
Abstract
Background
Observational data under real-life conditions in idiopathic pulmonary fibrosis (IPF) is scarce. We explored anti-fibrotic treatment, disease severity and phenotypes in patients with IPF from the Swedish IPF Registry (SIPFR).
Methods
Patients enrolled between September 2014 and April 2020 and followed ≥ 6 months were investigated. Demographics, comorbidities, lung function, composite variables, six-minute walking test (6MWT), quality of life, and anti-fibrotic therapy were evaluated. Agreements between classification of mild physiological impairment (defined as gender-age-physiology (GAP) stage 1) with physiological and composite measures of severity was assessed using kappa values and their impact on mortality with hazard ratios. The factor analysis and the two-step cluster analysis were used to identify phenotypes. Univariate and multivariable survival analyses were performed between variables or groups.
Results
Among 662 patients with baseline data (median age 72.7 years, 74.0% males), 480 had a follow up ≥ 6 months with a 5 year survival rate of 48%. Lung function, 6MWT, age, and BMI were predictors of survival. Patients who received anti-fibrotic treatment ≥ 6 months had better survival compared to untreated patients [p = 0.007, HR (95% CI): 1.797 (1.173–2.753)] after adjustment of age, gender, BMI, smoking status, forced vital capacity (FVC) and diffusion capacity of carbon monoxide (DLCO). Patients with mild physiological impairment (GAP stage 1, composite physiological index (CPI) ≤ 45, DLCO ≥ 55%, FVC ≥ 75%, and total lung capacity (TLC) ≥ 65%, respectively) had better survival, after adjustment for age, gender, BMI and smoking status and treatment. Patients in cluster 1 had the worst survival and consisted mainly of male patients with moderate-severe disease and an increased prevalence of heart diseases at baseline; Cluster 2 was characterized by mild disease with more than 50% females and few comorbidities, and had the best survival; Cluster 3 were younger, with moderate-severe disease and had few comorbidities.
Conclusion
Disease severity, phenotypes, and anti-fibrotic treatment are closely associated with the outcome in IPF, with treated patients surviving longer. Phenotypes may contribute to predicting outcomes of patients with IPF and suggest the patients’ need for special management, whereas single or composite variables have some limitations as disease predictors.
Similar content being viewed by others

Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive fibrosing interstitial lung disease (ILD) of unknown cause[1,2,3].The disease is characterized by an aberrant accumulation of fibrotic tissue in the lung parenchyma, resulting in extensive alterations of lung structure and function and leads finally to respiratory failure and death [2, 4, 5]. Long-term observational studies in clinically diverse IPF populations from all over the world are increasing [1, 6,7,8,9,10,11,12,13,14,15] and provide us with important information on disease behaviour, management, and effectiveness of approved treatments.
Forced vital capacity (FVC), diffusion capacity for carbon monoxide (DLCO), composite physiological index (CPI) [7, 16] and GAP (gender, age and physiology) stage have been used to define the severity of IPF and to predict mortality[7, 17, 18]. In a recent study, a six-minute walking test (6MWT) was proved to be important predictors for survival [19]. GAP stage 1 has been commonly used as a mild physiological impairment criterion [7]. However, the impact of these physiological variables on disease progression and mortality in patients with mild or more advanced disease is largely unknown. Furthermore, we have previously indicated potential gender differences in patients with IPF [20]. Thus, an unsupervised cluster analysis may provide novel insights into the phenotypes of IPF with potential prognostic significance. Progress in the management of IPF has been made with the introduction of two antifibrotics, pirfenidone and nintedanib, which have been shown to reduce the rate of disease progression [21, 22]. However, strict inclusion and exclusion criteria in clinical trials may limit the generalizability of the results in real clinical settings. For instance, patients with comorbidities, lower lung function, and concomitant medications have been commonly excluded from participation in randomized clinical trials [23,24,25]. Therefore, many questions remain about the generalizability of these findings to a wider IPF population.
Given the lack of knowledge on disease course and mid- to long-term outcomes in IPF, our aims were to explore characteristics, disease severity, phenotype, and anti-fibrotic treatments in patients with IPF under real-life conditions and to assess associations to mortality. We also wanted to ascertain whether further characterization may help patients with IPF and aid the development of personalized management and/or therapy. Additionally, we compared our data with other registries to highlight clinical and geographical variability.
Patients and methods
Study population
The Swedish IPF Registry (SIPFR) is a nationwide registry collecting comprehensive longitudinal data of IPF patients and implemented in 22 respiratory medicine units across Sweden [13, 19, 20]. The SIPFR also includes patients diagnosed before the registry was launched in 2014. The registry relies on a web-based platform (Granitics Unify Med, Granitic Ltd, Espoo, Finland) which allows secure data collection at each respective center. Data entries are made by nurses and physicians at each site, and the quality of the data is evaluated and improved by source data verification performed by the registry coordinator (LC). To be eligible for inclusion in the registry, the patient has to have a confirmed diagnosis of IPF according to the national and international guidelines [13, 26, 27] by a specialist in respiratory medicine either at a university hospital or a local hospital. The registry applies no explicit exclusion criteria, thereby reducing selection bias. We included all patients enrolled in the registry from Sep 2014 until April 2020, and the patients followed ≥ 6 months were enrolled in survival analyses. The outcome of death was defined as patients dying or receiving a lung transplant during the observation period. Patients who were alive at the last visit date during the follow-up period of this study were censored and classified as survivors. The primary survival time was calculated from the enrolment date, with baseline data. Secondary survival time was calculated from the diagnosis date, without matched baseline data.
Variables
Data covering demographics, self-reported comorbidities, lung function, 6MWT, radiology, quality of life (assessed with the King's Brief Interstitial Lung Disease Questionnaire (K-BILD)) and anti-fibrotic therapy were included [13]. Charlson Comorbidity Index (CCI) was calculated with an ad hoc modified formula, i.e. coronary artery disease, other cardiovascular diseases, diabetes, arterial hypertension, chronic obstructive pulmonary disease, and acid reflux gave one point each and history of cancer gave two points. The composite physiologic index (CPI) was calculated using the formula: CPI = 91.0—(0.65 × % predicted DLCO)—(0.53 × % predicted FVC) + (0.34 × % predicted FEV1). The gender-age-physiology (GAP) index was extrapolated for each patient with available data in the registry using the variables of the scoring system combining gender, age, and lung physiology (FVC and DLCO) and classified as GAP stage I (0–3 points), GAP stage II (4–5 points), or GAP stage III (6–8 points). Patients with smoking history included ex-smokers and/or current smokers. Patients were considered as "incident” cases if diagnosed within 6 months from inclusion, while patients with a diagnosis of more than 6 months from inclusion were considered as "prevalent". Each patient's IPF diagnosis was evaluated by "clinic radiological", "thoracoscopic biopsy", "open lung biopsy", or " multidisciplinary conference". Exposure was ascertained by the answers to self-reported questions, such as microbes, particles from the atmosphere, irritants, pollutants, allergens, and pathogens [28]. Data collected at 6 months prior to or after the consent date in this study was considered as baseline data.
Anti-fibrotic therapy
Treatment status was classified into an anti-fibrotic treatment group (receiving anti-fibrotic therapy after diagnosis ≥ 6 months) and untreated group (no treatment or anti-fibrotic therapy after diagnosis < 6 months) [10]. The anti-fibrotic treatment group was further classified into three groups: (1) patients only treated with nintedanib ≥ 6 months; (2) patients only treated with pirfenidone ≥ 6 months; (3) patients switched between pirfenidone (≥ 6 months) and nintedanib (≥ 6 months).
Classification of disease severity using different mild definitions
Disease severity was evaluated as mild and moderate to severe physiological impairment using different criteria [7]. We compared GAP criteria for mild physiological impairment (GAP stage 1) against other proposed criteria: FVC ≥ 75% (exploratory analysis for ≥ 90%, ≥ 80%, and ≥ 70%); DLCO ≥ 55% (exploratory analysis of ≥ 60%, ≥ 50%, and ≥ 45%), TLC ≥ 65% (exploratory analysis for ≥ 75%, ≥ 70%, and ≥ 60%), and CPI ≤ 45 (exploratory analysis CPI ≤ 30, CPI ≤ 40, CPI ≤ 50) exploring the agreement in classifications and relationship with disease outcomes (data not shown).
Cluster analysis on baseline SIPFR data with disease severity
A two-step cluster analysis was used to differentiate the patients into distinct phenotypes. Input variables for the cluster analysis were based on the basis of factor analysis, including baseline characteristics (age, gender, BMI), comorbidities (the number of comorbidities, CCI, acid reflux and cardiovascular diseases), and severity (cut-off level: GAP stage 1, FVC ≥ 75%, DLCO ≥ 55%, TLC ≥ 65% and CPI ≤ 45). The Kaiser–Meyer–Olkin (KMO) value of the scale (> 0.6) and the Bartlett test value of sphericity (p < 0.05) were used to determine the sampling adequacy for factor analysis. Cluster analysis was carried out by using a two steps process [29]. First, the number of clusters were pre-evaluated by Ward hierarchical cluster analysis and factor analysis. Then, the K-means cluster analysis was carried out by using the pre-specified number of clusters. The stepwise discriminant analysis was performed to identify variables discriminating amongst the clusters. For validation, we carried out the leave-one-out method to ensure the stability and repeatability of the cluster model.
Other statistical analyses
Descriptive analysis was performed with medians with interquartile ranges (IQR), or mean ± standard deviation (SD) for continuous variables, and counts with percentages for categorical variables. Missing data, primarily due to data not being registered, was not estimated but was removed from the denominator in calculation. Comparisons between groups were performed using t-test, ANOVA, Mann–Whitney U test, Chi-squared test, or pairwise comparison as appropriate. Univariable and multivariable Cox regression models were performed to investigate the relationships between baseline variables. All models were examined for assumptions of normality of the residuals and homogeneity of variance by examination of residual plots. Kaplan–Meier estimates and a log rank test for mortality were performed to calculate mortality by selected variables. The log-rank test was used to test the differences in survival between the two groups of patients. Comparisons with other IPF cohorts are descriptive and based on published data [1, 6, 9, 10]. All statistical analyses were performed using IBM’s SPSS Statistics version 21 (SPSS, Chicago, IL, USA), Stata 13.1 software package (StataCorp LP, College Station, TX, USA), and GraphPad Prism version 6.0 (GraphPad Software, San Diego, CA, USA). We considered p < 0.05 as statistically significant.
Results
SIPFR cohort
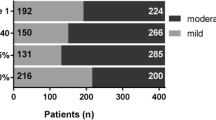
Included patients (n = 662, median age 72.7 years, males 74.0%) were collected between Sep. 2014 and Apr. 2020 (Table 1). Almost two thirds of patients reported a history of smoking, with approximately 60% of patients being ex-smokers, and 24 patients (4%) current smokers (Table 1). The time from IPF diagnosis to enrolment was 2 months. GAP stage was available for 384 patients, and the distribution of GAP stage was I (51.0%), II (40.9%) and III (8.1%). The median value of CCI was 4. The most frequently reported group of comorbidities were cardiovascular diseases, with 54.5% of patients reporting at least one cardiovascular disease (these included hypertension 35.6% of all patients, other cardiovascular diseases 31.6%, and ischaemic heart disease 20.2%) (Fig. 1). Approximately over 70% patients reported at least one comorbidity, and more than 40% of patients had two or three comorbidities at baseline (Fig. 1). According to the primary survival timeline, 480 patients were followed ≥ 6 months from enrolment date (median (interquartile range) 28 (15–46.5) months), while the secondary survival timeline, included 540 patients who had been followed ≥ 6 months from diagnosis date (20 (12–32) months).

Prevalence of comorbidities in the SIPFR. The number and percent of a single comorbidity, b the combination of comorbidities. COPD chronic obstructive pulmonary disease
During the follow-up time, 195 had died and 23 had undergone lung transplants. The increasing cumulative rate of death from the diagnosis date in one to five years was 7, 16, 30, 39, and 48%, respectively. The cumulative rate from the enrolment date in one to five years was 12, 32, 50, 62 and 78% (Fig. 2a). Figure 2 displays the different cumulative rates of death according to the GAP stage. A trend of shorter survival in male patients compared to women was observed (median: 35.0 vs. 44 months, log rank p = 0.067). In the univariate Cox analysis of baseline factors, decreased lung function, six-minute walking distance (6MWD), K-BILD, BMI, age, smoking history, CPI and GAP, were significant predictors of mortality (Table 2).

Kaplan–Meier analysis for survival in the cohort and in GAP stages. Kaplan–Meier analysis for mortality in the SIPFR cohort according to a time from the enrolment; b time from the diagnosis; c and d GAP stage GAP gender, age, physiology
Comparison with other IPF-registries
Comparison of the SIPFR with the Australian IPF Registry [6] Finnish IPF Registry [10] (FinnishIPF, n = 453), the German INSIGHTS-IPF Registry [9] (INSIGHTS, n = 623) and European IPF registry [1] (EurIPFreg, n = 525) is outlined in Table 3. Age and gender distributions were similar in all registries, whereas patients in SIPFR had lower BMI. Baseline lung functions in SIPFR were more preserved than in INSIGHTS- and EurIPF -registries, but worse than in AIPFR and the FinnishIPF. The 6MWD was similar in SIPFR and AIPFR, whereas the distance was greater than the one reported in the INSIGHTS- and EurIPF-registry. Only two registries presented data on TLC% at baseline. Swedish IPF registry presented a lower TLC% compared to EurIPFreg. The cumulative rate of death data from reports were available for SIPFR, AIPFR and FinnishIPF, with one-year mortality of 7% 5%, and 5%, respectively.
Anti-fibrotic therapy
Among the 540 patients with a follow up of ≥ 6 months from diagnosis, 347 (64.3%) received anti-fibrotic treatment for ≥ 6 months from diagnosis date, either with pirfenidone or nintedanib (33.9% and 26.3% respectively). A minor group of patients (4.1%) had switched treatment. Patients on anti-fibrotic therapy were younger compared to those who did not receive treatment (p = 0.018, Table 4). The median age at diagnosis of the “switched” group, “pirfenidone treated” group, and “nintedanib treated” group were 67.0 years, 72.0 years, and 72.0 years, respectively. However, the difference in age at diagnosis was not statistically significant (p = 0.056). Two thirds of patients (n = 218) had a smoking history (Table 4), with 3 current smokers receiving pirfenidone, 2 current smokers nintedanib, and 1 current smoker had switched treatment. The median age at diagnosis of the “switched” group, “pirfenidone treated” group, and “nintedanib treated” group were 67.0 years, 72.0 years, and 72.0 years, respectively. However, the difference in age at diagnosis was not statistically significant (p = 0.056). Two thirds of patients (n = 218) had a smoking history (Table 4), with 3 current smokers receiving pirfenidone, 2 current smokers nintedanib, and 1 current smoker had switched treatmentFVC % predicted and GAP stage did not differ between patients treated with anti-fibrotic and those who did not receive treatment (Table 4). GAP stage did not differ between nintedanib and pirfenidone treated patients (p = 0.807 and p = 0.116, respectively). Kaplan–Meier analysis showed improved survival in patients on anti-fibrotic therapy compared to untreated patients in all and in patients with GAP stage ≥ 2 ((log rank p = 0.037 and p = 0.034, Fig. 3a, b). When we separately analyzed the two anti-fibrotic drugs, we found that patients receiving nintedanib had better survival compared to untreated patients in all and in patients with GAP stage ≥ 2 (log rank p = 0.034 and p = 0.025, respectively, Fig. 3c, d). In addition, patients switching treatment also had a better survival compared to untreated patients (log rank p = 0.026, Fig. 3c). In the multivariate Cox regression analysis, patients with anti-fibrotic treatment still had a better prognosis than those without (p = 0.007, HR (95% CI): 1.797 (1.173–2.753)) after adjustment of age, gender, BMI, smoking status, FVC%, and DLCO%.

Kaplan–Meier analysis for survival in treatment. Kaplan–Meier analysis for mortality in the SIPFR cohort according to a, b patients with and without anti-fibrotic treatment in patients in all and GAP stage over 1; c, d patients with anti-fibrotic treatment (nintedanib, pirfenidone, switched treatment) and untreated in patients in all and GAP stage over 1
Classification of disease severity
Altogether 243 patients were followed ≥ 6 months, after exclusion of patients with missing data on FVC%, DLCO%, TLC%, CPI, and GAP stage. Mild physiological impairment defined by GAP stage 1 had a good agreement with CPI ≤ 45 (kappa value (k) = 0.62), and moderate agreement with DLCO ≥ 55% (k = 0.58), FVC ≥ 75% (k = 0.50), and TLC ≥ 65% (k = 0.47). Mild physiological impairment at baseline (DLCO ≥ 55%, TLC ≥ 65%, CPI ≤ 45, FVC ≥ 75% and GAP stage 1, respectively) was predictive of better survival compared to patients with moderate-severe disease in univariable analysis, as well as multivariable Cox analysis after adjustment of age, gender, BMI, smoking status and anti-fibrotic use (Table 5).
Cluster analysis
A two-step cluster analysis was performed with 15 variables selected on basis of baseline characteristics and severity (Table 6). Altogether, 164 patients were followed ≥ 6 months after exclusion of patients with missing data. Factor analysis showed the selected variables were suitable for further analysis, since the KMO measure of sampling adequacy was 0.612 and Bartlett's Test of sphericity demonstrated a significant difference (p < 0.001). Three clusters were identified in Fig. 4 A-D; patients in cluster 1 (n = 55) consisted mostly of heart diseases (96.4%), mainly male patients (87.3%) with moderate-severe disease at baseline; Cluster 2 (n = 70) was characterized by mild disease with more than 50% females and few comorbidities; Cluster 3 (n = 39) were younger, moderate-severe patients with few comorbidities. The discriminant analysis showed function 1 to mainly consist of the disease severity variables, while function 2 mainly contained comorbidity variables (Fig. 4e). Kaplan Meier analysis of clusters showed that patients in cluster 1 had a worst survival compared to cluster 2 and 3 (log rank p < 0.001 and p = 0.036), whereas patients in cluster 2 had the best survival compared to cluster 3 (log rank p = 0.017) (Fig. 4f). Multivariable Cox analysis showed that cluster 1 (HR: 3.154, 95%CI (1.855–5.364), p < 0.001) and cluster 2 (HR: 0.291, 95%CI (0.160–0.528, p < 0.001)) were predictors of survival, after adjustment of anti-fibrotic use.

Characteristics of clusters, distribution and survival. In a–d shown the basic characteristics of clusters; e The distribution in clusters, largest absolute correlation between each variable and any discriminant function in Function 1 (GAP stage 1, CPI ≤ 45% TLC ≥ 65%, DLCO ≥ 55%, males, LpSaO2, and 6MWD) and in Function 2 (CCI, the number of comorbidities, heart diseases, FVC ≥ 75%, age, smoking history, acid reflux and BMI); f Kaplan–Meier analysis for mortality in clusters
For validation, we carried out discriminant analysis by the leave-one-out method to ensure stability and repeatability of the model. This method showed that 95.7% of the originally grouped cases were correctly classified, and 90.9% of the cross-validated grouped cases were correctly classified.
Discussion
Similar to other IPF-registries, we demonstrate a heterogeneous patient cohort with respect to age, disease severity, and co-morbidities. The cumulative 1, 2, 3, 4 and 5 year mortality was 7, 16, 30, 39 and 48%, respectively. We were able to confirm that lung function, 6MWD and BMI are significant predictors of mortality [17, 18, 24, 30]. Patients receiving anti-fibrotic therapy had better survival than untreated patients in all and in GAP stage above 1. We investigated the agreement of the GAP stage with single and composite measures of physiological impairment and found that patients with mild physiological impairment have better survival than patients with moderate-severe disease. Three clusters were identified of which one, consisting of males with heart diseases, multiple comorbidities, and high GAP stage, had the worst survival.
One of the important findings in this study is a stratification for a standardized approach to disease severity. Potential stratifications of disease severity have been a widely discussed topic in the community for a long time. Heterogeneity in IPF is multidimensional. Although it is difficult to define the "best" definition of disease stratification, classification requires consideration of these disparate domains. Some of these characteristics have been incorporated in indexes of different domains such as the GAP-index and the composite physiological index, CPI. Patient registries give us the opportunity to include a heterogeneous group of patients with wide ranges of baseline physiology and disease severity. Our results showed that CPI ≤ 45, DLCO ≥ 55%, FVC ≥ 75%, and TLC ≥ 65%, agreed well with GAP stage 1 for staging of mild physiological impairment. This was a first study to define the mild physiological impairment by TLC% in a large scale of IPF patients. Moreover, we also showed that the presence of mild impairment at baseline was predictive of better survival compared to patients with moderate-severe disease on univariable as well as multivariable Cox analysis adjusting for age, gender, BMI, smoking status, and anti-fibrotic use.
To the best of our knowledge, no cluster analysis has been done on the IPF registry cohorts using longitudinal data so far. We report an explorative analysis of potential phenotypes of IPF patients in SIPFR, including our newly defined "mild" IPF classification, comorbidities, and demographic data. More than 40% of the patients had two or three comorbidities. Although we did not find a significant association between comorbidities and outcome in the univariate analysis, comorbidities showed high predictor importance in the cluster analysis. This may reflect the real-world IPF patient since single or composite variables have some limitations as disease predictors. Three clusters were identified, with GAP, comorbidities, and gender deemed important factors between clusters. Heart diseases and severity factors had high predictor importance value in our cluster analysis. As shown in other studies, IPF and heart disease may share several risk factors, and IPF has been associated with atherosclerosis [31,32,33]. The cluster comprising moderately to severe diseased males with heart diseases had worst survival, and mild disease cluster with less comorbidities had best survival. Thus, phenotypes may offer a novel multidimensional approach for predicting outcomes of patients with IPF and suggest patients’ need for special management.
Registries provide the opportunity to study disease progression in patients with anti-fibrotic treatment [10, 30]. In Sweden, anti-fibrotic drugs are completely reimbursed, which results in a large number of patients being on treatment [34]. Thus, approximately 65% of the patients received anti-fibrotic treatment in our study, which is considerably more than in Germany (44%) [9], Finland (26%) [10], and Australia (23%) [6]. The present study shows that patients on anti-fibrotic therapy appear to survive longer than untreated patients, a result similar to what other registries have reported [6, 8, 20]. In order to avoid a potential bias in mortality analysis, we showed that there were no significant differences in baseline lung function between anti-fibrotic treated and untreated groups. Furthermore, we adjusted the potential confounders (age, gender, BMI, smoking history, and anti-fibrotic use) at baseline to identify the association between low lung function parameters and mortality. The curves of antifibrotic use and untreated could be clearly distinguished in the Cox model. Although the effect driven by lung function decline was not included in the current baseline project, it is a focus in an upcoming project.
Interestingly, twenty-two patients had been followed ≥ 6 months, who received the switched antifibrotics treatment (Table 4). Reasons for switching antifibrotics is not a dedicated variable in the registry, resulting in a risk of missing disease progression as a cause to the switch. This might be the case for some of these patients in our dataset. In our experience, side effects make up the main reason and are reported for some of the patients in this group. Disease progression is, in our experience, a minor reason for switching treatment, simply because there are no defined definition of stable or progressive disease when it comes to the individual patient. It is important to clarify that our registry, like all other registries, is not designed to compare treatment effects. Differences in characteristics of the compared groups, non-randomization, other undetected confounders, and missing registry data are important factors that require a cautious interpretation of these results. For the purpose of studying treatment effects, well-powered randomized controlled trials are the only gold standard. Thus, lack of improved survival, or survival benefits, does not imply the absence or presence of a true, underlying difference between the groups. The favourable effect on survival of the “switched group” may only be hypothesis-generating and interpreted with caution since pirfenidone and nintedanib have different mechanisms of action. The idea of sequential treatment strategies in IPF has been discussed before, with few retrospective studies on small cohorts, supporting such strategy [35,36,37].
A number of limitations are worth noting. Firstly, no estimates were made for missing categorical and continuous data and missing data was not involved in further analysis. Secondly, while other studies and registries have highlighted the poorer prognosis in patients with pulmonary hypertension and/or lung cancer, our registry does not collect that type of data, potentially missing other explanatory variables for prognosis. Thirdly, prevalent patients, consisting of 35%, may have a slower disease progression [10, 11], increasing the risk of bias in the survival analysis. Finally, we considered the two timelines from diagnosis and enrolment and adjusted the confounders, but residual confounding might be possible and may have affected the regression analysis [38]. In addition, the effect of smoking on IPF behaviour was not deeply analysed, due to the small numbers of current smokers (n = 24). Only 19 of 24 (4%) patients had been followed ≥ 6 months. Hence, only smoking history (ex-and current smokers) were evaluated in this study. The influence of current smoking on the disease will require a larger cohort. Potential preventive effects of antifibrotics on hospitalizations and exacerbations and thus also on mortality were not analysed in this paper. Currently, data related to exacerbations in the Swedish IPF-registry is limited and needs further distinguishment and collection (e.g. distinguishing hospitalizations related to comorbidities from IPF related exacerbations).
Conclusion
We conclude that both disease severity and phenotype are closely associated with outcome in IPF which may be important for disease behaviour and follow-up. Survival was significantly higher in IPF patients with anti-fibrotic therapy, especially in patients with moderate-severe disease. Mild physiological impairments could be defined by TLC ≥ 65% in SIPFR. IPF patients with mild physiological impairment have better survival than patients with moderate-severe disease. Phenotypes may contribute to predicting outcomes of patients with IPF and suggest the patients’ need for special management, whereas single or composite variables have some limitations as disease predictors. Our results provide an insight into the characteristics, management, and outcome of IPF-patients in real life.
Role of the sponsors
The study funders/sponsors had no role in this study, including the design, collection, management, analysis, writing, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Availability of data and materials
No.
Abbreviations
- AIPFR:
-
The Australian idiopathic pulmonary fibrosis registry
- BIC:
-
The Schwarz Bayesian Criterion
- BMI:
-
Body mass index
- CCI:
-
The Charlson Comorbidity Index
- COPD:
-
Chronic obstructive pulmonary disease
- CPI:
-
Composite physiological index
- DLCO%:
-
Diffusing capacity of carbon monoxide, % of predicted
- EurIPFreg:
-
European idiopathic pulmonary fibrosis registry
- FEV1:
-
Forced expiratory volume in 1 s
- FEV1%:
-
Forced expiratory volume in 1 s, % of predicted
- FinnishIPF:
-
The Finnish idiopathic pulmonary fibrosis registry
- FVC:
-
Forced vital capacity
- FVC%:
-
Forced vital capacity, % of predicted
- GAP:
-
Gender-age-physiology index for idiopathic pulmonary fibrosis
- IPF:
-
Idiopathic pulmonary fibrosis
- IQR:
-
Interquartile ranges (IQR)
- INSIGHTS:
-
The German investigating significant health trends in idiopathic pulmonary fibrosis registry
- k:
-
Kappa value
- K-BILD:
-
King's brief interstitial lung disease health status questionnaire
- KMO:
-
The Kaiser–Meyer–Olkin (KMO) value of the scale
- LC:
-
The registry coordinator
- L-SpO2 :
-
Lowest oxygen saturation during 6-min walking test
- SIPFR:
-
The Swedish idiopathic pulmonary fibrosis registry
- TLC %:
-
Total lung capacity, % of predicted
- QoL:
-
Quality of life
- 6MWD:
-
6-Min walking distance
- 6MWT:
-
6-Min walking test
- UIP:
-
Usual interstitial pneumonia
References
Guenther A, Krauss E, Tello S, Wagner J, Paul B, Kuhn S, Maurer O, Heinemann S, Costabel U, Barbero MAN, et al. The European IPF registry (eurIPFreg): baseline characteristics and survival of patients with idiopathic pulmonary fibrosis. Respir Res. 2018;19:141.
Skold CM, Bendstrup E, Myllarniemi M, Gudmundsson G, Sjaheim T, Hilberg O, Altraja A, Kaarteenaho R, Ferrara G. Treatment of idiopathic pulmonary fibrosis: a position paper from a Nordic expert group. J Intern Med. 2017;281:149–66.
Martinez FJ, Chisholm A, Collard HR, Flaherty KR, Myers J, Raghu G, Walsh SL, White ES, Richeldi L. The diagnosis of idiopathic pulmonary fibrosis: current and future approaches. Lancet Respir Med. 2017;5:61–71.
Sgalla G, Iovene B, Calvello M, Ori M, Varone F, Richeldi L. Idiopathic pulmonary fibrosis: pathogenesis and management. Respir Res. 2018;19:32.
Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389:1941–52.
Jo HE, Glaspole I, Grainge C, Goh N, Hopkins PM, Moodley Y, Reynolds PN, Chapman S, Walters EH, Zappala C, et al. Baseline characteristics of idiopathic pulmonary fibrosis: analysis from the Australian Idiopathic Pulmonary Fibrosis Registry. Eur Respir J. 2017. https://doi.org/10.1183/13993003.01592-2016.
Jo HE, Glaspole I, Moodley Y, Chapman S, Ellis S, Goh N, Hopkins P, Keir G, Mahar A, Cooper W, et al. Disease progression in idiopathic pulmonary fibrosis with mild physiological impairment: analysis from the Australian IPF registry. BMC Pulm Med. 2018;18:19.
Behr J, Prasse A, Wirtz H, Koschel D, Pittrow D, Held M, Klotsche J, Andreas S, Claussen M, Grohe C, et al. Survival and course of lung function in the presence or absence of antifibrotic treatment in patients with idiopathic pulmonary fibrosis: long-term results of the INSIGHTS-IPF registry. Eur Respir J. 2020. https://doi.org/10.1183/13993003.02279-2019.
Behr J, Kreuter M, Hoeper MM, Wirtz H, Klotsche J, Koschel D, Andreas S, Claussen M, Grohe C, Wilkens H, et al. Management of patients with idiopathic pulmonary fibrosis in clinical practice: the INSIGHTS-IPF registry. Eur Respir J. 2015;46:186–96.
Kaunisto J, Salomaa ER, Hodgson U, Kaarteenaho R, Kankaanranta H, Koli K, Vahlberg T, Myllarniemi M. Demographics and survival of patients with idiopathic pulmonary fibrosis in the FinnishIPF registry. ERJ Open Res. 2019. https://doi.org/10.1183/23120541.00170-2018.
Tran T, Sterclova M, Mogulkoc N, Lewandowska K, Muller V, Hajkova M, Kramer MR, Jovanovic D, Tekavec-Trkanjec J, Studnicka M, et al. The European MultiPartner IPF registry (EMPIRE): validating long-term prognostic factors in idiopathic pulmonary fibrosis. Respir Res. 2020;21:11.
Zurkova M, Kriegova E, Kolek V, Lostakova V, Sterclova M, Bartos V, Doubkova M, Binkova I, Svoboda M, Strenkova J, et al. Effect of pirfenidone on lung function decline and survival: 5-yr experience from a real-life IPF cohort from the Czech EMPIRE registry. Respir Res. 2019;20:16.
Ferrara G, Carlson L, Palm A, Einarsson J, Olivesten C, Skold M. R SIPF: Idiopathic pulmonary fibrosis in Sweden: report from the first year of activity of the Swedish IPF-Registry. Eur Clin Res J. 2016. https://doi.org/10.3402/ecrj.v3.31090.
Wuyts WA, Dahlqvist C, Slabbynck H, Schlesser M, Gusbin N, Compere C, Maddens S, Kirchgaessler KU, Bartley K, Bondue B. Baseline clinical characteristics, comorbidities and prescribed medication in a real-world population of patients with idiopathic pulmonary fibrosis: the PROOF registry. BMJ Open Respir Res. 2018;5:e000331.
Wuyts WA, Dahlqvist C, Slabbynck H, Schlesser M, Gusbin N, Compere C, Maddens S, Lee YC, Kirchgaessler KU, Bartley K, Bondue B. Longitudinal clinical outcomes in a real-world population of patients with idiopathic pulmonary fibrosis: the PROOF registry. Respir Res. 2019;20:231.
Latsi PI, du Bois RM, Nicholson AG, Colby TV, Bisirtzoglou D, Nikolakopoulou A, Veeraraghavan S, Hansell DM, Wells AU. Fibrotic idiopathic interstitial pneumonia—the prognostic value of longitudinal functional trends. Am J Respir Crit Care Med. 2003;168:531–7.
Mogulkoc N, Brutsche MH, Bishop PW, Greaves SM, Horrocks AW, Egan JJ. Greater Manchester Pulmonary Fibrosis C: Pulmonary function in idiopathic pulmonary fibrosis and referral for lung transplantation. Am J Respir Crit Care Med. 2001;164:103–8.
Erbes R, Schaberg T, Loddenkemper R. Lung function tests in patients with idiopathic pulmonary fibrosis. Are they helpful for predicting outcome? Chest. 1997;111:51–7.
Pesonena I, Gao J, Kalafatisb D, Carlsona L, Skold M, Ferrara G. Six-minute walking test outweighs other predictors of mortality in idiopathic pulmonary fibrosis. A real-life study from the Swedish IPF registry. Res Med. 2020. https://doi.org/10.1016/j.yrmex.2020.100017.
Kalafatis D, Gao J, Pesonen I, Carlson L, Skold CM, Ferrara G. Gender differences at presentation of idiopathic pulmonary fibrosis in Sweden. BMC Pulm Med. 2019. https://doi.org/10.1186/s12890-019-0994-4.
Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, Cottin V, Flaherty KR, Hansell DM, Inoue Y, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–82.
King TE Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, Gorina E, Hopkins PM, Kardatzke D, Lancaster L, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–92.
Richeldi L. Treatments for idiopathic pulmonary fibrosis. N Engl J Med. 2014;371:783.
King TE Jr, Tooze JA, Schwarz MI, Brown KR, Cherniack RM. Predicting survival in idiopathic pulmonary fibrosis: scoring system and survival model. Am J Respir Crit Care Med. 2001;164:1171–81.
Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, King TE Jr, Lancaster L, Sahn SA, Szwarcberg J, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377:1760–9.
Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824.
Skold CM. Idiopatisk lungfibros, vårdprogram, Swedish Respiratory Society [Internet]. 2019. http://slmf.se/wp-content/uploads/2019/03/vp_ipf_19_web.pdf. Accessed 1 Feb 2021.
Racanelli AC, Kikkers SA, Choi AMK, Cloonan SM. Autophagy and inflammation in chronic respiratory disease. Autophagy. 2018;14:221–32.
Ilmarinen P, Tuomisto LE, Niemela O, Tommola M, Haanpaa J, Kankaanranta H. Cluster analysis on longitudinal data of patients with adult-onset asthma. J Allergy Clin Immunol Pract. 2017;5(967–978):e963.
Ley B, Ryerson CJ, Vittinghoff E, Ryu JH, Tomassetti S, Lee JS, Poletti V, Buccioli M, Elicker BM, Jones KD, et al. A Multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156:684-U658.
Oldham JM, Collard HR. Comorbid conditions in idiopathic pulmonary fibrosis: recognition and management. Front Med (Lausanne). 2017;4:123.
Nathan SD, Basavaraj A, Reichner C, Shlobin OA, Ahmad S, Kiernan J, Burton N, Barnett SD. Prevalence and impact of coronary artery disease in idiopathic pulmonary fibrosis. Respir Med. 2010;104:1035–41.
Hubbard RB, Smith C, Le Jeune I, Gribbin J, Fogarty AW. The association between idiopathic pulmonary fibrosis and vascular disease a population-based study. Am J Respir Crit Care Med. 2008;178:1257–61.
Pesonen I, Carlson L, Murgia N, Kaarteenaho R, Skold CM, Myllarniemi M, Ferrara G. Delay and inequalities in the treatment of idiopathic pulmonary fibrosis: the case of two Nordic countries. Multidiscip Respir Med. 2018;13:14.
Wuyts WA, Antoniou KM, Borensztajn K, Costabel U, Cottin V, Crestani B, Grutters JC, Maher TM, Poletti V, Richeldi L, et al. Combination therapy: the future of management for idiopathic pulmonary fibrosis? Lancet Respir Med. 2014;2:933–42.
Vianello A, Salton F, Molena B, Turato C, Graziani ML, Braccioni F, Frassani V, Sella D, Pretto P, Paladini L, et al. Nintedanib treatment for idiopathic pulmonary fibrosis patients who have been switched from pirfenidone therapy: a retrospective case series study. J Clin Med. 2020. https://doi.org/10.3390/jcm9020422.
Milger K, Kneidinger N, Neurohr C, Reichenberger F, Behr J. Switching to nintedanib after discontinuation of pirfenidone due to adverse events in IPF. Eur Respir J. 2015;46:1217–21.
O’Brien EC, Hellkamp AS, Neely ML, Swaminathan A, Bender S, Snyder LD, Culver DA, Conoscenti CS, Todd JL, Palmer SM, et al. Disease severity and quality of life in patients with idiopathic pulmonary fibrosis: a cross-sectional analysis of the IPF-PRO registry. Chest. 2020;157:1188–98.
Acknowledgements
We would like to thank all participants and physicians, study nurses and other clinical staff that are involv and contribute to the registry: Lena Aldenstam, Anne-Marie Andersson, Lars Andersson, Holger Becker, Alexander Bengtsson, Synnöve Bergentz, Christel Bergström, Anna Berséus, Helene Blomqvist, Kärstin Byström, Cristina Cretu, Margitha Dahl, Fredrik Dejby, Jonas Einarsson, Gunnel de Forest, Kristina Forsberg, Ingrid Gerhardsson, Lena Granbom, Christel Hannus, Kirsten Johansson, Helena Jonsson, Carl-Axel Karlsson, Eva-Marie Karlsson, Sara Klänge, Lise-Lotte Landenfelt Gestré, Leo Lazer, Håkan Lindberg, Carina Lundberg, Carina Modén, Pernilla Munthe-Granlund, Lennart Nilholm, Kenneth Nilsson, Marina Nilsson, Rikard Nilsson, Lina Niska, Cecilia Olivesten, Shumi Omar, Andreas Palm, Bo Pedersen, Lennart Persson, Anders Pettersson, Birgitta Pettersson, Olga Pettersson, Ewa Petterstedt, Anders Planck, Camilla Regnér, Rolf Rosin, Henrik Ryftenius, Pierre Sobrino, Jan Starlander, Senada Suljanovic, Emma Sundström, Anna Svensson, Camilla Thall, Anders Thylen, Pia Wallin, Ulla Waxne, Valentyna Yasinska, Milena Ymefors. The Swedish IPF registry is currently collecting data from the following hospitals: Karolinska University hospital, Skaraborgs hospital Skövde, Central hospital Kristianstad, Falun hospital, University hospital of Umeå, Sunderby hospital, Linköping university hospital, Uppsala university hospital, Gävle hospital, Östersund hospital, Skåne university hospital, NU hospital Trollhättan, Danderyd hospital, Sahlgrenska university hospital, Ängelholm hospital, Lindesberg hospital, Helsingborg hospital, Örebro university hospital, Höglands hospital Eksjö, Blekinge hospital Karlshamn, Ystad hospital, Trelleborg hospital. Special thanks to Prof Surinder Birring, King’s College London, London, UK, for his support with the K-BILD questionnaire. The research nurses Margitha Dahl, Helene Blomqvist, and Susanne Schedin are acknowledged for help with data handling. The authors would also like to thank Giovanni Ferrara, for participating in the initiation process of the Swedish IPF registry. Special thanks to Prof Surinder Birring, King’s College London, London, UK for his support with the K-BILD questionnaire.
Funding
Open Access funding provided by Karolinska Institute. Research based on the Swedish Idiopathic Pulmonary Fibrosis registry is funded by grants from the Swedish Heart–Lung Foundation, Karolinska Institutet and Karolinska University Hospital, Paul and Ragna Nyberg Foundation, and by investigator initiated grants from Roche and Boehringer Ingelheim.
Author information
Authors and Affiliations
Contributions
JG and MS are the guarantor of the manuscript and takes responsibility for the integrity of the data and the accuracy of the data analysis. JG, DK, LC, IP, and MS contributed to the design of the study; JG, DK, LC, CXL, ÅW, JM, and MS were involved in the analysis of the data; DK, LC, and IP contributed to the acquisition of the data as a study investigator; JG, DK, JM, and MS wrote the first draft of the manuscript; and LC, IP, CXL, and ÅW contributed to the development of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
The registry and its biobank have received ethical approval from the Stockholm’s Regional Ethical Committee (Ref No. 2014/1202-31/4 and Ref No. 2018/1449-31/1). The research was conducted strictly according to the principles of the Declaration of Helsinki. All patients have provided written informed consent prior to inclusion.
Consent for publication
Yes.
Competing interests
IP, DK, LC, JM and CMS received fees for lectures and advisory boards (CMS) from Roche. IP, LC, JM and CMS received fees for lectures and advisory boards (CMS) from Boehringer Ingelheim. CMS received research grants from Roche and Boehringer Ingelheim.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gao, J., Kalafatis, D., Carlson, L. et al. Baseline characteristics and survival of patients of idiopathic pulmonary fibrosis: a longitudinal analysis of the Swedish IPF Registry. Respir Res 22, 40 (2021). https://doi.org/10.1186/s12931-021-01634-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-021-01634-x