
Abstract
Background
Navafenterol (AZD8871) is a novel, long-acting, dual-pharmacology (muscarinic receptor antagonist and β2−adrenoceptor agonist) molecule in development for chronic obstructive pulmonary disease and asthma.
Methods
These two phase I, randomised, single-blind, multiple-ascending-dose studies evaluated inhaled navafenterol and placebo (3:1 ratio) in healthy, male, non-Japanese (study A; NCT02814656) and Japanese (study B; NCT03159442) volunteers. In each study, volunteers were dosed in three cohorts, allowing gradual dose escalation from 300 μg to 600 μg to 900 μg. The primary objective was to investigate the safety and tolerability of navafenterol at steady state. Pharmacokinetics were also assessed.
Results
Twenty-four volunteers completed each study (navafenterol, n = 6; placebo, n = 2 in each cohort). There were no deaths, serious adverse events (AEs) or treatment-emergent AEs (TEAEs) leading to discontinuation of navafenterol. The most frequent TEAEs were vessel puncture-site bruise (placebo, n = 2; navafenterol 900 μg; n = 3) in study A and diarrhoea (placebo, n = 1; navafenterol 300 μg, n = 2; navafenterol 900 μg, n = 3) in study B. No dose-response relationship was observed for TEAEs. There was a dose-dependent increase in mean heart rate on day 16 in both studies. The pharmacokinetics of navafenterol were similar between non-Japanese and Japanese volunteers.
Conclusions
Multiple ascending doses of navafenterol were well-tolerated and the safety and pharmacokinetics of navafenterol were similar in non-Japanese and Japanese volunteers. The findings support navafenterol clinical development.
Trial registration
ClinicalTrials.gov; Nos.: NCT02814656 and NCT03159442; URL: www.clinicaltrials.gov.
Similar content being viewed by others


Introduction
Chronic obstructive pulmonary disease (COPD) and asthma cause substantial morbidity and mortality worldwide; in 2015, there were 3.2 million deaths from COPD and 0.4 million from asthma [1], and both diseases ranked amongst the top 15 causes of disability [2]. Current treatment for both diseases involves a step-wise approach, and bronchodilator therapy is a key component of this. In COPD, combination treatment with a long-acting muscarinic receptor antagonist (LAMA) and long-acting β2-adrenoceptor agonist (LABA) is recommended as a step-up treatment for patients whose COPD is not well managed on initial treatment with LAMA, LABA, or LABA/inhaled corticosteroids (ICSs) [3]. Additionally, combined LAMA/LABA treatment is recommended as initial treatment for patients with a substantial symptom burden [3]. Triple therapy with LAMA/LABA/ICS is an option for patients with persistent symptoms and exacerbation risk [3]. Triple therapy is also used in the management of asthma, with the LAMA tiotropium recommended as add-on therapy for patients aged ≥12 years whose asthma is not well controlled with ICS/LABA therapy [4].
Navafenterol (AZD8871) is a novel chemical entity possessing long-acting, dual-pharmacology (muscarinic receptor antagonist and β2−adrenoceptor agonist [MABA]) activities in a single molecule. Preclinical pharmacological characterisations of navafenterol and an earlier compound in the series, LAS190792, have been described previously [5, 6]. The key structural difference between these two compounds is in the region linking the antimuscarinic and β2-adrenoceptor functional regions, which is an N-phenylcarbamate in navafenterol and a benzotriazole in LAS190792 [6]. The linker in navafenterol, in contrast to that in LAS190792, tips the balance of activities toward M3 antagonism, resulting in a dual bronchodilator with fewer of the secondary effects associated with β-adrenoceptor agonism [6]. The β2-adrenoceptor activity of LAS190792 is markedly higher than that of navafenterol and both compounds have low β1-adrenoceptor activity [6].
MABAs could represent an alternative therapeutic approach in COPD and, when combined with an ICS, provide simplified formulation development and a single pharmacokinetic (PK) profile compared with LAMA/LABA combinations [7]. Through its dual pharmacological activity, it is anticipated that navafenterol would offer greater efficacy than single-mechanism LAMAs or LABAs, and similar or potentially greater efficacy than free- or fixed-dose combination therapies, with an equivalent or superior safety and tolerability profile. In the first-in-human study, single ascending doses of navafenterol of 50, 200, 400, 900, 1800 and 2100 μg in patients with mild asthma were well tolerated with no safety concerns raised [8]. Clinically meaningful and sustained bronchodilation was observed with doses from 200 to 2100 μg [8]. Navafenterol is intended to be developed for registration worldwide and so it is important to study its effects in different populations since differences in the PK, safety and tolerability, or pharmacodynamic effects of drugs are sometimes observed between individuals of different ethnicities [9]. Here, we present safety, tolerability and PK data for navafenterol after single and repeat dosing in healthy volunteers from different ethnic backgrounds.
Materials and methods
Study design
These were two phase I, single-centre, randomised, single-blind, placebo-controlled, multiple–ascending-dose studies of inhaled navafenterol in healthy, male, non-Japanese (study A; NCT02814656) and Japanese (study B; NCT03159442) volunteers. The primary objective of each study was to investigate the safety and tolerability of navafenterol at steady state. The secondary objective was to characterise the PK of navafenterol and its metabolites, LAS191861 and the pharmacologically inactive LAS34850, after single and multiple doses of navafenterol and to assess the time required to reach steady state, the degree of accumulation and the time-dependency. Exploratory pharmacodynamic endpoints (lung function and pupillometry) were also evaluated (e-Appendix 1).
Both studies were conducted at PAREXEL EPCU, Northwick Park Hospital, London, UK. The final protocols and informed consent forms were approved by the local ethics committee (e-Appendix 1). The studies were performed in accordance with the Declaration of Helsinki, the International Council for Harmonisation/Good Clinical Practice guidelines, applicable regulatory requirements and the AstraZeneca policy on Bioethics. Volunteers provided voluntary, written informed consent before taking part in study procedures.
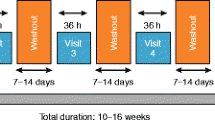
Volunteers were admitted the day prior to the first dose (day − 1) and remained on-site until day 20, 96 h following administration of the final dose on day 16 (Fig. 1). In each cohort, volunteers received a single dose of navafenterol or placebo on day 1, followed by a 4-day wash-out period and one dose/day from days 5 to 16. Evaluation and characterisation of PK were conducted after administration of a single dose (PK monitoring for 5 days), then the multiple dose study began with the same subjects so the 20-day residence comprised both single dose and multiple dose studies in the same subjects. Volunteers were randomised 3:1 to receive navafenterol or placebo, both administered via a variant of the Genuair™/Pressair®Footnote 1 dry powder inhaler adapted internally to deliver a single dose of inhalation powder. Devices containing placebo or navafenterol had identical external appearances. There were three cohorts (one for each dose level) and each volunteer only participated in one cohort. The study design allowed a gradual escalation of dose from cohort to cohort with intensive monitoring to ensure the safety of volunteers.

Study design for (a) study A and (b) study B
Volunteers in cohort 1 received a single dose of navafenterol 300 μg or placebo. Dose selection for cohorts 2 and 3 was determined by a Safety Review Committee following review of data from the previous cohort. In order to proceed to the next dose level, a minimum of 5 volunteers on active treatment must have completed dosing. The doses selected by the safety review committee for cohorts 2 and 3 were 600 and 900 μg, respectively. Predefined stopping criteria are summarised in e-Appendix 1.
Volunteers
Males aged 18–55 (study A) or 20–55 (study B) years were eligible for inclusion. In study B, Japanese volunteers were defined as those born in Japan with two Japanese biological parents and four Japanese grandparents and who had not lived outside of Japan for more than 5 years or had a significant change in lifestyle or diet since leaving Japan. Inclusion and exclusion criteria and study restrictions are reported in e-Appendix 1.
Assessments
Safety and tolerability
Safety and tolerability assessments included adverse events (AEs), physical examination, vital signs, clinical laboratory assessments (including serum glucose and potassium i-STAT measurements), 12-lead digital and safety local electrocardiograms (ECGs), and 2-lead real-time telemetry. Assessment timings are outlined in e-Appendix 1.
Pharmacokinetics
Blood samples for PK analysis were collected at pre-dose and at 15, 30 and 45 min, and 1, 1.5, 2, 3, 4, 6, 8, 12, 16, 24, 36, 48, 72 and 96 h post-dose on days 1 and 16 and also at pre-dose on days 6 to 15 (both studies) and at the follow-up visits (days 26 and 30, study B only). The plasma concentrations of both navafenterol and its metabolites were assessed using validated bioanalytical assays with a lower limit of quantification of 2 pg/mL (navafenterol and LAS191861) or 25 pg/mL (LAS34850). The PK parameters assessed included: observed maximum concentration (Cmax); time to reach Cmax (tmax); terminal half-life (t½λz); area under the concentration-time curve (AUC) from time zero extrapolated to infinity (AUC0-∞), AUC from time zero to the time of last quantifiable concentration (AUClast), and AUC from time zero to 24 h post-dose (AUC0–24); accumulation ratio for Cmax (Rac [Cmax], estimated as the ratio of Cmax on day 16 to that on day 1); accumulation ratio for AUC0–24 (Rac [AUC0–24], estimated as the ratio of AUC0–24 to that on day 1); and the metabolite to parent ratio for Cmax (MRCmax) and AUC0–24 (MRAUC0–24).
Statistical analysis
Due to the exploratory nature of the studies, the sample size was not based on formal statistical considerations. It was planned to randomise 24 volunteers in each study (8 volunteers per cohort, 6 receiving navafenterol and 2 receiving placebo). No formal statistical hypothesis testing or corrections for multiplicity were performed.
Demographic and baseline data were summarised by treatment (navafenterol dose), but placebo data from the three cohorts were pooled. Safety data were analysed descriptively for the safety population (all volunteers who received at least 1 dose of investigational drug [navafenterol or placebo] and for whom any post-dose safety data were available).
PK data were analysed in the PK population (all volunteers in the safety population who received at least 1 dose of navafenterol, had at least one of the parameters Cmax, AUC or AUClast evaluable for navafenterol, and were not affected by factors such as protocol deviations). Dose proportionality, time dependency and accumulation were assessed.
Details of the statistical analysis of the PK and pharmacodynamics are provided in e-Appendix 1.
Results
Volunteer demographics and baseline characteristics
Twenty-four volunteers completed each study (8 volunteers per cohort: 6 received navafenterol, 2 received placebo; Fig. 2). In study B, 1 volunteer (navafenterol 300 μg) withdrew prior to the first dose and was replaced by a reserve volunteer as per the protocol. There were no other discontinuations. Demographics and baseline characteristics were generally similar between treatment groups in each study and, with the exception of race and ethnicity, were similar between the studies (Table 1).

Patient disposition and flow in (a) study A and (b) study B. PK = pharmacokinetic; PP = per protocol. aOne volunteer withdrew prior to the first dose and was replaced by a reserve volunteer as per the protocol
Safety
Adverse events
There were no deaths, serious AEs or treatment-emergent AEs (AEs with onset after the first dose [TEAEs]) leading to discontinuation of navafenterol in either study. TEAEs occurring in ≥2 volunteers in any treatment group in either study are presented in Table 2; all TEAEs were of mild severity in the placebo group and of mild-to-moderate severity in the navafenterol groups. The most frequent TEAEs overall were vessel puncture-site bruise (placebo, n = 2; navafenterol 900 μg, n = 3) in study A and diarrhoea (placebo, n = 1; navafenterol 300 μg, n = 2; navafenterol 900 μg, n = 3) in study B. TEAEs considered related to treatment, as assessed by the site investigator, were headache (navafenterol 300 μg, n = 2), oropharyngeal pain (placebo, n = 1) and cough (placebo, n = 1) in study A and headache (navafenterol 900 μg, n = 1), diarrhoea (navafenterol 300 μg, n = 2), chest discomfort (navafenterol 300 μg, n = 1), thirst (navafenterol 300 μg, n = 1), increased hepatic enzymes (navafenterol 600 μg, n = 1) and somnolescence (placebo, n = 1) in study B. No dose-response relationship was observed for any TEAEs.
Clinical laboratory assessments
Overall, there were no clinically significant haematology, clinical chemistry or urinalysis findings in either study. One volunteer had alanine aminotransferase values above the upper limit of normal (navafenterol 600 μg; study B); this did not meet the criteria of Hy’s Law and was reported as a TEAE. There were no clinically relevant trends in serum glucose or potassium concentrations over time; results on day 16 are presented in Fig. 3.

Mean change from baseline on day 16 (safety population) in glucose i-STAT concentration in (a) study A and (b) study B, and mean change from baseline in potassium i-STAT concentration in (c) study A and (d) study B. SD = standard deviation
ECG, telemetry and vital signs
No trends or dose response were observed in mean change from baseline in Fridericia’s corrected QT interval (QTcF) after multiple dosing and no QTcF outlier values were observed. There were no clinically significant changes in other ECGs or telemetry and no significant vital sign abnormalities. Based on vital signs data, collected at pre-dose on all dosing days and also at 1, 2, 4, 12 and 24 h post dose on Days 1, 5, 7, 10, 12 and 16, an increase of heart rate was observed over time, with a trend towards stabilisation after 7–10 days of repeated administration of navafenterol, after PK steady state was achieved. Based on digital ECGs, a dose dependent increase was registered in heart rate at day 16 in both studies (Fig. 4).

Mean change from baseline in heart rate over time at day 16 (safety population) in (a) study A and (b) study B. aexcept for navafenterol 600 μg at 12 h post dose on day 16, n = 5. bpm = beats per minute; SD = standard deviation
Physical examination
Physical examination findings in both studies, reported as TEAEs, occurring in ≥2 volunteers in any treatment arm were vessel puncture/catheter site bruise and contact dermatitis (Table 2).
Pharmacokinetics
Both studies showed rapid absorption of navafenterol on day 1 (median tmax range: 1.01–1.52 h; Table 3; Fig. 5a and b), with similar results after multiple dosing (Table 4). Steady state for navafenterol was achieved after approximately 8 and 10 days of dosing in study A (Fig. 5c) and study B (Fig. 5d), respectively. At steady state, after tmax was achieved, plasma concentrations declined in a biexponential manner, with mean t½λz of 62.4–70.2 h in non-Japanese volunteers and 209.2–250.7 h in Japanese volunteers on day 16. Increases in exposure (Cmax and AUC) with increasing dose were generally dose proportional, although there was some statistical evidence of greater than dose-proportional increases in exposure of navafenterol after multiple dosing in study B, with the 95% confidence intervals for the slope estimates excluding unity on day 16 (e-Table 1). No significant time dependency was observed for navafenterol in either study (e-Table 2). Both studies showed some evidence of accumulation of navafenterol with multiple dosing (Rac [Cmax] range 0.90–2.37; Rac [AUC0–24] range 1.39–3.06; Table 4).

Geometric mean plasma concentration-time profiles of navafenterol (pharmacokinetic population) on days 1 and 16 in (a) study A and (b) study B, and pre-dose concentrations from days 6 to 16 in (c) study A and (d) study B. Note that a longer sampling scheme was used in study B (samples collected up to 336 h following the last dose) than in study A (samples collected up to 96 h following the last dose). SD = geometric standard deviation
The metabolite to parent ratio on day 16 was similar between studies for LAS191861 (MRCmax: study A, ~ 11–14%; study B, ~ 9–13%; MRAUC0–24: study A, ~ 23–28%; study B, ~ 19–29%) and LAS34850 (MRCmax: study A, ~ 136–230%; study B, ~ 145–373%; MRAUC0–24: study A, ~ 396–431%; study B, ~ 282–550%).
Pharmacodynamic results are provided in e-Appendix 2.
Discussion
Overall, these first safety, tolerability and PK studies of the dual pharmacology MABA, navafenterol, in healthy volunteers identified no safety concerns. There was no dose-response relationship in the pattern of TEAEs, clinical laboratory assessments (including serum glucose and potassium) or ECG results (including QTc interval) at the collective level or in individual volunteers, with the exception of a dose-dependent response effect on heart rate at steady state (day 16). In both studies, dosing remained below prespecified human exposure limits and the maximum tolerated dose was not reached. These two studies were performed in healthy volunteers and were not, therefore, designed to examine the efficacy of navafenterol. No notable trends or abnormalities were observed in exploratory lung function parameters in either study.
The increase in heart rate observed following multiple dosing with navafenterol in both studies is one of the potential effects expected for navafenterol in healthy volunteers naïve to LABA bronchodilators, based on the β2-adrenoceptor agonist component of its dual bronchodilator effect [10]. This effect was not noted in a 14-day study of patients (n = 42) with COPD treated with navafenterol and there were no other significant changes in vital signs, ECG results, or clinical laboratory tests in this population [11]. However, given the small number of participants in studies A and B, this effect does require further monitoring in future studies.
Race and ethnicity can affect drug exposure and response. A review of new drugs approved by the FDA between 2008 and 2013 found that one-fifth demonstrated differences in exposure and/or response across racial/ethnic groups [9]. In the current studies, there was no indication of a difference in exposure of navafenterol between healthy male non-Japanese and Japanese volunteers. Steady state for navafenterol was achieved after 8 days of multiple dosing in non-Japanese volunteers and after 10 days of multiple dosing in Japanese volunteers. Due to a longer than anticipated t½λz of navafenterol, the t½λz values were generally calculated over a period of less than the desired three half-lives and this may have led to unreliable estimates. The substantially longer t½λz observed for navafenterol in Japanese volunteers compared with non-Japanese volunteers was the result of a longer PK sampling period following the last dose in study B (336 h) compared with study A (96 h). Based on the time to achieve steady state and the accumulation, the ‘effective’ half-life was substantially shorter, with approximate values of 48 h in both studies. The t½λz was consistent across the dose range in both studies. The PK of navafenterol appeared to be linear with dose and time.
Conclusion
Multiple ascending doses of navafenterol (300, 600 and 900 μg) were well-tolerated in healthy male volunteers. No safety concerns were identified and stopping criteria were not met in either study. The safety and PK of navafenterol were similar in non-Japanese and Japanese volunteers. The findings of these studies support further clinical development of navafenterol.
Availability of data and materials
Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca’s data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
Notes
Registered trademarks of the AstraZeneca group of companies; for use within the USA as Pressair® and Genuair™ within all other licensed territories
Abbreviations
- AE:
-
Adverse event
- AUC:
-
Area under the concentration-time curve from time zero extrapolated to infinity
- AUC0–24 :
-
Area under the concentration-time curve from time zero to 24 h post-dose
- AUClast :
-
Area under the concentration-time curve from time zero to the time of last quantifiable concentration
- Cmax :
-
Maximum concentration
- COPD:
-
Chronic obstructive pulmonary disease
- ECG:
-
Electrocardiogram
- GOLD:
-
Global Initiative for chronic obstructive lung disease
- LABA:
-
Long-acting β2 adrenoceptor agonist
- LAMA:
-
Long-acting muscarinic receptor antagonist
- MABA:
-
Muscarinic receptor antagonist and β2 adrenoceptor agonist
- MRAUC0–24 :
-
Metabolite to parent ratio for AUC0–24
- MRCmax :
-
Metabolite to parent ratio for Cmax
- PK:
-
Pharmacokinetic
- QTcF:
-
Fridericia’s corrected QT interval
- Rac (AUC0–24):
-
Accumulation ratio for AUC0–24
- Rac (Cmax):
-
Accumulation ratio for Cmax
- t½λz :
-
Terminal half-life
- TEAE:
-
Treatment-emergent adverse event
- tmax :
-
Time to reach maximum concentration
References
GBD 2015 Chronic Respiratory Disease Collaborators. Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Respir Med. 2017;5(9):691–706.
GBD 2015 Chronic Respiratory Disease Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388(10053):1545–602.
Global Initiative for Chronic Obstructive Lung Disease. Global Strategy for the Diagnosis, Management and Prevention of COPD. 2019. https://goldcopd.org/wp-content/uploads/2018/11/GOLD-2019-v1.6-FINAL-08Nov2018-wms.pdf. Accessed 12/03/2018.
Global Initiative for Asthma. 2018 GINA Report, Global Strategy For Asthma Management and Prevention. 2018. http://ginasthma.org/2018-gina-report-global-strategy-for-asthma-management-and-prevention/. Accessed 20/04/2018.
Aparici M, Carcasona C, Ramos I, et al. Pharmacological preclinical characterization of LAS190792, a novel inhaled bifunctional muscarinic receptor antagonist /β2-adrenoceptor agonist (MABA) molecule. Pulm Pharmacol Ther. 2017;46:1–10.
Aparici M, Carcasona C, Ramos I, et al. Pharmacological profile of AZD8871 (LAS191351), a novel inhaled dual M3 receptor antagonist/ β2-adrenoceptor agonist molecule with long-lasting effects and favorable safety profile. J Pharmacol Exp Ther. 2019;370(1):127–36.
Hughes AD, Jones LH. Dual-pharmacology muscarinic antagonist and β2 agonist molecules for the treatment of chronic obstructive pulmonary disease. Future Med Chem. 2011;3(13):1585–605.
Singh D, Astbury C, Jimenez L, et al. A randomized placebo and active controlled trial of AZD8871 a novel dual acting bronchodilator in COPD patients. Eur Respir J. 2017;50(Suppl. 61):PA1798.
Ramamoorthy A, Pacanowski MA, Bull J, Zhang L. Racial/ethnic differences in drug disposition and response: review of recently approved drugs. Clin Pharmacol Ther. 2015;97(3):263–73.
Salpeter SR, Ormiston TM, Salpeter EE. Cardiovascular effects of beta-agonists in patients with asthma and COPD: a meta-analysis. Chest. 2004;125(6):2309–21.
Singh D, Fuhr R, Jimenez L, et al. A randomized trial of dual-acting bronchodilator AZD8871 for chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2019;199(10):1282–4.
Battisti WP, Wager E, Baltzer L, et al. Good publication practice for communicating company-sponsored medical research: GPP3. Ann Intern Med. 2015;163(6):461–4.
Acknowledgements
We thank the volunteers and the site staff who participated in these studies. This study was previously presented at the American Thoracic Society International Conference, San Diego, CA, USA (May 18–23, 2018) and the European Respiratory Society Congress, Paris, France (September 15–19, 2018).
About this supplement
This article has been published as part of Respiratory Research, Volume 21 Supplement 1, 2020: Results from the Phase I study programme for navafenterol (AZD8871), a novel inhaled dual pharmacology bronchodilator (MABA). The full contents of the supplement are available at https://respiratoryresearch.biomedcentral.com/articles/supplements/volume-21-supplement-1.
Funding
These studies were funded by AstraZeneca. Sponsorship for these studies and article processing charges were funded by AstraZeneca. Employees of, or contractors to, AstraZeneca (VB, LJ, UW-H, CA, BS, M-PM, AL, AA and IP) were involved in various aspects of the conception and design of the study, acquisition of data and analysis and interpretation of data, and input into manuscript development. All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analyses. The sponsor did not place any restriction on authors about the statements made in the final article. The sponsor reviewed the publication to ensure medical and scientific accuracy and protection of intellectual property. Navafenterol is an investigational product with no approved indication. Article processing charges were funded by AstraZeneca. Medical writing support, under the direction of the authors, was provided by Suzanne McAllister, PhD, on behalf of CMC Connect, a division of McCann Health Medical Communications Ltd., Macclesfield, UK and by David Candlish, Jt. Hons. BSc, of inScience Communications, part of the SpringerNature group, Chester, UK, funded by AstraZeneca, in accordance with Good Publication Practice (GPP3) guidelines [12].
Author information
Authors and Affiliations
Contributions
VB, MA, LJ, UW-H, CA, BS, AL and AA were involved in study design, data analysis and manuscript preparation. M-PM and IP were involved in data analysis and manuscript preparation. All named authors meet the International Committee of Medical Journal Editors criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval to the version to be published. IP takes responsibility for (is the guarantor of) the content of the manuscript, including the data and analysis.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The final protocols and informed consent forms were approved by the local ethics committee (e-Appendix 1). The studies were performed in accordance with the Declaration of Helsinki, the International Council for Harmonisation/Good Clinical Practice guidelines, applicable regulatory requirements and the AstraZeneca policy on Bioethics. Volunteers provided voluntary, written informed consent before taking part in study procedures.
Consent for publication
Not applicable.
Competing interests
VB was an employee of AstraZeneca at the time these studies were conducted and is now a current employee of IQVIA. LJ, UW-H, CA, BS, AL and IP are employees of AstraZeneca, and may own stock or stock options. AA was an employee of AstraZeneca at the time these studies were conducted and is now a current employee of Eloxx Pharmaceuticals. M-PM was a contractor to AstraZeneca at the time these studies were conducted. MA is an employee of PAREXEL. AstraZeneca provided funding to PAREXEL for the conduct of these studies.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1 : e-Appendix 1
. Methods. e-Appendix 2. Results. E-Table 1 Assessment of Dose Proportionality of Navafenterol in Studies A and B. E-Table 2 Assessment of Time-Dependency of Navafenterol in Studies A and B.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Balaguer, V., Albayaty, M., Jimenez, E. et al. Navafenterol (AZD8871) in healthy volunteers: safety, tolerability and pharmacokinetics of multiple ascending doses of this novel inhaled, long-acting, dual-pharmacology bronchodilator, in two phase I, randomised, single-blind, placebo-controlled studies. Respir Res 21 (Suppl 1), 212 (2020). https://doi.org/10.1186/s12931-020-01474-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-020-01474-1