
Abstract
Workplaces with elevated organic dust levels such as animal feed barns also commonly have elevated levels of gasses, such as CO2. Workers exposed to such complex environments often experience respiratory effects that may be due to a combination of respirable factors. We examined the effects of CO2 on lung innate immune responses in mice co-exposed to the inflammatory agents lipopolysaccharide (LPS) and organic dust. We evaluated CO2 levels at the building recommended limit (1000 ppm) as well as the exposure limit (5000 ppm). Mice were nasally instilled with dust extracts or LPS and immediately put into chambers with a constant flow of room air (avg. 430 ppm CO2), 1000 ppm, or 5000 ppm CO2 enriched air. Results reveal that organic dust exposures tended to show decreased inflammatory responses with 1000 ppm CO2 and increased responses at 5000 ppm CO2. Conversely, LPS with addition of CO2 as low as 1000 ppm tended to inhibit several inflammatory markers. In most cases saline treated animals showed few changes with CO2 exposure, though some changes in mRNA levels were present. This shows that CO2 as low as 1000 ppm CO2 was capable of altering innate immune responses to both LPS and organic dust extracts, but each response was altered in a different fashion.
Similar content being viewed by others
Background
Organic dust exposures in animal feed operations has long been shown to be detrimental to the health of workers in these facilities [1, 2]. Experimental approaches to determining the factors present in these dusts that may be responsible for respiratory inflammation have yielded considerable data, particularly on the role of microbial products such as endotoxins like lipopolysaccharides (LPS) and proteoglycans, and others as potential causes of these lung problems [3, 4]. Such dust exposures however are often in the context of facilities with a variety of elevated gases. Ammonia and hydrogen sulfide have been implicated in causing lung problems or being immuno-modulatory [5, 6]. However, carbon dioxide (CO2) has often been overlooked despite significant data showing routine elevations of CO2 in animal feed operations [7].
Carbon dioxide levels in animal confinement operations often exceed standards including the American Society of Heating, Refrigeration and Air-conditioning Engineers (ASHRAE) recommended limit (1000 ppm) as well as the Occupational Safety and Health Administration (OSHA) 8 h time weighted average (TWA) limit of 5000 ppm. CO2 at levels as low as 1000 ppm have been shown to induce cognitive changes in humans [8, 9]. Indeed, elevated CO2 is a broader problem, and common in many facilities such as schools [9], daycares [10, 11], prisons [12], cars [13], airplanes [14], and many other locations [15].
Previously we have shown that innate immune responses to organic dust extracts were changed when co-exposed to CO2 at 5000 ppm [16]. These changes were at the protein and mRNA levels and in most cases were indicative of enhanced inflammation. Subsequent to this study others showed that neutrophils were altered by similar levels of CO2 [17]. At present however these are the only studies we are aware of to address this issue in lung immunology at these low levels. There have been several studies related to behavior [18] and at higher levels of CO2 where there is induction of hypercapnic acidosis in animals and cell cultures [19,20,21]. Results for these latter studies have been somewhat mixed, particularly in situations of LPS exposure versus more complex challenges such as tobacco extracts [21, 22]. These studies also do not address CO2 at levels applicable to most work environments and crowded spaces [23, 24].
The aims of the current study were to extend the previous co-exposure model to assess lower levels of CO2 exposure while at the same time testing both a defined single inflammatory stimuli (LPS) and a complex one (organic dust extracts). We hypothesize that the lung alters innate immune responses when presented with a combined exposure of an inflammatory agent and CO2 as low as 1000 ppm.
Methods
Animals
Six to seven week old female C57BL/6 mice were purchased from Charles River Laboratories (Montreal, Quebec, Canada). C57BL/6 mice were selected for purposes of comparability to other papers on organic dust exposure and female mice to be comparable to our previous study on CO2 exposure [16]. Mice were housed in standard cages in a controlled room with temperature of 23 °C ± 2 °C and a 12-h light/dark cycle. Mice were fed standard laboratory chow and water ad libitum. Mice were acclimatized in their first week in facility for 2 h per day for 5 days to whole body plethysmography chambers (part 601-1425-001; DSI, Minneapolis, MN) ventilated by a Buxco Finepointe whole body plethysmography 4-site system (DSI, Minneapolis Minnesota).
Mice were randomly assigned to 9 experimental groups (5 mice/group): each of control (saline) with room air (avg 430 ppm CO2), control (saline) with 1000 ppm CO2, or control (saline) with 5000 ppm CO2; LPS with room air (avg 430 ppm CO2), LPS with 1000 ppm CO2, or LPS with 5000 ppm CO2; or Organic Dust (HDE) with room air (avg 430 ppm CO2), Organic Dust (HDE) with1000 ppm CO2, or Organic Dust (HDE) with 5000 ppm CO2 exposure. All intranasal treatments (saline, LPS, and HDE) were given between 8:00 and 8:30 AM to minimize any potential circadian rhythm differences between treatments and then mice were immediately placed in Buxco Finepoint whole body plesthysmography 4-site system (DSI, Minneapolis, Minnesota) with 1000 ppm or 5000 ppm CO2 concentrations, or room air for 6 h. At end of the experiment mice were euthanized.
Organic dust extracts, LPS, and control treatments
Intranasal treatment with organic dust extracts (HDE), LPS, or control (saline) were done immediately prior to the 6-h gas exposure in plethysmography chambers (see CO2 Delivery and Whole Body Plethysmography).
Organic dust extracts (HDE) were produced from settled dust samples collected from swine confinement facilities. These dust extracts have been characterized previously for muramic acid, endotoxin, and protein [25], and the bacterial composition has been previously described [26]. HDE extracts were prepared as described previously [27] by mixing 1 g dust with 10 ml HBSS (without calcium, Sigma, St. Louis, MS) and incubated for 1 hr at room temperature before centrifugation twice, and subsequent filter sterilization through a Nalgene 0.2 μM SFCA membrane syringe filter (ThermoFisher, Rochester, NY), resulting in a solution of approximately 0.105 g/ml dust. To prevent any variation due to dust used or extraction procedure, all extract samples in this study were created from a single extraction of the dust sample. Dust extracts were given to mice at a final concentration of 12.5% vol/vol or 0.005 g/ml dust delivered in 40 μl dropwise to the nares, and mice allowed to inhale under light (1%) isoflurane anesthesia.
LPS from Escherichia coli 0111:B4 (lot 095M4163V, Sigma, St. Louis) was diluted in HyClone HBSS (1X) (HyClone Laboratories; Logan, UT) to 0.1 µg, and delivered in 40 μl to the nares, and mice allowed to inhale under light (1%) isoflurane anesthesia. Control groups were given 40 μl HBSS in the same manner under light anesthesia.
CO2 delivery and whole body plethysmography
All mice upon intranasal treatment were placed immediately into whole body plethysmography (WBP) chambers (DSI, Minneapolis, MN) for the 6-h exposure under positive airflow of 5 psi. The bias flow air consisted of either room air (avg 430 ppm CO2), or air from tanks consisting of room air enriched for CO2 to 1000, or 5000 ppm CO2 (Praxair; Mississauga, Canada). Biasflow was constant through the exposure period to prevent any accumulation of CO2 due to respiration from animals. Mice were provided with food and water in exposure chambers for the duration of exposures. CO2 readings of the treatment room provided an average CO2 level of 430 ppm as measured using a Q-Trak Indoor Air Quality Monitor 7575 (TSI; Shoreview, MN).
Readings from the WBP chambers were captured every 2 s for the entire 6 h duration of exposure. Results were averaged for 20 min intervals using the Finepointe system software (DSI, Minneapolis, MN). Measures of PenH, peak inspiratory volume, peak expiratory volume, volume per minute, and breath frequency were taken.
Bronchoalveolar lavage
Lungs were lavaged as detailed earlier [27]. Briefly, lungs were washed three times with 0.5 ml HBSS each time. Bronchioalveolar lavage (BAL) fluid was centrifuged at 1000×g for 10 min, and supernatant transferred to new tubes and stored at − 80 °C. Cell pellets were resuspended in HBSS and stained with trypan blue (Life Technologies, Grand Island, NY) and counted using a hemocytometer. Cells were resuspended to 100 μl in HBSS and adhered to glass slides (Fisher Scientific; Pittsburgh, PA) via cytocentrifugation for 10 min at 10,000×g (Cytopin, Shandon Elliott, Great Britain). Cells were dried overnight and fixed and stained using Diff Quik kit (Siemens Healthcare Disagnostics, Newark, DE) and mounted using MM24 mounting media (Leica Biosystems; Buffalo Grove, IL). A differential count of 100 cells was made based on morphological assessment of cells and expressed as absolute cell numbers.
Lung collection
After BAL collection lungs were excised. The right lung was tied off at the primary bronchus, removed, flash frozen in liquid nitrogen, and stored at −80 °C. The left lung was slowly inflated with 200 μl of 4% PFA (Sigma; St. Louis, MO) and stored in 4% PFA overnight, followed by 100% ethanol. The fixed lung was embedded the next day in paraffin in a Intelsint RVG/1 tissue processor (Intelsint; Turin, Italy) followed by mounting in paraffin blocks using a Tissue Tek II tissue embedder (Sakura Finetek; Nagano, Japan).
Lung histology
Lung sections from paraffin embedded lungs were sectioned in 5 μM slices on a Microm 350S microtome (Microm, Germany). Tissue sections were then stained with Hemotoxylin and Eosin using the Histology Core Facility’s Protocol (https://healthsciences.usask.ca/facility-services/histology-core-facility.php) and mounted using Surgipath MM24 (Leica Biosystems, Richmond, IL). Sections were imaged using an Aperio CS2 virutual microscopy system (Leica Biosystems, Concord ON Canada) and examined for any notable changes to lung structure.
ELISA
Quantification of MCP-1, MIP-2, IL-6, KC, IL-1β, TNF-α, IL-4, IL-5, IL-13, IL-33, and IL-10 in BAL fluid was determined by Luminex xMAP multiplex ELISA (Procartaplex, ThermoFisher) according to manufacturer’s specifications. Samples were read using a Bioplex 200 system (Bio-Rad, Hercules, California) and Bioplex Manager Software (Bio-Rad).
RNA purification and RT-PCR analysis
RNA was purified from lung tissue samples using a Qiagen RNeasy Plus Mini kit (Qiagen, Chatsworth CA) according to the manufacturer’s instructions for tissue extraction. Briefly, 20–25 mg of lung tissue was put into 350 μl of RLT lysis buffer and 2.0 mm zircon beads (BioSpec, Bartlesville, OK). Samples were placed in a Biospec Mini-BeadBeater-24 (BioSpec) and run twice for 2 min, with a pause between runs of 5 min where samples were held on ice to prevent heat buildup. Supernatant was removed to DNA eliminator column, and the remainder of purification was done according to the kit protocol. RNA was quantified by Take3 plate (Biotek, Winooski, VT) in a Synergy HT plate reader (BioTek). cDNA synthesis was done using the iScript Reverse Transcription Supermix for RT-qPCR (Bio-Rad, Hercules, CA) with 300 ng of template mRNA. Samples were incubated at 25 °C for 5 min, 46 °C for 20 min, and finally at 95 °C for 1 min in a Bio-Rad CFX96 Touch Real-Time PCR Detection System (BioRad). RT-PCR was done using probes for TLR2 (Mm00442346_m1), TLR4 (Mm445273_m1), Hsp72 (Mm01159846_S1), A20/TNFAIP3 (Mm00437121_m1) (Life Technologies, Grand Island, NY). Ribosomal RNA (Life Technologies) was used as an endogenous control in all reactions. PCR was conducted using a Bio-Rad CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Reactions were all carried out in duplicate and performed 2 min at 50 °C, 10 min at 95 °C, then 40 cycles of 15 s at 95 °C and 1 at 60 °C min each using ddPCR Supermix for Probes kit (Bio-Rad). Relative comparison of selected targets to the ribosomal endogenous control cycle threshold (CT) value was analyzed with the ΔΔCt method.
Statistical analysis
Data was analyzed using GraphPad Prism 6 (GraphPad Software, San Diego, CA). Error bars represent mean ± SEM. For values outside the assay limit of detection (LOD) either the LOD/2 or a minimum value below the lowest attained value was designated. Statistical significance was determined using one-way ANOVA with follow-up Tukey test for multiple comparisons. If the assumption of equal variance using the Brown’s Forsythe test was not met, the data was log transformed followed by one-way ANOVA and multiple comparison tests. For all tests, a p-value ≤ 0.05 was considered significant for differences between groups.
Results
Plethysmography data
Monitoring of breathing parameters of mice in exposure chambers showed changes in several parameters over the 6 h of exposure, across all treatments (Fig. 1).

Plethysmography data: Mice were monitored during exposure period and PenH, peak inspiratory volume, peak expiratory volume, volume per minute, and breath frequency. Each result represents the average value over the 6 h exposure. Statistical significance denoted by “a” (treatment vs respective saline control), “b” (1000 ppm CO2 vs room air treatment), “c” (5000 ppm CO2 vs room air treatment) and “d” (5000 ppm CO2 vs 1000 ppm CO2 treatment). N = 5 mice/group. Minimum, maximum, mean (+ symbol) and median (line) are denoted for each treatment
Animals given saline showed elevations in peak inspiratory and expiratory flow (Fig. 1c, d), tidal volume, total volume (Fig. 1e), and minute volume (Fig. 1g) at 1000 ppm CO2, and tidal volume and total inspiratory time (Figs. 1e and f) at 5000 ppm. Animals treated with HDE showed significant increased peak inspiratory flow, p < 0.05 (Fig. 1c) and tidal volume, p < 0.01 (Fig. 1e) over room air at 5000 ppm CO2, whereas at 1000 ppm only tidal volume was elevated (p < 0.05). LPS treatment to animals showed increases in tidal volume over room air co-exposure (p < 0.05) at 1000 and 5000 ppm CO2 co-exposure. CO2 meanwhile resulted in a decrease in LPS-induced increase in PenH, (p < 0.01) (Fig. 1a).
Comparing within and between treatments there was increased tidal volume with all co-treatments with CO2 compared to treatments with no CO2.
None of the treatments appeared to have any significant effect on breathing frequency (Fig. 1b), total inspiratory time (Fig. 1f), or minute volume (Fig. 1g).
Bronchoalveolar lavage cells
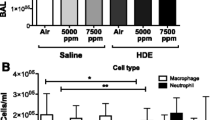
Cell counts from BAL fluid showed several patterns (Fig. 2). First, mice given saline intranasally showed no significant changes in BAL cell composition or number with increased CO2 (Fig. 2a).

Effects of CO2 on BAL fluid cells. Mice were treated intranasally with saline, organic dust (HDE), or lipopolysaccharide (LPS) and then exposed to either room air (~ 500 ppm), 1000 ppm (1 K), or 5000 ppm (5 K) CO2. Values given are based on number of cells in BAL fluid multiplied by the fraction of monocytes/macrophages (black bars) or neutrophils (open bars) in (a) saline, (b) HDE or (c) LPS treated animals. Bar graphs represent averages and error bars the SEM. N = 4–5 mice/group. Statistical significance denoted by “a” (treatment vs respective saline control), “b” (1000 ppm vs room air treatment), “c” (5000 ppm vs room air treatment) and “d” (5000 ppm vs 1000 ppm treatment) and error bars denoting SEM
LPS treatment showed an increase in macrophages/monocytes compared to saline control. This increase was significant to treatment at a similar CO2 level but only at 5000 ppm CO2 co-exposure (p < 0.05). No significant increases in neutrophils were noted between saline and LPS treatments, except between Saline + 5000 ppm CO2 and LPS + 5000 ppm CO2 (p < 0.01).
HDE exposure (Fig. 2c) on its own or with room air was not able to induce a significant rise in monocytes/macrophages. However, co-exposure of HDE with 1000 ppm CO2 was able to induce an non-significant increased numbers of cells over saline given at a similar level of CO2 (p = 0.0732), which was significantly increased with 5000 ppm co-exposure (p < 0.05). Levels of neutrophils were elevated with HDE, but not significantly different as compared to saline. However, co-exposure of HDE with 5000 ppm CO2 caused a substantial increase of the neutrophil population as compared to HDE and room air (p < 0.001), which was also significantly different from the HDE and 1000 ppm CO2 co-exposure (p < 0.01), and from the saline and 5000 ppm CO2 co-exposure (p < 0.0001).
ELISAs
As shown in Figs. 3–1 and –2, the expression of cytokines appeared to follow three general patterns. First, no difference in cytokines was noted when CO2 was co-exposed with saline.


Protein expression in BAL fluid. Values given are in OD reading (a) MCP-1, b MIP-2, c KC, d IL-6, e IL-1B, f TNF-a, g IL-5, h IL-4, i IL-33, j IL-13, and k IL-10. Mice were treated intranasally with saline, organic dust (OD), or lipopolysaccharide (LPS) intranasally and then exposed to either room air (~ 500 ppm), 1000 ppm (1 K), or 5000 ppm (5 K) CO2. BAL fluid was centrifuged, cells removed and tested by ELISA. Results show reductions in KC and MIP-2 with LPS treated animals plus CO2, and an increase in all three for OD plus 5000 ppm CO2. Bar graphs represent averages and error bars the SEM. N = 3–5 mice/group. by “a” (treatment vs respective saline control), “b” (1000 ppm vs room air treatment), “c” (5000 ppm vs room air treatment) and “d” (5000 ppm vs 1000 ppm treatment)
For LPS, increased CO2 provoked a decrease in cytokines/chemokines as compared to co-exposure with room air. Co-delivery of 1000 ppm CO2 with LPS resulted in significant decreases in IL-6 (p < 0.01), KC (p < 0.001), TNF-α (p < 0.01), and IL-1β (p < 0.01) as compared to co-exposure with room air. Similar trends were noted at 5000 ppm CO2 co-exposure with LPS with reductions in IL-6 (p < 0.05), KC (p < 0.001), and TNF-α (p < 0.01) as compared to room air co-exposure to LPS. Cytokine/chemokines were not significantly different between co-exposure of LPS to CO2 at 1000 ppm and 5000 ppm as compared to saline co-exposures at these same CO2 levels.
The situation was more complex with HDE. Compared to co-exposure with room air, at 1000 ppm CO2 co-exposure HDE showed no significant differences between cytokines/chemokines though there was some apparent drop in production. However at 5000 ppm CO2 co-exposure there was a significant rise in MCP-1 and MIP-2 (p < 0.05) compared to HDE on room air. There was also a rise in MCP-1 (p < 0.001), MIP-2 (p < 0.01), and IL-1β (P < 0.01) at 5000 ppm codelivery as compared to co-delivery of 1000 ppm CO2. This suggested an increase in inflammatory response at higher CO2 delivery as opposed to the reductions seen with all CO2 co-delivery levels when CO2 was co-exposed with LPS. Further, with HDE co-exposure with 1000 ppm CO2 there was a tendency to reduced cytokines whereas 5000 ppm CO2 co-delivery with HDE showed increases in cytokines, suggesting differential responses with increased CO2 concentrations when co-exposed with HDE.
RT-PCR
We looked for evidence of changes to signaling in lung cells first at the receptor level with two of the receptors most commonly associated with response to HDE, TLR2 and TLR4, the latter of which is critical to responses to LPS.
TLR2 (Fig. 4a), which is implicated in HDE induced inflammation [28, 29] showed no significant change with HDE co-exposure with any level of CO2. However, LPS with room air induced a significantly large increase in TLR2 mRNA compared to saline and room air (p < 0.05), and this increase was inhibited by addition of 1000 ppm CO2 (p < 0.01) and 5000 ppm CO2 (NS) co-exposures.

mRNA expression in mouse lung tissue. Values given are fold change in mRNA compared to a control value. mRNA harvested from lungs of mice given saline, organic dust (OD), or lipopolysaccharide (LPS) intranasally and then exposed to either room air (approximately 500 ppm), 1000 ppm (1 K), or 5000 ppm (5 K) CO2. Tissue was tested for the receptor TLR2 (a), TLR4 (b), cytoplasmic protein A20 (c) and Hsp1A (d). Results show reductions in TLR2, and Hsp1A with LPS treated animals plus CO2, and general decreases in A20 and Hsp1A for OD plus CO2. Hsp1A was elevated with CO2 delivery to control, to date the only protein mRNA found to do this. Bar graphs represent averages and error bars the SEM. N = 3–5 mice/group. by “a” (treatment vs respective saline control), “b” (1000 ppm vs room air treatment), “c” (5000 ppm vs room air treatment) and “d” (5000 ppm vs 1000 ppm treatment)
TLR4 (Fig. 4b) showed no significant change in mRNA expression across all LPS and HDE treatments and CO2 co-exposures.
With little work having been done on CO2 signaling pathways we next looked at a negative regulator of the TLR/NF-kB pathway, TNFAIP3 (A20) (Fig. 4c), which has been implicated by others in the prevention of asthma in rural populations [30]. While there was no increase in mRNA production with LPS exposure, there was an increase in A20 mRNA expression with HDE compared with control (p < 0.01). Interestingly, for HDE, this increase was inhibited by CO2 co-delivery at 1000 ppm (p < 0.05), and less inhibitory at 5000 ppm.
mRNA expression of Hsp72 (Hsp1A) was also tested (Fig. 4d).This protein has been shown to have effects on MAPK and NF-kB signaling and thus may alter signaling in these two key pathways. CO2 at 5000 ppm co-exposure with saline was able to induce some Hsp72 mRNA increase, but not a significantly compared to saline and room air. When co-exposed with room air, HDE induced a similar non-significant Hsp72 increase while LPS induced a significant increase (p < 0.01). However, Hsp72 mRNA was decreased when HDE or LPS stimuli were co-exposed with either of the two CO2 concentration (p < 0.05 for LPS + CO2 treated groups).
Histology
Tissue sections (Fig. 5) overall showed little in the way of overt changes due to inflammation. This may be in part due to the limited time frame of exposure, and longer exposures are likely to produce more significant changes. No consistent overt differences were noted in mice for saline, LPS, or HDE treatments compared to their similarly treated but CO2 co-exposed duplicates.

Histological examination of mouse lungs. Left lungs of mice were fixed in 4% PFA and processed for paraffin blocks. 5 μm tissue slices were cut and stained with hematoxylin and eosin and imaged. Images at 100× are included for saline (a–c), HDE (d–f), and LPS (g–i). Changes in inflammation were apparent between saline, HDE, and LPS, treatments, but CO2 co-treatment appeared to have little effect
Discussion
In high intensity animal production operations, there are buildups of both particulates in the air as well as a variety of gasses, mostly as a result of the animals housed there. Given the problems of respiratory effects in exposed workers [1, 31] numerous studies have tried to recreate these conditions in animal models [2] to determine responsible agents for health effects. Given that CO2 elevation is common in many environments, not just in barns, we previously looked at the effects of human workplace safety limit levels (5000 ppm) of CO2 on mice treated with barn dust extract to try to devise a more holistic model of barn exposure inflammation. The results from this study suggested that we look at both a more simple innate inflammatory molecule (LPS), as well as CO2 concentrations that are more reflective of a number of environments.
The first questions was if breathing parameters showed any changes, as might be expected from an elevation in ambient CO2 levels. Martrette et al. have reported potential stress after long repeated exposures to elevated CO2 in rats (700 ppm, 6 hr/day over 15 days) [18], whereas Rasid et al. have shown changes in minute volume associated with stressful procedures [32]. In the present study there were no significant changes to minute volume for any of the treated animals over the 6 h course of our studies, suggesting the treatments were reasonably tolerated. Similarly, breath frequency was unaffected across all treatments and CO2 levels. Tidal volume appeared to be the most sensitive of the tested parameters to treatments and CO2 exposure. Changes however appeared to be due to presence of CO2 alone, as the secondary treatment had no effect. This is unsurprising as volume of air moved in and out of the lung should logically increase to counteract elevated CO2. Interestingly, within the HDE exposed animals the peak expiratory flow, peak inspiratory flow, and tidal volume measures increased with increasing ppm co-exposure to CO2, suggesting that when co-exposed, the increasing doses of carbon dioxide may be resulting in increases in airways responsiveness. While this would fit with the barn dust extract and CO2 treatment being one of the most inflammatory treatments tested here, the changes were nearly immediate within the monitoring time (within 40 min, data not shown) which may suggest very early inflammatory mechanisms could be playing a role. Overall however we would expect minimal changes to breathing due to the short time frame of the exposure.
Looking at cellular levels in the lung extracellular space we see that similar to our previous study [16], increasing CO2 appeared to have no effect on saline treated animals. HDE by comparison showed a trend to increases in total cells and a trend to increased monocytes with CO2 co-treatment, even at levels as low as 1000 ppm CO2, and by 5000 ppm a significant increase in total BAL cells and neutrophils in the lung. This general increased inflammatory profile is similar to our previous report [16]. A further similarity to that report was that in lung tissue histology (Fig. 5) there was no discernable changes seen with any of the treatments with or without added CO2. Given that this was a 6 h exposure, we did not expect to see significant histological changes. We suspect a more chronic exposure may yield different results though, given other changes in cytokines and chemokine expression.
Past work by our lab has shown that CO2 co-exposure did cause significant changes at the molecular level, increasing inflammatory responses to organic dusts. As LPS is a common constituent of agricultural dusts it was thought that LPS exposures and co-exposures may respond similarly to HDE. Our results showed a relatively common pattern across several cytokines with regards to LPS as compared to saline, showing significant increase in inflammatory cytokine production with LPS exposure. However, for the proinflammatory cytokines, the LPS with room air response tended to be greater than the HDE with room air response. These responses changed with co-exposure to CO2. For LPS, there were significant decreases in cytokines with both levels of CO2 co-exposures. By contrast, when HDE was co-exposed with CO2, at 1000 ppm there was a decreased cytokine response compared to the room air HDE + CO2 co-exposure, however, at 5000 ppm HDE + CO2 co-treatment the cytokines were greater as compared to the co-exposures to room air and 1000 ppm CO2. These results are similar to the BAL cellular responses, particularly with well-known monocyte and macrophage chemoattractors such as MCP-1, MIP-2, KC, TNF-a, and IL-1β [33, 34]. IL-1β should warrant particular attention given past work in ex-vivo systems with neutrophil microparticles that shows increased levels of IL-1β per microparticle with increasing CO2 up to 4000 ppm [17]. While we did not see this with unstimulated animals, we do see this increased IL-1B with HDE co-exposure. The similar results shown with human and mouse neutrophils in response to CO2 suggests the mouse model could reflect similar patterns. Their further proof of iNOS activation provides yet another avenue of exploration in future studies.
Of the cytokines tested TNF-α, MCP-1, MIP-2 and KC are all are transcriptionally controlled by NF-kB and AP-1 [35,36,37,38], suggesting possible targets for future work on mechanisms of CO2 inflammation. Very little is currently known about a CO2 “sensor” or signaling pathway in cells. Several studies have reported effects on certain proteins such as ERK and JNK activation [39]. The most promising results for immunological effects have looked at changes to the IKKa and NF-kB alternate pathway signaling (P100 and RelB) in response to CO2 exposure [40, 41]. These results implicate the NF-kB pathway as being important. For this reason, we decided to examine expression of receptors and regulators of this pathway associated with organic dust exposure and LPS. TLR2 and TLR4 are two of the most heavily implicated receptors associated with HDE exposure [3, 28, 29, 42]. TLR4 is also the primary receptor associated with LPS detection [43]. Interestingly we can see that TLR4 mRNA expression tended to be similar for all treatments. TLR2 on the other hand appears to exhibit a pattern of expression that follows a pattern similar to that seen with many of the proinflammatory cytokines we tested for, though other than for LPS + 1000 ppm CO2, the changes were not significant. As TLR2 can bind to LPS [44], the reduction in TLR2 mRNA may suggest a mechanism for reduced LPS responses with higher CO2 but would not explain the increased indicators of inflammation seen with HDE. An examination at the mRNA and protein level of TLR2, TLR4, as well as MD-2 and CD14 [43, 45, 46], all of which are involved in the binding of LPS to TLR2 and TLR4 [47] could assist in further delineating pathway involvements.
More recently an important upstream regulator of NF-kB signaling, TNFAIP3 (A20), was found to be vital in development of protection to asthma in rural children exposed to LPS [30]. While this may suggest that farm work may be protective against some respiratory problems, those working in concentrated animal feed operations receive high endotoxin exposure but show increased susceptibility to a variety of respiratory problems, including asthma [30]. Thus, we examined A20 mRNA expression in response to CO2. While there was little expression of A20 when exposed to LPS alone, there was a significant increase in expression with exposure to HDE, but this was inhibited by CO2 co-exposure, particularly at higher levels. While this result needs to be explored further it does suggest one way in which elevated CO2 may enhance HDE innate immune responses, by reducing expression of the inhibitor A20. Extrapolating to the previously mentioned study [30] such an A20 reduction may result in a non-protective exposure, if A20 is as critical to inhibiting future atopic responses as has been reported. This association of CO2 with worse asthma [48] and wheeze has been shown in several daycare and school studies [10, 11]. It is possible that CO2 exerts changes on innate immunity through NF-kB, but would further suggest that this may be done (or additionally modified) through further upstream pathways that may induce NF-kB (TLRs) or regulate other such inputs and NF-KB such as A20. Studies looking at blocking such regulatory pathways and their effect on NF-kB in this context would be a useful next step.
Finally, we looked at a very general heat shock protein, Hsp72 (HspA1). This particular protein has been shown to be produced in response to a broad array of stress stimuli [49]. Links between HspA1 and control of a more recently examined player in dust inflammation, HMGB1, made us interested in its induction. Interestingly, there were significant reductions in HspA1 when LPS was co-exposed with CO2, even at levels as low as 1000 ppm CO2, and a trend in HDE treated animals in the same direction. As localization of HspA1 is a strong indicator of whether it is a pro or anti-inflammatory agent, longer exposures will need to be done in the future, looking at protein expression and localization. Our initial mRNA results suggest that addition of a CO2 co-exposure appears to reduce HspA1 mRNA when given in conjunction with an inflammatory stimulus.
While this work shows interesting results related to co-exposures to inflammatory agents, additional work is necessary. This work extends previous findings [16] and uses well quantified and described organic dust samples. The work is limited to describing effects in female mice and similar studies need to be replicated in male animals. Further applicability to humans should also be taken with some reservation given established differences in lung anatomy between mice and what this may mean to parameters such as lung function [50]. Future studies with proteomic assessment of these inflammatory measures, assist in determining the breadth and scope of changes CO2 induces. Assessment of NF-kB signaling, including use of its alternate signaling pathway should be conducted. There is of course the need to assess chronic exposures, as well as situations of removing CO2 co-exposure at the start or end of an exposure period, to see if there is a time in which this increased CO2 exposure is critical to alteration of innate immunity. Several of these parameters are part of ongoing work in our lab.
While this study was to look at exposures common in animal feed facilities, there is ample evidence that people are exposed in a number of environments to elevated CO2 that exceed 1000 ppm [15]. This work highlights that immunological exposures may not just be a factor of the introduced inflammatory agent, but also the environmental context in which it is given. The importance of CO2 in respiratory effects may be particularly important where indications of respiratory symptoms in children such as wheeze, asthma [10, 11], and respiratory infection [51] could be exacerbated by increased CO2 exposure in their environment in addition to more commonly tested irritants. We show here that expression of mRNA of some innate immune receptors may be altered with just CO2 alone, but that response to inflammation can be significantly altered by these indoor CO2 levels at 1000 ppm or higher. Interestingly, our current study suggests that these responses will not necessarily be modified in the same manner across different inflammatory agents. This highlights the further need to deduce signaling pathways or commonalities in responses to certain immunological insults to better predict the impact of CO2. If response to an infection, or progression of certain chronic diseases (ex. COPD, asthma) can be impacted by elevated CO2, the possibility exists of ameliorating some effects of these same illnesses by improving ventilation.
Availability of data and materials
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Schiffman SS, Studwell CE, Landerman LR, Berman K, Sundy JS. Symptomatic effects of exposure to diluted air sampled from a swine confinement atmosphere on healthy human subjects. Environ Health Perspect. 2005;113:567–76.
Poole JA, Romberger DJ. Immunological and inflammatory responses to organic dust in agriculture. Curr Opin Allergy Clin Immunol. 2012;12:126–32.
Bauer C, et al. Myeloid differentiation factor 88–dependent signaling is critical for acute organic dust-induced airway inflammation in mice. Am J Respir Cell Mol Biol. 2013;48:781–9.
Schneberger D, Aulakh G, Channabasappa S, Singh B. Toll-like receptor 9 partially regulates lung inflammation induced following exposure to chicken barn air. J Occup Med Toxicol. 2016;11:1.
Donham KJ, Cumro D, Reynolds S. Synergistic effects of dust and ammonia on the occupational health effects of poultry production workers. J Agromedicine. 2002;8:57–76.
Li L, et al. Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. FASEB J. 2005;19:1196–8.
Reeve KA, Peters TM, Anthony TR. Wintertime factors affecting contaminant distribution in a swine farrowing room. J Occup Environ Hyg. 2013;10:287–96.
Satish U, et al. Is CO2 an indoor pollutant? Direct effects of low-to-moderate CO2 concentrations on human decision-making performance. Environ Health Perspect. 2012;120:1671–7.
Fisk WJ. The ventilation problem in schools: literature review. Indoor Air. 2017;27:1039–51.
Carreiro-Martins P, et al. CO2 concentration in day care centres is related to wheezing in attending children. Eur J Pediatr. 2014;173:1041–9.
Carreiro-Martins P, et al. Effect of indoor air quality of day care centers in children with different predisposition for asthma. Pediatr Allergy Immunol. 2016;27:299–306.
Hoge CW, et al. An epidemic of pneumococcal disease in an overcrowded, inadequately ventilated jail. N Engl J Med. 1994;331:643–8.
Hudda N, Fruin SA. Carbon dioxide accumulation inside vehicles: the effect of ventilation and driving conditions. Sci Total Environ. 2018;610–611:1448–56.
Waters MA; Bloom TF, Grajewski B, Deddens J. Measurements of indoor air quality on commercial transport aircraft. In The 9th International Conference on Indoor Air Quality and Climate. pp. 782–787 (NIOSH, 2002).
Jacobson TA, et al. Direct human health risks of increased atmospheric carbon dioxide. Nat Sustain. 2019;2:691–701.
Schneberger D, DeVasure JM, Bailey KL, Romberger DJ, Wyatt TA. Effect of low-level CO2 on innate inflammatory protein response to organic dust from swine confinement barns. J Occup Med Toxicol. 2017;12:1.
Thom SR, Bhopale VM, Hu JP, Yang M. Increased carbon dioxide levels stimulate neutrophils to produce microparticles and activate the nucleotide-binding domain-like receptor 3 inflammasome. Free Radic Biol Med. 2017;106:406–16.
Martrette JM, et al. Effects of prolonged exposure to CO2 on behaviour, hormone secretion and respiratory muscles in young female rats. Physiol Behav. 2017;177:257–62.
Laffey JG, Kavanagh BP. Carbon dioxide and the critically ill—too little of a good thing? Lancet. 1999;354:1283–6.
Wang N, et al. Elevated CO2 selectively inhibits interleukin-6 and tumor necrosis factor expression and decreases phagocytosis in the macrophage. FASEB J. 2010;24:2178–90.
Nichol AD, et al. Hypercapnic acidosis reduces oxidative reactions in endotoxin-induced lung injury. Anesthesiology. 2010;113:116–25.
Abolhassani M, Guais A, Chaumet-Riffaud P, Sasco AJ, Schwartz L. Carbon dioxide inhalation causes pulmonary inflammation. Am J Physiol Cell Mol Physiol. 2009;296:L657–65.
Occupational Safety and Health Adminstration. OSHA Technical Manual (OTM)—section III: Chapter 2: Indoor Air Quality Investigation. United States Department of Labor. 1999.
Petty, S. Summary of Ashrae’s Position on Carbon Dioxide (Co2) Levels in Spaces. Refrig. Air Cond; 2002. 6
Poole JA, et al. Muramic acid, endotoxin, 3-hydroxy fatty acids, and ergosterol content explain monocyte and epithelial cell inflammatory responses to agricultural dusts. J Toxicol Environ Heal Part A. 2010;73:684–700.
Boissy RJ, et al. Shotgun pyrosequencing metagenomic analyses of dusts from swine confinement and grain facilities. PLoS ONE. 2014;9:e95578.
Romberger DJ, Bodlak V, Von Essen SG, Mathisen T, Wyatt TA. Hog barn dust extract stimulates IL-8 and IL-6 release in human bronchial epithelial cells via PKC activation. J Appl Physiol. 2002;93:289–96.
Bailey KL, et al. Toll-like receptor 2 is upregulated by hog confinement dust in an IL-6-dependent manner in the airway epithelium. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1049-54.
Poole JA, et al. Toll-like receptor 2 regulates organic dust-induced airway inflammation. Am J Respir Cell Mol Biol. 2011;45:711–9.
Schuijs, M. J. et al. Farm dust and endotoxin protect against allergy through A20 induction in lung epithelial cells. Science (80-. ). 349, 1106–1110 (2015).
von Essen SG, Banks DE. Life-long exposures on the farm, respiratory symptoms, and lung function decline. Chest. 2009;136:662–3.
Raşid O, Chirita D, Iancu AD, Stavaru C, Radu DL. Assessment of routine procedure effect on breathing parameters in mice by using whole-body plethysmography. J Am Assoc Lab Anim Sci. 2012;51:469–74.
Patton LM, Saggart BS, Ahmed NK, Leff JA, Repine JE. Interleukin-1 beta-induced neutrophil recruitment and acute lung injury in hamsters. Inflammation. 1995;19:23–9.
Vieira SM, et al. A crucial role for TNF-α in mediating neutrophil influx induced by endogenously generated or exogenous chemokines, KC/CXCL1 and LIX/CXCL5: RESEARCH PAPER. Br J Pharmacol. 2009;158:779–89.
Orlichenko LS, et al. Transcriptional regulation of CXC-ELR chemokines KC and MIP-2 in mouse pancreatic acini. Am J Physiol Gastrointest Liver Physiol. 2010;299:G867–76.
Shyy JY, et al. The cis-acting phorbol ester "12-O-tetradecanoylphorbol 13-acetate"-responsive element is involved in shear stress-induced monocyte chemotactic protein 1 gene expression. Proc Natl Acad Sci U S A. 1995;92:8069–73.
Nicholson WJ, Slight J, Donaldson K. Inhibition of the transcription factors NF-κB and AP-1 underlies loss of cytokine gene expression in rat alveolar macrophages treated with a diffusible product from the spores of Aspergillus fumigatus. Am J Respir Cell Mol Biol. 1996;15:88–96.
Redhu NS, Saleh A, Halayko AJ, Ali AS, Gounni AS. Essential role of NF-kB and AP-1 transcription factors in TNF-α-induced TSLP expression in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2011;300:479–85.
Welch LC, et al. Extracellular signal-regulated kinase (ERK) participates in the hypercapnia-induced Na, K-ATPase downregulation. FEBS Lett. 2010;584:3985–9.
Keogh CE, et al. Carbon dioxide-dependent regulation of NF-κB family members RelB and p100 gives molecular insight into CO2-dependent immune regulation. J Biol Chem. 2017;292:11561–71.
Cummins EP, et al. NF- B Links CO2 sensing to innate immunity and inflammation in mammalian cells. J Immunol. 2010;185:4439–45.
Charavaryamath C, Keet T, Aulakh GK, Townsend HG, Singh B. Lung responses to secondary endotoxin challenge in rats exposed to pig barn air. J Occup Med Toxicol. 2008;3:24.
Poltorak A, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 Gene. Science. 1998;282:2085–8.
Yang R-B, et al. Toll-like receptor-2 mediates lipopolysaccharide-induced cellular signalling. Nature. 1998;395:284–8.
Shimazu R, et al. MD-2, a molecule that confers lipopolysaccharide responsiveness on toll-like receptor 4. J Exp Med. 1999;189:1777–82.
van Bergenhenegouwen J, et al. TLR2 & Co: a critical analysis of the complex interactions between TLR2 and coreceptors. J Leukoc Biol. 2013;94:885–902.
Dziarski R, Wang Q, Miyake K, Kirschning CJ, Gupta D. MD-2 enables Toll-like receptor 2 (TLR2)-mediated responses to lipopolysaccharide and enhances TLR2-mediated responses to Gram-positive and Gram-negative bacteria and their cell wall components. J Immunol. 2001;166:1938–44.
Mi YH, Norbäck D, Tao J, Mi YL, Ferm M. Current asthma and respiratory symptoms among pupils in Shanghai, China: influence of building ventilation, nitrogen dioxide, ozone, and formaldehyde in classrooms. Indoor Air. 2006;16:454–64.
Williams JHH, Ireland HE. Sensing danger–Hsp72 and HMGB1 as candidate signals. J Leukoc Biol. 2008;83:489–92.
Irvin CG, Bates JHT. Measuring the lung function in the mouse: the challenge of size. Respir Res. 2003;4:1.
Kovesi T, et al. Indoor air quality and the risk of lower respiratory tract infections in young Canadian Inuit children. Can Med Assoc J. 2007;177:155–60.
Acknowledgements
The authors would like to thank Dr. Mabood Quereshi for helping with blood gas analysis on our mice, and Dr. Todd Wyatt for provision of dust samples.
Funding
We would also like to thank the University of Saskatchewan College of Medicine Research Award (CoMRAD) for providing the financial support for the presented study (awarded to “Effect of low dose Carbon Dioxide on barn dust and endotoxin induced Lung Inflammation”).
Author information
Authors and Affiliations
Contributions
DS conceived the study and study design, performed treatments and most assays, collected and analyzed data. UP helped with data collection, analysis, and animal treatment. BT helped with data collection, sample processing and analysis of data and helped editing the manuscript. SK supervised the project and helped with data analysis, statistical measures, and in writing and editing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All experiments and procedures were approved by and conducted according to the protocols of the University of Saskatchewan Committee on Animals Care Assurance (AUP 20160104).
Consent for publication
Not applicable.
Competing interests
The authors declare they have no actual or potential competing financial interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Schneberger, D., Pandher, U., Thompson, B. et al. Effects of elevated CO2 levels on lung immune response to organic dust and lipopolysaccharide. Respir Res 22, 104 (2021). https://doi.org/10.1186/s12931-021-01700-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-021-01700-4