
Abstract
Background
Idiopathic pulmonary fibrosis (IPF) is an incurable disease characterized by progressive lung fibrosis ultimately resulting in respiratory failure and death. Recurrent micro-injuries to the alveolar epithelium and aberrant alveolar wound healing with impaired re-epithelialization define the initial steps of the pathogenic trajectory. Failure of timely alveolar epithelial repair triggers hyper-proliferation of mesenchymal cells accompanied by increased deposition of extracellular matrix into the lung interstitium.
Methods
We previously isolated fibrosis-specific mesenchymal stem cell (MSC)-like cells from lung tissue of patients with interstitial lung diseases. These cells produced factors bearing anti-fibrotic potential and changed their morphology from mesenchymal to epithelial upon culture in an epithelial cell (EC)-specific growth medium. Here, we set out to molecularly characterize these MSC-like cell-derived ECs using global gene expression profiling by RNA-sequencing. Moreover, we aimed at characterizing disease-specific differences by comparing the transcriptomes of ECs from IPF and non-IPF sources.
Results
Our results suggest that differentially expressed genes are enriched for factors related to fibrosis, hypoxia, bacterial colonization and metabolism, thus reflecting many of the hallmark characteristics of pulmonary fibrosis. IPF-ECs showed enrichment of both pro- and anti-fibrotic genes, consistent with the notion of adaptive, compensatory regulation.
Conclusions
Our findings support the hypothesis of a functional impairment of IPF-ECs, which could possibly explain the poor clinical outcome of IPF that roughly compares to those of advanced-stage cancers. Our study provides a valuable resource for downstream mechanistic investigation and the quest for novel therapeutic IPF targets.
Similar content being viewed by others


Background
Idiopathic pulmonary fibrosis (IPF) is an incurable interstitial lung disease (ILD) characterized by progressive fibrosis and worsening dyspnea, ultimately leading to respiratory failure and death [1, 2]. The prognosis of IPF is markedly poor, with survival rates comparing to those of advanced-stage cancers [2, 3]. Although the underlying pathomechanisms are incompletely understood, it is generally believed that ongoing damage to the alveolar epithelium triggers a secondary fibrotic response in lung-resident fibroblasts that further impinges on lung function [4, 5]; formal support for this concept for IPF pathogenesis came from mechanistic studies in mice [6, 7]. The appreciation of IPF as a predominantly fibrotic, rather than inflammatory, condition [2, 8] has paved the way for new anti-fibrotic medicines including nintedanib [9] and pirfenidone [10].
Recently, we succeeded in isolating MSC-like cells from peripheral human fibrotic lung tissue. The MSC-like cells met the defining criteria of mesenchymal stem cells (MSCs) [11]. Specifically, the cells stained positive for the surface markers CD44, CD90 and CD105, and were able to differentiate into various cell types of the mesenchymal lineage, including adipocytes, osteocytes and chondrocytes. Furthermore, the cells expressed the pluripotency-associated markers Oct-3/4 and Nanog, thus indicating their potential stemness. Of note, MSC-like cells changed their mesenchymal look to a bona fide (cobblestone-like) epithelial morphology when cultured in epithelial cell (EC)-specific growth medium. This distinct change in morphology was paralleled by induction of E-cadherin (CD324) expression, a canonical marker of ECs [5].
Here, we sought to determine the transcriptomic differences between these MSC-like cell-derived ECs from IPF and non-IPF sources. We further aimed at delineating the phenotypic properties of these ECs, following the goal to elucidate a possible functional involvement of these cells in IPF, which may help to explain the poor clinical outcome of IPF as compared to other types of ILDs.
Methods
Isolation of primary lung-resident MSC-like cells
Characteristics of the study population are specified in Table 1. MSC-like cells were isolated from lung bronchoscopic or surgical biopsies of IPF (n = 5) and non-IPF ‘control’ patients (n = 5) between November 2015 and November 2016 as previously described [5]. Briefly, the harvested lung tissue was chopped into pieces (~ 1 mm3) which were then cultured in tissue flasks under standard conditions (37 °C, 21% O2, 5% CO2) in DMEM supplemented with fetal bovine serum (10%), penicillin (20 U/l), streptomycin (20 μg/ml), and amphotericin B (2.5 μg/ml). MSC-like cells showed sprouting-like growth emanating from the tissue samples and finally formed a confluent cell monolayer around the tissue piece. The non-adherent cell fraction was washed away over the course of the culture through repeated exchange of medium. After 5 days, the tissue pieces were removed and the medium was exchanged for the EC-specific growth medium Cnt-17 (CELLnTEC Advanced Cell Systems AG, Bern, Switzerland). Total RNA was extracted after 7 days of culture in Cnt-17.
RNA extraction
Total RNA was extracted from ECs (bulk populations) using the MicroElute Total RNA Kit (Omega Bio-Tek, Norcross, GA). RNA was quantified using the QuantiFluor RNA System (Promega, Madison, WI) and quality-controlled on the Bioanalyzer instrument using the RNA 6000 Pico Kit (both from Agilent, Santa Clara, CA).
RNA-sequencing
Fifty ng total RNA were used for library generation using the TruSeq Stranded mRNA Library Prep Kit High Throughput (Illumina, San Diego, CA), and 15 PCR cycles were performed. The Standard Sensitivity NGS Fragment Analysis Kit (Advanced Analytical, Ames, IA) was used to assess the quality of the libraries (123 ± 30 nmol/L average concentration and 321 ± 8 bp average library size). 1.8 pM of pooled sample was used for cluster generation on the NextSeq 500 instrument (Illumina). Single-read 76 bases (plus 8 + 8 bases for indexing) were sequenced using the NextSeq 500 High Output Kit 75-cycles (Illumina). Primary analysis of sequencing data was conducted using RTA version 2.4.11 and bcl2fastq v2.20.0.422 (both from Illumina).
qPCR validation
Total RNA was isolated from additional samples of an independent cohort and gene expression levels were determined using TaqMan® Gene Expression Assays (Thermo Fisher Scientific, Waltham, MA). The following target genes were analyzed: CXCL8 (Hs00174103 m1), ICAM1 (Hs00164932 m1), PTGS2 (Hs00153133 m1), LIF (Hs01055668 m1), IL7R (Hs00902334 m1), and GAPDH/housekeeper (Hs03929097_g1). Reactions were performed at 50 °C for 2 min (1 cycle), 95 °C for 10 min (1 cycle), and 95 °C for 15 s followed by 60 °C for 1 min (40 cycles). Relative expression data are shown as -ΔCt of target gene-GAPDH.
Statistical and bioinformatic considerations
All analyses were done using the R statistical software (R Core Team 2018, www.R-project.org). The Bioconductor package ‘edgeR’ (version 3.24.3) was used for the identification of differentially expressed genes. Exploratory analyses including Principal Component Analysis (PCA) were performed using the package ‘ade4’. Pathway enrichment analyses and visualization were done using the package ‘clusterProfiler’ (version 3.10.1) [12]. Differentially expressed gene overrepresentation in various functional annotation databases including Gene Ontology (GO, http://geneontology.org), Kyoto Encyclopedia of Genes and Genomes (KEGG, www.genome.jp/kegg/pathway.html) and DisGeNET (www.disgenet.org/rdf) was investigated using hypergeometric tests as implemented in clusterProfiler.
Results
Transcriptomic profiling of ECs
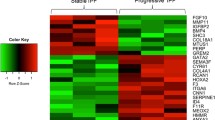
We isolated primary MSC-like cells from bronchoscopic or surgical biopsies of patients with IPF (n = 5) and non-IPF lung disease (‘control’; n = 5) (Table 1). While we previously showed that MSC-like cells exhibit EC morphology when cultured in an EC-specific growth medium [5], we here set out to molecularly characterize these ECs using global expression profiling by RNA-sequencing (Fig. 1a). Despite quite some variation between samples, PCA separated IPF from non-IPF samples on the two first principal components (Fig. 1b), indicating disease-specific transcriptomic differences. A differential expression analysis revealed the differential expression of 199 genes between IPF and non-IPF (FDR-adjusted p-values < 0.05) (Fig. 1c). Of note, epithelial markers including EPCAM, CDH1 (encoding E-cadherin/CD324) and keratins displayed only minor, non-statistically significant differences between IPF and non-IPF (FDR-adjusted p-values > 0.05) (Fig. 1d).

RNA-Sequencing of MSC-Like Cell-Derived ECs. a Conceptual and experimental design of the study. b PCA scores of ECs from IPF and non-IPF sources. c Heatmap representation and hierarchical cluster analysis of global gene expression in IPF versus non-IPF samples. A total of 199 genes were differentially regulated in IPF versus non-IPF samples. d Expression of epithelial-related markers in ECs from IPF and non-IPF sources. Abbreviations used: CPM, counts per million (reads mapped); EC, epithelial cell; IPF, idiopathic pulmonary fibrosis; MSC, mesenchymal stem cell; PCA, Principal Component Analysis
Principal signatures of ECs from an IPF source
We annotated the up- (n = 80) and downregulated genes in IPF (n = 119) (Supplementary Table 1) using functional enrichment analysis on the biological processes of the GO database. We found that IPF-ECs were enriched in various pathways potentially related to IPF pathogenesis including signatures specific for hypoxia, response to microbes, and organ development (Fig. 2a). Conversely, pathways related to ion homeostasis/transport as well as glycosylation were enriched for downregulated genes (Fig. 2b). We specifically analyzed the differentially expressed genes with respect to gene sets/pathways that likely play a role in the development and/or progression of IPF – ‘response to hypoxia’, ‘response to molecule of bacterial origin’ and ‘lung morphogenesis’ (Fig. 2c). Between 4 and 9 differentially expressed genes could be allocated to these pathways (of which some were overlapping between the signatures) and included adhesion molecules (ICAM1), chemokines (CXCL8), growth factors (CSF2), growth factor receptors (TGFBR2), matrix metalloproteinases (MMP10), and stem cell-related factors (SOX11, SHH, LIF). Notably, some of the genes were previously reported to be involved in pulmonary fibrosis pathogenesis [13,14,15]. We next performed functional enrichment analysis of the differentially expressed genes using the KEGG pathway database. Although inflammation is no longer considered the main driving force behind IPF development and progression [2, 8], this analysis showed enrichment of various immune-related pathways in IPF-ECs, including the TNF, IL-17, and JAK-STAT signaling pathways (p < 0.05) (Fig. 3a+b). It should be noted, though, that these signatures largely involved the same differentially expressed genes as the enriched GO categories (Fig. 2). Moreover, quite some variation was observed within the IPF and non-IPF groups (Fig. 3a), consistent with the notion of significant interpatient variability. Using the DisGeNET database to analyze the differentially expressed genes with regards to IPF hallmark characteristics, we found evidence that ECs from an IPF source (‘IPF-ECs’) expressed fibrosis-associated genes, suggestive of persistent ‘imprinting’ from the original IPF milieu ex vivo (Fig. 4). We next set out to validate key genes from the RNA-sequencing experiment using qPCR analysis of independent IPF and control patients (Supplementary Table 2). As shown in Supplementary Figure 1A, four out of five key genes (CXCL8, ICAM1, LIF, PTGS2) could be validated as overexpressed in IPF, with the remaining gene (IL7R) showing enrichment in IPF samples as well without reaching statistical significance. Moreover, comparing these key genes to the publically available GSE134692 dataset [16], we found that three of the genes (CXCL8, LIF, PTGS2) showed the same direction of regulation among IPF and non-IPF samples, whereas ICAM1 and IL7R were differentially regulated between the datasets (Supplementary Figure 1B). This suggested both similarities and characteristic differences between the datasets, thus corroborating our findings but also highlighting the distinct nature of our ex vivo model in comparison to primary explanted tissue. Moreover, the increasing appreciation of senescence as a major contributor to IPF development [17] prompted us to specifically investigate the KEGG pathway ‘cellular senescence’. However, of the 160 genes allocated to this pathway, only two (CXCL8 and TGFBR2) were found to be overlapping with our dataset, suggesting that this cellular process plays only a minor part in IPF-ECs. Taken together, these data suggest that the molecular signature of MSC-like cell-derived ECs may at least partially reflect the biological situation in the IPF-affected lung, including the hallmark characteristics (i) compromised gas exchange, (ii) aberrant bacterial colonization/dysbiosis, and (iii) developmental remodeling/repair.

Functional Pathway Annotation Using GO Enrichment Analysis. Functional enrichment analysis of upregulated (a) and downregulated pathways (b) in IPF-ECs using GO annotations. (c) In-depth analysis of three upregulated pathways that likely play a role in IPF pathogenesis. Abbreviations used: CPM, counts per million (reads mapped); EC, epithelial cell; GO, Gene Ontology; IPF, idiopathic pulmonary fibrosis

Functional Pathway Annotation Using KEGG Enrichment Analysis. a Heatmap analysis of functional pathway enrichment in IPF versus non-IPF samples using KEGG annotations. b In-depth analysis of five immune-related pathways that demonstrate upregulation in ECs from an IPF source. Abbreviations used: CPM, counts per million (reads mapped); EC, epithelial cell; IPF, idiopathic pulmonary fibrosis; KEGG, Kyoto Encyclopedia of Genes and Genomes

Expression Pattern of Fibrosis-Associated Genes in ECs. Comparative bar plot representation of the expression of 16 fibrosis-associated genes in IPF versus non-IPF samples. The gene panel was retrieved from the DisGeNET database. Abbreviations used: CPM, counts per million (reads mapped); EC, epithelial cell; IPF, idiopathic pulmonary fibrosis
Functional pathway analysis reveals IPF-specific signaling circuits
We moved on to interrogate the interconnection of the differentially regulated pathways and biological processes using the R package ‘clusterProfiler’. While pathways enriched for downregulated genes in IPF-ECs did not notably cluster (Fig. 5a), activated pathways showed functional enrichment mostly within three clusters that we termed ‘development’, ‘hypoxia and response to bacteria’, and ‘metabolism’ (Fig. 5b). Within the cluster ‘development’, several organ-specific developmental pathways were identified, suggesting overlapping mechanisms of tissue remodeling occurring in the IPF lung and during embryonic development. In addition, enrichment for development-related genes and pathways may indicate stem cell potential of the ECs – in turn necessary for epithelium-regenerating capacity. While the cluster ‘metabolism’ may reflect the metabolic requirements of a disease with a proliferative character, the cluster ‘hypoxia and response to bacteria’ may be more specific for IPF and reflect the typical conditions in the IPF-affected lung, including low oxygen levels and dysbiosis. Moreover, the pathway ‘collagen metabolic process’ within the same cluster may indicate the overt fibrotic response in the diseased lung characterized at least in part by massive collagen matrix deposition. Collectively, comprehensive analysis of the interrelation of pathways revealed distinct functional clusters that are plausibly implicated in the pathogenesis of IPF.

Analysis of Pathway Interrelation. Network analysis of functional pathways downregulated (a) and upregulated (b) in ECs from an IPF source using the R package ‘clusterProfiler’. Significant clustering was manually delineated and the clusters were named based on their integrated biological processes (B). Abbreviations used: EC, epithelial cell; IPF, idiopathic pulmonary fibrosis
Discussion
In this article, we provide a comprehensive description of the transcriptomic landscape of MSC-like cell-derived ECs from IPF versus non-IPF sources. Results from this study suggest that IPF-ECs (i) remain imprinted from the in vivo IPF milieu (hypoxia and dysbiosis), (ii) are more fibrotic than ECs from non-IPF sources, (iii) are enriched in various immune-related pathways, and (iv) carry signatures associated with organ development and cellular stemness. We therefore propose a distinct functional failure of these cells in IPF, which may help to explain the poor clinical outcome of IPF patients.
It is important to note that ECs were experimentally generated using a specific cell culture method and that their in vivo relevance is yet to be determined. Overlapping findings with the GSE134692 dataset based on primary tissue specimens from lung transplant recipients and donors [16] suggest, however, that our model may be able to capture the principal signatures of the IPF-affected lung including upregulation of CXCL8, LIF, and PTGS2. CXLC8 (encoding interleukin 8) is an inflammatory mediator attracting neutrophils and contributing to pathogen clearance, but may also confer secondary fibrotic tissue damage [18, 19]. In contrast, PTGS2 (encoding cyclooxygenase-2) [20, 21] and LIF [22] are believed to counteract fibrosis through various mechanisms including ferroptosis and immune modulation. Their upregulation as seen here may therefore indicate compensatory anti-fibrotic activity of the MSC-like cell-derived ECs that warrants future investigation.
IPF is an age-related disease characterized by progressive and irreversible scarring [23, 24]. The predominant occurrence in the elderly population is in line with the idea that cellular senescence drives, and is ultimately responsible for, pulmonary fibrosis [17]. While senescent fibroblasts have been shown to be fibrogenic and causally involved in disease pathogenesis in the bleomycin-injury IPF model [17], other studies have suggested a role for alveolar EC senescence in triggering lung remodeling and fibrosis [7, 25]. Mechanistically, adoption of a senescent phenotypic state may result from mutations in the enzyme telomerase reverse transcriptase, shortened telomeres, or other telomere dysfunctions [7, 26]. The senescence of functional (ECs) and/or structural (fibroblasts) lung cells is thought to be ‘counteracted’ by increased proliferation of non-senescent fibroblasts, with little success, finally resulting in excessive matrix deposition and fibrosis/scarring [27]. Indeed, we have looked at signatures of cellular senescence without detecting relevant enrichment of senescence-associated genes in IPF-ECs. We speculate that this may be explainable by their derivation from lung-resident stem-like cells rather than alveolar ECs or fibroblasts.
Autophagy, i.e., the on-demand degradation and recycling of intracellular components and organelles, is yet another fundamental cell process that is deregulated in IPF. Diminished also during aging, the autophagy flux is typically reduced to insufficient levels in IPF, which in turn accelerates cellular senescence and provokes pro-fibrotic myofibroblast differentiation [28, 29]. As these effects appear to be mediated through TGFβ signaling, upregulation of TGFBR2 in IPF-ECs may indicate IPF-specific perturbation in the autophagy cascade.
Our study is based on fresh, native samples from a rare patient cohort and hence provides a valuable resource for IPF research. It is important, however, to mention also the limitations of our study: First, our study is based on a limited number of cases, which precludes that robust, general conclusions can be drawn. Second, the reference cohort was based on patients with various kinds of non-IPF lung diseases, rather than healthy individuals. Third, our study is descriptive, meaning that causal links or rational therapeutic targets cannot be established. Fourth, our study is ultimately based on a single technological platform, i.e., RNA-sequencing, and does not report protein-level data.
Conclusion
Taken altogether, we here provide a comprehensive, molecular characterization of IPF-ECs generated ex vivo from IPF patients. Our results suggest that the transcriptome of IPF-ECs is clearly distinct from non-IPF-ECs and dominated by signatures whose underlying biological processes are plausibly involved in IPF pathogenesis, such as hypoxia, bacteria, metabolism and development. Further research is warranted to decipher the suitability of ECs as a cell-based medicinal product for anti-fibrotic, epithelium-regenerating treatment of IPF.
Availability of data and materials
Processed RNA-sequencing data have been deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible through GEO series accession number GSE151673.
Abbreviations
- EC:
-
Epithelial cell
- FDR:
-
False discovery rate
- GO:
-
Gene ontology
- ILD:
-
Interstitial lung disease
- IPF:
-
Idiopathic pulmonary fibrosis
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- MSC:
-
Mesenchymal stem cell
- PCA:
-
Principal component analysis
References
Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389:1941–52.
Raghu G. Idiopathic pulmonary fibrosis: lessons from clinical trials over the past 25 years. Eur Respir J. 2017;50.
Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183:431–40.
El Agha E, Kramann R, Schneider RK, Li X, Seeger W, Humphreys BD, Bellusci S. Mesenchymal stem cells in fibrotic disease. Cell Stem Cell. 2017;21:166–77.
Hostettler KE, Gazdhar A, Khan P, Savic S, Tamo L, Lardinois D, Roth M, Tamm M, Geiser T. Multipotent mesenchymal stem cells in lung fibrosis. PLoS One. 2017;12:e0181946.
Sisson TH, Mendez M, Choi K, Subbotina N, Courey A, Cunningham A, Dave A, Engelhardt JF, Liu X, White ES, et al. Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am J Respir Crit Care Med. 2010;181:254–63.
Naikawadi RP, Disayabutr S, Mallavia B, Donne ML, Green G, La JL, Rock JR, Looney MR, Wolters PJ. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight. 2016;1:e86704.
Fernandez IE, Eickelberg O. New cellular and molecular mechanisms of lung injury and fibrosis in idiopathic pulmonary fibrosis. Lancet. 2012;380:680–8.
Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, Cottin V, Flaherty KR, Hansell DM, Inoue Y, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–82.
King TE Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, Gorina E, Hopkins PM, Kardatzke D, Lancaster L, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–92.
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop D, Horwitz E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–7.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7.
Nakao A, Hasegawa Y, Tsuchiya Y, Shimokata K. Expression of cell adhesion molecules in the lungs of patients with idiopathic pulmonary fibrosis. Chest. 1995;108:233–9.
Jia G, Chandriani S, Abbas AR, DePianto DJ, N'Diaye EN, Yaylaoglu MB, Moore HM, Peng I, DeVoss J, Collard HR, et al. CXCL14 is a candidate biomarker for hedgehog signalling in idiopathic pulmonary fibrosis. Thorax. 2017;72:780–7.
Li M, Krishnaveni MS, Li C, Zhou B, Xing Y, Banfalvi A, Li A, Lombardi V, Akbari O, Borok Z, Minoo P. Epithelium-specific deletion of TGF-beta receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J Clin Invest. 2011;121:277–87.
Sivakumar P, Thompson JR, Ammar R, Porteous M, McCoubrey C, Cantu E 3rd, Ravi K, Zhang Y, Luo Y, Streltsov D, et al. RNA sequencing of transplant-stage idiopathic pulmonary fibrosis lung reveals unique pathway regulation. ERJ Open Res. 2019;5.
Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. 2017;8:14532.
Carre PC, Mortenson RL, King TE Jr, Noble PW, Sable CL, Riches DW. Increased expression of the interleukin-8 gene by alveolar macrophages in idiopathic pulmonary fibrosis. A potential mechanism for the recruitment and activation of neutrophils in lung fibrosis. J Clin Invest. 1991;88:1802–10.
Russo RC, Garcia CC, Teixeira MM, Amaral FA. The CXCL8/IL-8 chemokine family and its receptors in inflammatory diseases. Expert Rev Clin Immunol. 2014;10:593–619.
Hodges RJ, Jenkins RG, Wheeler-Jones CP, Copeman DM, Bottoms SE, Bellingan GJ, Nanthakumar CB, Laurent GJ, Hart SL, Foster ML, McAnulty RJ. Severity of lung injury in cyclooxygenase-2-deficient mice is dependent on reduced prostaglandin E (2) production. Am J Pathol. 2004;165:1663–76.
Kong Z, Liu R, Cheng Y. Artesunate alleviates liver fibrosis by regulating ferroptosis signaling pathway. Biomed Pharmacother. 2019;109:2043–53.
Welc SS, Flores I, Wehling-Henricks M, Ramos J, Wang Y, Bertoni C, Tidball JG. Targeting a therapeutic LIF transgene to muscle via the immune system ameliorates muscular dystrophy. Nat Commun. 2019;10:2788.
Mora AL, Rojas M, Pardo A, Selman M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat Rev Drug Discov. 2017;16:755–72.
Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol. 2014;9:157–79.
Tian Y, Li H, Qiu T, Dai J, Zhang Y, Chen J, Cai H. Loss of PTEN induces lung fibrosis via alveolar epithelial cell senescence depending on NF-kappaB activation. Aging Cell. 2019;18:e12858.
Liu T, Gonzalez De Los Santos F, Zhao Y, Wu Z, Rinke AE, Kim KK, Phan SH. Telomerase reverse transcriptase ameliorates lung fibrosis by protecting alveolar epithelial cells against senescence. J Biol Chem. 2019;294:8861–71.
Huang C, Xiao X, Yang Y, Mishra A, Liang Y, Zeng X, Yang X, Xu D, Blackburn MR, Henke CA, Liu L. MicroRNA-101 attenuates pulmonary fibrosis by inhibiting fibroblast proliferation and activation. J Biol Chem. 2017;292:16420–39.
Araya J, Kojima J, Takasaka N, Ito S, Fujii S, Hara H, Yanagisawa H, Kobayashi K, Tsurushige C, Kawaishi M, et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2013;304:L56–69.
Sosulski ML, Gongora R, Danchuk S, Dong C, Luo F, Sanchez CG. Deregulation of selective autophagy during aging and pulmonary fibrosis: the role of TGFbeta1. Aging Cell. 2015;14:774–83.
Acknowledgements
RNA-sequencing was performed at the Genomics Facility Basel of the ETH Zurich. Calculations were performed at sciCORE (http://scicore.unibas.ch/) scientific computing center at University of Basel.
Funding
This project was supported by a Sinergia grant from the Swiss National Science Foundation (SNF, grant number 160704, to TG) and a grant from the Swiss Society for Pulmonology (SGP)/Lungenliga.
Author information
Authors and Affiliations
Contributions
Conception and design: MB, FB, MHB, MT, LK, AG, TG, PK, KEH Collection and/or assembly of data: PK, KEH Data analysis and interpretation: MB, FB, MHB, JR, TG, PK, KEH Designed and generated the figures: MB, FB Wrote the first draft of the manuscript: MB, FB Wrote the final version of the manuscript: MB, FB, MHB, TG, PK, KEH Approved the paper for publication: MB, FB, MHB, MT, JR, LK, AG, TG, PK, KEH.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Patients undergoing diagnostic lung biopsy at the University Hospital Basel consented in written form to the use of their tissue for research. The Ethical Review Board of the University Hospital Basel approved the study under the number EKBB 05/06.
Consent for publication
Not applicable.
Competing interests
There are no conflicts of interest to declare. There is also no non-author involvement in the preparation of the manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Supplementary Table 1.
List of Differentially Expressed Genes. A total of 199 genes were differentially expressed in MSC-like cell-derived ECs from IPF versus non-IPF patients, with 80 genes upregulated and 119 genes downregulated in IPF-ECs. Abbreviations used: CPM, counts per million (reads mapped); EC, epithelial cell; FC, fold change; FDR, false discovery rate; IPF, idiopathic pulmonary fibrosis; MSC, mesenchymal stem cell.
Additional file 2: Supplementary Table 2.
Patient Characteristics of qPCR Validation Cohort (n = 16). Abbreviations used: IPF, idiopathic pulmonary fibrosis; ILD, interstitial lung disease.
Additional file 3: Supplementary Figure 1.
qPCR Validation of RNA-Sequencing Results and Comparison to a Published Dataset. (A) TaqMan-based qPCR analysis of 5 key genes in independent cohorts of IPF (n = 9) and non-IPF patients (n = 7; one patient overlapping with the RNA-sequencing analysis). (B) Expression levels of the 5 key genes in IPF and non-IPF patients from the GSE134692 dataset and two-tiered comparison to our data (same direction of regulation – yes/no). Abbreviations used: CPM, counts per million (reads mapped); IPF, idiopathic pulmonary fibrosis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Boesch, M., Baty, F., Brutsche, M.H. et al. Transcriptomic profiling reveals disease-specific characteristics of epithelial cells in idiopathic pulmonary fibrosis. Respir Res 21, 165 (2020). https://doi.org/10.1186/s12931-020-01414-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-020-01414-z