species of interest (SOI) are analyzed independently with 4 steps described in Fig. 1B:
-
i.
First, the number of reads associated to each SOI and SPC are counted and normalized per million of bacterial reads. These normalized numbers of reads are then compared to a detection threshold, specific to each species, to determine if SOI and SPC can be considered detected;
-
ii.
The second step consists in the calculation of the absolute concentration of genome equivalent (GEq) of the SOI (CSOI) or minimal detectable concentration of genome equivalent of SOI (CminSOI) based on the known SPC concentration (≈ 1.7 × 104 GEq/mL) and the genome size of SOI and SPC. As the link between the concentration of GEq and the colony forming unit has not been established for each SOI, we used a unique metagenomic threshold (MT) of 5.3 × 103 GEq/mL, defined as the concentration in genome equivalent of SOI above which infection can be suspected [15];
-
iii.
In the third step, calculated CSOI or CminSOI are compared to MT;
-
iv.
The last step consists in reporting the results of detection.
The rules of interpretation (Fig. 1B) define 4 possible outcomes:
-
i.
Suspected colonization, when a SOI is detected and quantified below metagenomics threshold (MT 5.3 × 103 GEq/mL);
-
ii.
Suspected infection, when a SOI is detected and quantified at or above MT or when SOI is detected but not the SPC;
-
iii.
Absence of SOI detection, when the SOI is not detected and the calculated CminSOI is < MT;
-
iv.
Not interpretable, when both SOI and SPC are not detected or when calculated CminSOI is > MT.
The second part of the report provided the list of the 25 species with the highest read counts. In this analysis, the concentrations in GEq/mL are calculated without correction for genome size and validation filters are not applied. Therefore, this list provides an indication of the patient’s respiratory microbiota and may suggest an atypical infection by an organism not belonging to the pneumonia panel.
Statistical analysis
Continuous demographic and clinical characteristics of patients are presented as median (range). Qualitative parameters are reported as percentages. The performance of mNGS test was analyzed with sensitivity, specificity, positive predictive value and negative predictive value, culture being used as the reference.
Results
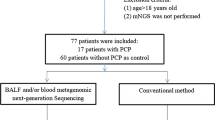
From July 2017 to November 2018, we enrolled 57 patients with HAP/VAP suspicion in ICU of Annecy-Genevois Hospital. We collected 60 BAL samples; among them, 41 (68%) were properly stored and had a sufficient volume to perform mNGS. After retrospective analysis by the adjudication committee, 32 of these 41 samples were linked to a confirmed HAP/VAP episode and were therefore selected for the final analysis (Fig. 2).

Flow chart
The patients characteristics are summarized in Table 2.
Results from BAL culture (gold standard method)
22/32 BALF were positive by culture (69%), 7 with only one bacterial species ≥ 104 CFU/mL and 15 with several micro-organisms (Table 3). S. pneumoniae, S. aureus, E. coli and H. influenzae were the most common species responsible for early-onset pneumonia; E. coli, S. aureus and S. marcescens were the main species in late-onset VAP.
Results from mNGS
The challenges of this study are threefold. First, confirming results from the reference method using NGS. Second, diagnosing potentially pathogenic bacteria that may have gone undetected in culture. Third, providing diagnoses for patients in cases where cultures failed to identify the pathogen.
mNGS data of culture-positive BALF
In 9/22 culture-positive BALF, mNGS and culture results were fully concordant (Table 3). In 12/22 samples, mNGS confirmed the culture-based detections above the clinical threshold but revealed additional species, some of which belonged to pneumonia panel (BAL 8, 29, 35, 48). The only BALF in which culture-positivity for a pathogen (S. aureus) was not confirmed by mNGS was BAL 59. The number of reads assigned to this species was below the mNGS detection threshold defined for S. aureus, which did not allow reliable quantification of the pathogen load. The performance of mNGS was even higher when only pneumonia panel SOIs were analyzed. In this case, mNGS confirmed culture results for 17/22 BALFs. Three samples (BAL 8, 29, 35) had both true positive and false positive SOI detections, one sample (BAL 48) had a false positive SOI detection and in one sample (BAL 59) a false negative (H. influenzae) and a false positive SOI (S.aureus) were identified. The Additional file 1: Table S1 provides a more detailed breakdown of all the results (quantification, …).
mNGS data of culture-negative BALFs
Among 10 BALF considered culture-negative (Table 4), 5 were mNGS-positive (BAL 20, 33, 34, 43, 45) for pulmonary pathogen like E. coli or H. influenzae and others species which do not belong to the classical pneumonia pathogen.
Quality control and sample processing control
In 23/32 samples SPC were detected. When detection failed, the negative identification of pathogens cannot be validated, sample calibration was not possible and quantification in GEq/mL of the bacteria was not achievable.
Assignment error
In almost all samples E. cloacae and M. tuberculosis were detected above the metagenomics threshold. These identifications suggest a potential assignment error. Verification methods were applied to all samples, demonstrating the absence of detection of these bacteria. MetaPhlAn and BLAST tools used to confirm mNGS results did not validate the identification of E. cloacae in any sample.
Time to results (mNGS)
In our study, the mean laboratory turnaround time was above 45 h: 6 h were needed for sample preparation and 39 h for Illumina sequencing. Duration of bioinformatics analysis and report generation steps depended on the number of reads generated for each sample and the number of threads available for data analysis. Typically, it ranged between 30 min and 6 h.
Performance of mNGS in detecting SOI
The detection by mNGS of SOI likely to be the source of infection is compared with that of microbial culture for all 19 SOI in the 32 BALF samples (total data population = 608), using a 2 × 2 confusion matrix [25]. Results considered uninterpretable due to the absence of detection of both SOI and SPC (Fig. 1) were counted as negative detections and their quantities are indicated in brackets in Table 5. mNGS showed 96.2% sensitivity and a specificity of 97.8% (Table 5) using culture results as a reference. The positive and negative predictive value were 65.8% and 97.8%, respectively.
Discussion
The purpose of this study was to evaluate the feasibility of mNGS for the direct detection of organisms in BAL fluid without using specific tests for different organisms. The recent COVID-19 pandemic and the occurrence of multiple bacterial superinfections complicating viral infection in ICU have underscored the significance of rapid diagnostics for tailoring treatments and avoiding the indiscriminate use of broad-spectrum antibiotics. While mNGS is not novel approach and has been explored by others, this study holds merit as it contributed to the currently limited knowledge in this field.
Our main encouraging result was that all respiratory pathogens identified by the culture were also detected by mNGS method. To be considered as a diagnosis test, it is essential that no false negative is detected when using a new method. 96% of species positive in culture exceeded the positivity threshold in the mNGS analysis. Only one false negative case is reported, the pathogen in question was detected by mNGS but the result was considered non interpretable due to an insufficient number of sequencing reads. The other main results of interest are the discrepancies between mNGS and culture results, mNGS helps identifying the pneumonia etiology in patients without microbiological diagnosis. In this study, in 7 patients, mNGS revealed putative HAP/VAP-causing agents that were either undetected or reported under the clinical significance threshold by culture tests. Some of these species were typical respiratory pathogens (e.g. E. coli and H. influenzae) while the others did not belong to the respiratory panel (e.g. Prevotella spp., Neisseria spp. or Gardnerella vaginalis). The detection of microorganisms by mNGS can reflect normal microbiota, colonizers, infection-causing microorganisms or sample contamination. When mNGS is used to detect pathogens in normally sterile liquids, such as urine, cerebrospinal or synovial fluids, the assignment of clinical significance for detected organisms is more straightforward [26, 27]. For BALF samples, the interpretation is more complex because the presence of a microorganism does not necessarily reflect an infection. In five out of 10 VAP cases with sterile BALF or BALF with cultured bacteria present under the clinical significance threshold, several bacterial species were considered mNGS-positive (above the MT threshold). In two of these cases (BAL 20, 45), these species belonged to the pneumonia panel (E. coli, H. influenzae). But in two others samples without microbiological diagnosis, R. pickettii was found above the threshold. We cannot exclude the possibility of reagent contamination by R. pickettii DNA [28,29,30] or an infection. Species of the genus Ralstonia are recognized as emerging gram-negative nosocomial pathogens causing bacteremia, VAP and meningitis. In a mixed infection, one bacterial species could potentially promote the pathogenicity of another. While coinfections with several pathogens in the context of VAP have been described using culture tests [31], there is no consensus on whether in such situations bacteria found below the clinically significance thresholds (< 104 CFU/mL) should be considered as pneumonia etiological agents. This is even less evident with mNGS that identifies a wider spectrum of bacteria per sample as compared to culture. For example, previous studies found Mycoplasma in many BALF from patients with VAP but these were mainly commensal species and not M. pneumoniae [31, 32]. This is a typical illustration of a bacterial taxon, not cultured with standard techniques but detected by mNGS, that could play a role in the pathogenicity of VAP [33]. It would be interesting to evaluate in a prospective clinical study the impact of real time mNGS results where the main objectives would be the clinical course of patients in order to determine the status of these newly diagnosed pathogens. One group could use the strategy of a prompt antibiotic de-escalation guided by mNGS results and the other the standard of care method of antibiotic adaptation. The impact of this technique on antibiotic utilization and antibiotic outcomes is unknown. It is possible that this method will lead to MORE antibiotic use rather than lest because more organisms are identified, some of which may may just be colonizers.
Twelve out of the 32 patients received antibiotics before BAL sampling: for 5 of them (BAL 13, 29, 31, 49, 56), pneumonia pathogens exceeded the clinical significance threshold in both culture and mNGS analyses. In other two cases (BAL 20, 45), bacteria detected by mNGS were (theoretically) susceptible to the previously administered antibiotics, which may explain their low abundance or absence in the culture. For example, the BAL 45 was mNGS-positive for H. influenzae and the BAL 20 for E. coli while the culture did not detect these pathogens, most likely because these patient received an efficient antibiotherapy on these bacteria before sampling (cefotaxime for patient 45 and piperacillin–tazobactam for patient 20).
The issue of the relative versus absolute quantification of microbiota members in respiratory samples assessed by culture-free methods was raised by Emonet et al. [34] and tackled in a metataxonomic (16S-targeted metagenomics) analysis of respiratory samples combined with panbacterial qPCR assay [31]. In our study, as suggested earlier [35], B. subtilis was added as a sample processing control (SPC) to BALF at a defined concentration to enable identification of possible analytical failures and extrapolate the absolute abundance of pathogens. Our study is one of the first to use SPC to provide quantitative mNGS data for respiratory pathogens, expressed as GEq/mL. However, the correlation between GEq/mL and CFU/mL has not yet been clearly established and its rather conservative interpretation remains to be prospectively validated. Currently, most published studies comparing culture and mNGS give raw mNGS results (detection or not). Specie detected by mNGS were considered positive without confirmation tests like MetaPhlAn and BLAST tools. Clinical interpretation is therefore difficult, and mNGS results can simply be regarded as a description of the lung microbiome with pathogen and non-pathogen species. No guidelines have been established to assist clinicians in interpreting positive mNGS results and the final clinical diagnosis was determined by several experienced clinicians, leading to unavoidable subjective biases due to the lack of unified standards [36, 37]. Some authors have established rules to mitigate subjectivity in the interpretation of mNGS results. For example, Langelier et al. classified microbes as confirmed pathogens if (1) both clinical testing and mNGS identified the microbe, (2) there was existing literature evidence of pathogenicity in the lungs, and (3) the score was as least twofold greater than that of any other microbe of the same type (virus, bacteria, or fungus) identified in the patient [38] and Jin et al. considered results positive when the relative abundance of pathogens detected by mNGS at the genus level was greater than or equal to 30% [39]. Our study is therefore one of the first to use an SPC to provide objective quantitative results and assist in interpretation using a pathogenicity threshold [40].
Currently, the scope of mNGS application in clinical microbiology is confined to situations where cultures fail to identify pathogens, especially in infections associated with (prior) usage of antibiotics. This is also applicable to identification of slow-growing or difficult-to-grow pathogens such as M. tuberculosis, Legionella spp., viruses or fungi; infections by these micro-organisms might be currently underestimated [26, 41].
This proof-of-concept study has several limitations. First, the application of mNGS was possible only for 41 BAL fluids among the 60 collected. Then, 9/41 (23%) were excluded due to pneumonia misdiagnosis. HAP/VAP diagnosis remains complex even for experts in the field; physicians may come to different conclusions about whether a given patient meets criteria for pneumonia especially due to difficulties in interpretation of chest radiographies in the ICU. Secondly, it does not provide antimicrobial susceptibility predictions thus limiting potential impact on antibiotic use. Thirdly, the mNGS process currently lacks standardization, complicating interpretation of results. One of the main preanalytical mNGS issues is that BAL samples contain many human cells. Therefore, removal of the overwhelming human DNA is critical for increasing the sensitivity of microbial signals in mNGS datasets [34]. An advantage of the mNGS pipeline used in our study is that combination of saponin and DNAse treatment remove extracellular DNA and DNA from damaged cells (dead bacteria), providing a clinically relevant bacterial detection. Bioinformatics analysis of mNGS data also constitutes a challenge. In our dataset, E. cloacae and M. tuberculosis were identified in nearly all samples above the mNGS threshold. However, MetaPhlAn and BLAST tools used to confirm results did not validate the identification of E. cloacae in any sample. These false identifications suggest that some bacterial genomic sequences from the reference database could be contaminated with those of human origin, resulting in misclassification of human reads as bacterial. M. tuberculosis, whose detection was not checked with BLAST or MetaPhlAn, could also be confused with other non-pathogenic mycobacteria. A second assessment was conducted using mNGS, utilizing Oxford Nanopore MinION and another database named WIMP. It does not find this E. cloacae and M. tuberculosis in any of the samples, thus confirming a potential error of assignation. Therefore, the quality of the reference database is of paramount importance for reliable taxonomic assignments and accurate pathogens detection. In our protocol, the turnaround time was 45–51 h, which included 39 h of Illumina MiSeq sequencing. Current sequencing technologies and mNGS pipelines allow an even faster time to result of < 30 h [42] with Illumina or ~ 6 h with Oxford Nanopore [43], allowing identification of pathogens in a timeframe that is compatible with the clinical practice. Reducing the time to pathogen identification, which was not the objective of our study, not only improves patient care but also contributes to a more rational use of antibiotics. But some might argue that this faster time to result is already achieved with the advent of PCR in clinical practice. However, in cases like the Filmarray Pneumonia Panel, implementing technical changes for new bacteria detection is time-consuming. To obtain authorization for clinical use, the process takes nearly a year. Additionally, the acid nucleic purification used in PCR employs a non-specific extraction of all acid nucleic (micro-organism and human DNA) using magnetic beads. To the opposite, the DNAse and the comparison of the size of sequenced genomes allows differentiation between dead and alive bacterial genomes, providing clinically relevant information.
This method is suitable in reference laboratory where the technical expertise and staffing required for performing mNGS are available. However, it is challenging to envision conducting these analyses in frontline hospital laboratories. Also, transporting samples to a reference laboratory could potentially increase the turn-around time, diminishing the method’s appeal. To avoid this, we could consider locally performing the technical processes of DNA extraction and libraries preparation, then transferring the data to bioinformatics reference laboratory with an on-call service to obtain results as quickly as possible.
In critically ill or severely immunocompromised patients, mNGS has the potential to improve clinical care and outcome by providing timely and efficient pathogen identification. Further studies are needed to determine the clinical impact of mNGS as an adjunct or even alternative to currently used diagnostic tests for HAP/VAP.
Conclusion
We demonstrated technical feasibility and clinical proof-of-concept for the use of mNGS for diagnosing HAP/VAP in the ICU setting. Our results matched well those of culture tests and identified pathogens potentially implicated in pneumonia cases without etiologic diagnosis. Although the costs related to wet lab and bioinformatics resources currently limit the use of mNGS in clinical routine, this methodology is complementary to conventional approaches in antimicrobial decision-making in patients with infections, which may further improve clinical outcomes.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ARDS:
-
Acute respiratory distress syndrome
- BAL:
-
Bronchoalveolar lavage
- BALF:
-
Bronchoalveolar lavage fluid
- CFU:
-
Colony forming unit
- COPD:
-
Chronic obstructive pulmonary disease
- CT:
-
Culture threshold
- DNA:
-
Deoxyribonucleic acid
- GEq:
-
Genome equivalent
- HAP:
-
Hospital-acquired pneumonia
- ICU:
-
Intensive care unit
- mNGS:
-
metagenomics Next Generation Sequencing
- MT:
-
Metagenomic threshold
- PCR:
-
Polymerase Chain Reaction
- RRT:
-
Renal replacement therapy
- SOI:
-
Species of interest
- SPC:
-
Sample processing control
- VAP:
-
Ventilator associated pneumonia
References
Melsen WG, Rovers MM, Groenwold RHH, Bergmans DCJJ, Camus C, Bauer TT, et al. Attributable mortality of ventilator-associated pneumonia: a meta-analysis of individual patient data from randomised prevention studies. Lancet Infect Dis. 2013;13(8):665–71.
Timsit JF, Zahar JR, Chevret S. Attributable mortality of ventilator-associated pneumonia. Curr Opin Crit Care. 2011;17(5):464–71.
Piskin N, Aydemir H, Oztoprak N, Akduman D, Comert F, Kokturk F, et al. Inadequate treatment of ventilator-associated and hospital-acquired pneumonia: Risk factors and impact on outcomes. BMC Infect Dis. 2012;12(1):1–9.
Evans L, Rhodes A, Alhazzani W, Antonelli M, Coopersmith CM, French C, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock 2021. Intensive Care Med. 2021;47(11):1181–247.
Musher DM. Bacterial coinfection in COVID-19 and influenza pneumonia. Am J Respir Crit Care Med. 2021;204(5):498–500.
Torres A, Niederman MS, Chastre J, Ewig S, Fernandez-Vandellos P, Hanberger H, et al. International ERS/ESICM/ESCMID/ALAT guidelines for the management of hospital-acquired pneumonia and ventilator-associated pneumonia: Guidelines for the management of hospital-acquired pneumonia (HAP)/ventilator-associated pneumonia (VAP) of the European Respiratory Society (ERS), European Society of Intensive Care Medicine (ESICM), European Society of Clinical Microbiology and Infectious Diseases (ESCMID) and Asociación Latinoamericana del Tórax (ALAT). Eur Respir J. 2017;50(3):1700582.
Trouillet JL, Chastre J, Vuagnat A, Joly-Guillou ML, Combaux D, Dombret MC, et al. Ventilator-associated pneumonia caused by potentially drug-resistant bacteria. Am J Respir Crit Care Med. 1998;157(2):531–9.
Bogaerts P, Hamels S, de Mendonca R, Huang TD, Roisin S, Remacle J, et al. Analytical validation of a novel high multiplexing real-time PCR array for the identification of key pathogens causative of bacterial ventilator-associated pneumonia and their associated resistance genes. J Antimicrob Chemother. 2013;68(2):340–7.
Lee SH, Ruan SY, Pan SC, Lee TF, Chien JY, Hsueh PR. Performance of a multiplex PCR pneumonia panel for the identification of respiratory pathogens and the main determinants of resistance from the lower respiratory tract specimens of adult patients in intensive care units. J Microbiol Immunol Infect. 2019;52(6):920–8.
Gastli N, Loubinoux J, Daragon M, Lavigne JP, Saint-Sardos P, Pailhoriès H, et al. Multicentric evaluation of BioFire FilmArray Pneumonia Panel for rapid bacteriological documentation of pneumonia. Clin Microbiol Infect. 2020;27:1308–14.
Peiffer-Smadja N, Bouadma L, Mathy V, Allouche K, Patrier J, Reboul M, et al. Performance and impact of a multiplex PCR in ICU patients with ventilator-associated pneumonia or ventilated hospital-acquired pneumonia. Crit Care. 2020;24(1):366.
Federal Register. Infectious disease next generation sequencing based diagnostic devices: microbial identification and detection of antimicrobial resistance and virulence markers; draft guidance for Industry and Food and Drug Administration Staff; Extension of Comment Period; 2016. https://www.federalregister.gov/documents/2016/08/11/2016-19109/infectious-disease-next-generation-sequencing-based-diagnostic-devices-microbial-identification-and. Accessed 17 Oct 2023.
Ruppé E, Greub G, Schrenzel J. Messages from the first international conference on clinical metagenomics (ICCMg). Microbes Infect. 2017;19(4–5):223–8.
Le Dorze M, Gault N, Foucrier A, Ruppé E, Mourvillier B, Woerther PL, et al. Performance and impact of a rapid method combining mass spectrometry and direct antimicrobial susceptibility testing on treatment adequacy of patients with ventilator-associated pneumonia. Clin Microbiol Infect. 2015;21(5):468.e1-6.
Hauser S, Lazarevic V, Tournoud M, Ruppé E, Allexant ES, Guigon G, et al. A metagenomics method for the quantitative detection of pathogens causing ventilator-associated pneumonia. Microbiol Spectr. 2023. https://doi.org/10.1128/spectrum.01294-23.
Pezzulo AA, Kelly PH, Nassar BS, Rutland CJ, Gansemer ND, Dohrn CL, et al. Abundant DNase I-sensitive bacterial DNA in healthy porcine lungs and its implications for the lung microbiome. Appl Environ Microbiol. 2013;79(19):5936–41.
Tournoud M, Ruppé E, Perrin G, Schicklin S, Guigon G, Mahé P, et al. Clinical metagenomics bioinformatics pipeline for the identification of hospital-acquired pneumonia pathogens antibiotic resistance genes from bronchoalveolar lavage samples. 2020 p. 2020.02.26.966309. https://www.biorxiv.org/content/10.1101/2020.02.26.966309v1. Accessed 19 Dec 2021.
Wood DE, Salzberg SL. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014;15(3):R46.
Davis JJ, Wattam AR, Aziz RK, Brettin T, Butler R, Butler RM, et al. The PATRIC Bioinformatics Resource Center: expanding data and analysis capabilities. Nucleic Acids Res. 2020;48(D1):D606–12.
Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016;44(14):6614–24.
Sichtig H, Minogue T, Yan Y, Stefan C, Hall A, Tallon L, et al. FDA-ARGOS is a database with public quality-controlled reference genomes for diagnostic use and regulatory science. Nat Commun. 2019;10:3313.
Peng Y, Leung HCM, Yiu SM, Chin FYL. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28(11):1420–8.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10.
Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, et al. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods. 2015;12(10):902–3.
Tharwat A. Classification assessment methods. ACI. 2021;17(1):168–92.
Wilson MR, Sample HA, Zorn KC, Arevalo S, Yu G, Neuhaus J, et al. Clinical metagenomic sequencing for diagnosis of meningitis and encephalitis. N Engl J Med. 2019;380(24):2327–40.
Schmidt K, Mwaigwisya S, Crossman LC, Doumith M, Munroe D, Pires C, et al. Identification of bacterial pathogens and antimicrobial resistance directly from clinical urines by nanopore-based metagenomic sequencing. J Antimicrob Chemother. 2017;72(1):104–14.
Büchler AC, Lazarevic V, Gaïa N, Girard M, Eckstein F, Egli A, et al. Mycobacterium chelonae infection identified by metagenomic next-generation sequencing as the probable cause of acute contained rupture of a biological composite graft-a case report. Int J Mol Sci. 2021;23(1):381.
Nasir N, Sayeed MA, Jamil B. Ralstonia pickettii bacteremia: an emerging infection in a tertiary care hospital setting. Cureus. 2019;11(7): e5084.
Waugh JB, Granger WM, Gaggar A. Incidence, relevance and response for Ralsfonia respiratory infections. Clin Lab Sci. 2010;23(2):99–106.
Emonet S, Lazarevic V, Leemann Refondini C, Gaïa N, Leo S, Girard M, et al. Identification of respiratory microbiota markers in ventilator-associated pneumonia. Intensive Care Med. 2019;45(8):1082–92.
Nolan TJ, Gadsby NJ, Hellyer TP, Templeton KE, McMullan R, McKenna JP, et al. Low-pathogenicity Mycoplasma spp. alter human monocyte and macrophage function and are highly prevalent among patients with ventilator-acquired pneumonia. Thorax. 2016;71(7):594–600.
Bousbia S, Papazian L, Saux P, Forel JM, Auffray JP, Martin C, et al. Repertoire of intensive care unit pneumonia microbiota. PLoS ONE. 2012;7(2): e32486.
Emonet S, Lazarevic V, Pugin J, Schrenzel J, Ruppé E. Clinical metagenomics for the diagnosis of hospital-acquired infections: promises and hurdles. Am J Respir Crit Care Med. 2017;196(12):1617–8.
Hauser et al. A metagenomics method for the quantitative detecti.pdf.
Shi XQ, Tian L, Huang ZH, Song WT, Wu JB, Chen LM. Metagenomic next-generation sequencing vs. conventional detection methods for detecting the pulmonary infections. Eur Rev Med Pharmacol Sci. 2023;27(10):4752–63.
Liang M, Fan Y, Zhang D, Yang L, Wang X, Wang S, et al. Metagenomic next-generation sequencing for accurate diagnosis and management of lower respiratory tract infections. Int J Infect Dis. 2022;122:921–9.
Langelier C, Zinter MS, Kalantar K, Yanik GA, Christenson S, O’Donovan B, et al. Metagenomic sequencing detects respiratory pathogens in hematopoietic cellular transplant patients. Am J Respir Crit Care Med. 2018;197(4):524–8.
Jin X, Li J, Shao M, Lv X, Ji N, Zhu Y, et al. Improving suspected pulmonary infection diagnosis by bronchoalveolar lavage fluid metagenomic next-generation sequencing: a multicenter retrospective study. Microbiol Spectr. 2022;10(4): e0247321.
Gadsby NJ, Dunn JJ, Johnson CL, McQuillan T, McHugh MP, Templeton KE, et al. Discordance between semi-quantitative nucleic acid detection of bacteria and quantitative bacteriology in sputum from patients with pneumonia. J Infect. 2023;86(6):607–9.
Ruppé E, Lazarevic V, Girard M, Mouton W, Ferry T, Laurent F, et al. Clinical metagenomics of bone and joint infections: a proof of concept study. Sci Rep. 2017;7(1):7718.
Kolb M, Lazarevic V, Emonet S, Calmy A, Girard M, Gaïa N, et al. Next-generation sequencing for the diagnosis of challenging culture-negative endocarditis. Front Med (Lausanne). 2019;6:203.
Gu W, Deng X, Lee M, Sucu YD, Arevalo S, Stryke D, et al. Rapid pathogen detection by metagenomic next-generation sequencing of infected body fluids. Nat Med. 2021;27(1):115–24.
Acknowledgements
We thank the patients who agreed to participate to this study. We thank the clinical teams who cared for the patients and the laboratory teams who realized culture and bioinformatics analysis.
Funding
An unrestricted grant from bioMérieux (Marcy l’Étoile, France) was used to financially support the metagenomics analysis but this study was independently designed, executed, analyzed and reported by the authors.
Author information
Authors and Affiliations
Contributions
MH collected and analyzed the results from mNGS data and wrote the manuscript, SH developed the bioinformatics, performed the mNGS analyses and wrote the manuscript, AL collected the samples, contributed to the conception and design of study, SB performed the bacteriological analyses, KL, ESA, JS also contributed to the conception and design. JS, VL, OB and all authors read, critically reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study protocol was approved by the French institutional ethics committee (CPP Sud-Méditerranée, Marseille, 2017-A00253-50, FRANCE). All patients or their relatives received an information letter and a non-objection form for participation was signed.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Comparison of the results of bacterial quantification by culture and mNGS in 32 BALFs. Only species identified by culture (present above or below the clinical threshold of 104 CFU/mL) and/or those exceeding metagenomics threshold (5.3 × 103 GEq/mL) in the mNGS analysis are presented. In bold: pathogen detections above the clinical threshold for culture or above the mNGS positivity threshold. Species not belonging to the pneumonia panel are indicated between square brackets. Dotted lines indicate no detection. False positive (FP) and true positive (TP) SOI (pneumonia panel) are indicated for mNGS using culture data as a reference. NI, non-interpretable quantification of SOI; indicated only for SOIs identified by culture (below or above clinical threshold). FP* and NI*: corresponding SOIs were detected by culture under the clinical threshold. “ > MT” means that SOI was detected but SPC was undetected (see “Methods”).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Heitz, M., Levrat, A., Lazarevic, V. et al. Metagenomics for the microbiological diagnosis of hospital-acquired pneumonia and ventilator-associated pneumonia (HAP/VAP) in intensive care unit (ICU): a proof-of-concept study. Respir Res 24, 285 (2023). https://doi.org/10.1186/s12931-023-02597-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-023-02597-x