
Abstract
Background
Several registries of idiopathic pulmonary fibrosis (IPF) have been established to better understand its natural history, though their size and duration of follow-up are limited. Here, we describe the large European MultiPartner IPF Registry (EMPIRE) and validate predictors of long-term survival in IPF.
Methods
The multinational prospective EMPIRE registry enrolled IPF patients from 48 sites in 10 Central and Eastern European countries since 2014. Survival from IPF diagnosis until death was estimated, accounting for left-truncation. The Cox proportional hazards regression model was used to estimate adjusted hazard ratios (HR) of death for prognostic factors, using restricted cubic splines to fit continuous factors.
Results
The cohort included 1620 patients (mean age at diagnosis 67.6 years, 71% male, 63% smoking history), including 75% enrolled within 6 months of diagnosis. Median survival was 4.5 years, with 45% surviving 5 years post-diagnosis. Compared with GAP stage I, mortality was higher with GAP stages II (HR 2.9; 95% CI: 2.3–3.7) and III (HR 4.0; 95% CI: 2.8–5.7) while, with redefined cut-offs, the corresponding HRs were 2.7 (95% CI: 1.8–4.0) and 5.8 (95% CI: 4.0–8.3) respectively. Mortality was higher with concurrent pulmonary hypertension (HR 2.0; 95% CI: 1.5–2.9) and lung cancer (HR 2.6; 95% CI: 1.3–4.9).
Conclusions
EMPIRE, one of the largest long-term registries of patients with IPF, provides a more accurate confirmation of prognostic factors and co-morbidities on longer term five-year mortality. It also suggests that some fine-tuning of the indices for mortality may provide a more accurate long-term prognostic profile for these patients.
Similar content being viewed by others

Background
Idiopathic pulmonary fibrosis (IPF) is a specific form of chronic fibrosing interstitial pneumonia of unknown cause, occurring primarily in older adults [1]. IPF is a severe disease characterised by progressive worsening in lung function and associated with poor prognosis. While IPF is ultimately fatal, with an estimated median survival of 2–5 years, its clinical course is variable with a rapid decline in lung function in some patients and slower progression in others [2, 3]. Although it is generally assumed that IPF is a rare disease, more recent studies suggest that the incidence might be higher than previously thought [4].
In recent years, two anti-fibrotic drugs - pirfenidone and nintedanib - have been approved for the treatment of IPF in Europe based on large randomised clinical trials [5,6,7,8,9]. Registries of patients with IPF with extended follow-up are necessary to evaluate the effectiveness and safety of these IPF treatments in the real-world setting [10, 11]. Multicentre IPF registries, important to collect such data and describe the epidemiology, the natural course and the clinical management of IPF, have already been implemented in Germany, the United States, Sweden, Australia, the United Kingdom, and Greece [12,13,14,15,16,17]. However, the existing IPF registries are relatively small in sample size and restricted to a single country, with only limited data from Central and Eastern Europe [12, 18]. Moreover, the validation of valuable prognostic tools such as the GAP index in other populations has been constrained by the relatively small cohort sizes and short follow-up [19,20,21].
We use the large European MultiPartner IPF Registry (EMPIRE) to address these limitations. We describe the clinical characteristics of these patients and assess predictors of survival over a long follow-up period spanning over 6 years.
Methods
The European MultiPartner IPF registry (EMPIRE)
EMPIRE is a multinational, observational longitudinal registry designed to describe the characteristics and outcomes of patients with IPF in 11 Central and Eastern European countries. A detailed description of the registry is provided in the (Additional file 1: Appendix). Briefly, this registry was initiated in 2014 and has been enrolling patients at 48 sites from ten countries as of October 2018 (Additional file 1: Figure S1). The registry was approved according to national regulations in each participating country and has ethical approval to operate in all participating centres. Enrollment of patients and data collection are in compliance with the ethical principles detailed in the Declaration of Helsinki. To be eligible to participate in the registry, patients have to be at least 18 years of age with a diagnosis of IPF according to the diagnostic criteria of IPF based on the international guidelines [1] as assessed by a face-to-face multidisciplinary discussion. This includes patients with prevalent IPF diagnosed before, and incident patients diagnosed after registry initiation in September 2014. Even though most IPF patients are diagnosed at the age of 45 years or older, the minimum age of 18 years allows inclusion of patients with familial IPF. All participating patients must provide written informed consent. Follow-up visits are every 3 or 6 months, following standard clinical practice at each centre. Patient data are collected by the treating physician in a structured and non-interventional manner. Additional file 1: Table S1, lists a selection of variables collected in EMPIRE. Patients are followed in the registry until death or lung transplantation.
Study cohort
The study cohort included all patients with IPF enrolled in the EMPIRE registry who had information on baseline pulmonary function on TLCO, FVC, and FEV1, and on follow-up. Patients were followed from IPF diagnosis until death from any cause, lung transplant or last follow-up visit.
Statistical analysis
Descriptive statistics were used to summarize the demographic and clinical characteristics of IPF patients at enrollment. For the analysis of survival, the Kaplan–Meier method was used to estimate survival over time from diagnosis until death from any cause. Because the registry includes both patients with newly and previously diagnosed IPF, we accounted for left truncation arising from the delay between diagnosis and enrollment into the registry by placing patients in the risk set only from their time of enrollment. For example, a patient enrolled 1 year after diagnosis and followed up in the registry for 3 years was considered to enter the risk set at 12 months and was right censored at 48 months [22].
The Cox proportional hazards regression model was used to identify independent prognostic factors of survival up to 5 years after IPF diagnosis. Continuous factors, including age, FVC and TLCO were analysed using Cox proportional hazards regression model with restricted cubic splines, using four knots. This model fits a curve using a cubic polynomial function separately within five mutually exclusive intervals, imposing that the curves join at the boundaries to generate a smooth continuous function. Prognostic factors were also classified by categories from the GAP index, namely age at diagnosis as ≤60, 61–65 and > 65 years, FVC as < 50, 50–75, and > 75% of the predicted value, and TLCO as ≤35, 36–55, > 55% of the predicted value [23]. The results of the spline analyses were used to redefine cut-points for prognostic variables that make up the GAP index. Finally, an analysis based on the three stages of the GAP index was performed to compare the mortality of these patients. As the GAP index was designed to predict 3-year mortality, we also assessed survival up to 3 years after IPF diagnosis. All models were adjusted for all other independent prognostic factors in multivariate analyses. Independent prognostic factors included age at diagnosis, sex, predicted FVC, predicted TLCO, pulmonary hypertension, lung cancer, and long-term oxygen therapy. Data were analysed using SAS (Version 9.4) and R (version 3.5.0).
Results
The registry enrolled 2789 patients between 1996 and 2018. We excluded 1169 patients, primarily those who only had a baseline visit (305) and those with no or partial lung function data (650), described in Additional file 1: Table S2. Thus, the final study cohort included 1620 patients with IPF who were enrolled as of October 12, 2018 (Additional file 1: Figure S2). A majority of the patients (74.9%) were enrolled into the registry within 6 months after diagnosis (“incident” subjects), with 25.1% considered “prevalent” subjects at the time of enrollment. The mean time from IPF diagnosis to enrollment was 9.7 months.
Table 1 displays the baseline characteristics of the cohort and separately for the incident and prevalent patients. The mean age at diagnosis was 67.6 years, with most patients male (71.4%) and 62.9% with a history of smoking. The mean duration of symptoms prior to diagnosis was 1.6 years. Pulmonary function tests at enrollment showed a mean predicted FEV1 of 86.5%, FVC of 78.7%, TLCO of 46.3%, and a mean 6-min walk distance of 388.2 m. The distribution of GAP stage was I (47.7%), II (43.8%) and III (8.5%). Approximately 40% of all patients were receiving or had received pharmacological treatment for IPF at or prior to enrollment, which included a history of anti-fibrotic medication as well as other immunosuppressive combined regimen, but does not include medications received after enrollment. The incident and prevalent subjects were generally similar.
The mean and median follow-up from enrollment were 1.6 and 1.3 (maximum 6.7) years respectively. There were 357 deaths during follow-up. One-year survival from diagnosis was 89.2%, while it was 65.5 and 46.4% at 3 and 5 years, respectively, with a median survival of 4.5 years.
Table 2 presents the adjusted hazard ratios (HR) of mortality from the time of IPF diagnosis for the significant independent predictors of this outcome. In addition to the known factors in the GAP stage (sex, age, FVC and TLCO), the presence of pulmonary hypertension (HR 2.04; 95% CI: 1.46–2.86), lung cancer (HR 2.55; 95% CI: 1.33–4.92), and long-term oxygen therapy (HR 1.49; 95% CI: 1.19–1.88) at the time of diagnosis were all found to increase the risk of death during follow-up.
Figure 1 displays the mortality fitted by cubic splines for the continuous forms of age, FVC and TLCO. For age at diagnosis, the increase in mortality starts at 65 years and is gradual until 90 (Fig. 1a). For FVC, the increase starts when FVC is 85% of predicted and increases gradually with decreasing FVC, reaching a four-fold increase when FVC reaches 30% of predicted (Fig. 1b). For TLCO, the increase starts gradually at 60% predicted, reaching a plateau of a 3-fold increase at 40% predicted, remaining constant up to 10% predicted (Fig. 1c). These analyses suggest a readjustment of some cut-points for the prognostic variables that make up the GAP index, namely age at diagnosis as ≤65, 66–74 and > 75 years, and TLCO as ≤55 and > 55% of the predicted value (Table 3).

Smooth cubic spline curve of the adjusted hazard ratio (solid line) and 95% CIs (dotted lines) of death, estimated by Cox proportional hazards model, as a function of: a) age at IPF diagnosis; b) % predicted FVC; and c) % predicted TLCO
For the GAP index, the crude hazard ratio of mortality for stage II is 3.32 (95% CI: 2.59–4.26) and for stage III it is 5.12 (95% CI: 3.60–7.28), compared to stage I (Table 4). When adjusted for pulmonary hypertension, lung cancer and long-term oxygen therapy, the hazard ratio of mortality for stage II is 2.89 (95% CI: 2.25–3.73) and for stage III it is 3.97 (95% CI: 2.76–5.71), compared to stage I. The results for 3-year mortality were similar (Additional file 1: Table S3). When the cut-offs were readjusted, along with weights proportional to the hazard ratios, the corresponding adjusted hazard ratio of mortality for stage II is 2.66 (95% CI: 1.80–3.94) and for stage III it is 5.79 (95% CI: 4.03–8.31), compared to stage I (Table 4).
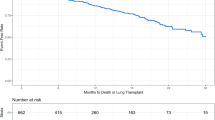
The Kaplan-Meier survival curves according to the standard GAP stage classification show clear separations (Fig. 2a), though these are somewhat more distinct with the redefined cut-points, i.e. the survival curve for stage II rises (Fig. 2b).

Kaplan–Meier survival functions for the cohort of 1620 patients with IPF from the time of their IPF diagnosis over a 5-year follow-up period, according to the a) original GAP stage classification and b) with redefined cut-points
Discussion
Using a cohort of over 1600 patients with IPF from the EMPIRE registry, its long-term follow-up allowed for accurate estimates of mortality after diagnosis, finding that only 46% of patients survive to 5 years after diagnosis. Even though the GAP index was designed to predict mortality in the first 3 years after diagnosis, this study confirms the prognostic value of the GAP stages on mortality at 5 years post-diagnosis. It also suggests that some minor redefinition of the categories of age and TLCO could improve the prognostic accuracy of the GAP index. We also found that pulmonary hypertension, lung cancer and long-term oxygen were associated with higher mortality, above and beyond the effect of GAP.
IPF registries have been implemented in Germany, the United States, Sweden, Australia, the United Kingdom, and Greece [12,13,14,15,16,17]. However, large registries based on multiple countries are scarce. EMPIRE includes complete data on over 1600 patients from ten countries in Central and Eastern Europe. Patients enrolled in the EMPIRE registry have similar patient characteristics to those reported in the previous literature [24,25,26,27]. The EMPIRE registry data confirm a more favourable prognosis after IPF diagnosis for female sex, younger age, greater predicted FVC, and greater predicted TLCO at diagnosis, all factors of the GAP index. Additionally, IPF patients with pulmonary hypertension, lung cancer, or those on long-term oxygen therapy have poorer prognosis. These findings are consistent with the data reported in previous studies, although the higher mortality with pulmonary hypertension was not shown in all other studies [2, 28,29,30,31]. Indeed, the recent study by Kreuter et al. was based on 272 subjects and found that PH was not independently associated with mortality (HR 1.1; CI 0.7–1.7), though the upper confidence limit cannot rule out a HR of 1.7, while our much larger study found a HR of 2.0 (CI 1.5–2.9). Pulmonary hypertension and lung cancer add to the symptoms and decline in pulmonary function, and other physiological consequences of IPF which can explain the increased risk of death among IPF patients with these co-morbidities [32]. The association between long-term oxygen therapy and mortality likely characterizes severity of disease [33].
Previous studies reporting survival according to the GAP staging present only crude analyses [34, 35]. When we adjusted the effects from the GAP stages for pulmonary co-morbidity, we noted that effect of the stage III group on mortality was reduced compared with stage I. This corroborates the recent trend to introduce co-morbidity in prognostic indices [36]. Our cubic spline analyses of the continuous predictors age at diagnosis, FVC, and TLCO also showed that the GAP categories may merit a reassessment in future indices. For example, we found no difference in mortality (HR 1.01; 95% CI: 0.70–1.47) between patients < 60 years of age (score = 0 in GAP) and those 61–65 years (score = 1). We also found that the effect on mortality was of the same magnitude with TLCO ≤ 35% (score = 2) and 36–55% (score = 1) predicted, which has not been reported before. When these were redefined, the corresponding adjusted hazard ratio of mortality for stages II and III were 2.7 and 5.8 respectively, while for the standard stages II and III it was 2.9 and 4.0, compared to stage I. These results suggest that it may be useful to re-examine the commonly used thresholds in large cohorts of patients with IPF, while incorporating pulmonary co-morbidity.
This study has several strengths, including using data from the EMPIRE multinational IPF registry which enrols a large, well-defined and diverse IPF patient population from Central and Eastern Europe, with sufficient numbers in relevant subgroups such as age < 60 years or TLCO ≤ 35% predicted. Our study included 419 (26%) patients with TLCO ≤ 35% predicted, which likely provides a more accurate representation of the association with mortality in this group, compared to smaller studies. Indeed, the largest study providing mortality data only had 94 patients with TLCO ≤ 35% predicted [37]. Also, the long follow-up until death allowed to identify predictors of long-term survival after IPF diagnosis. A major strength of the registry is the large proportion of patients (75%) who were included within 6 months of diagnosis, thus avoiding potential biases due to prevalent cohorts. Finally, the spline analysis of the continuous predictors allowed a more accurate understanding of these prognostic predictors of mortality, and to verify the assumptions of linear relationships made by other studies.
Limitations include differences with patients who chose not to participate, a limiting factor regarding generalizability. Approximately 60% of the patients did not have pharmacological treatment reported at enrollment, though this included patients diagnosed at enrollment and thus might not have had an IPF-related medication history yet. Finally, the exclusion of patients with missing data on pulmonary function and follow-up decreased the size of the study cohort, though the cohort of 1620 patients was still larger than in most studies in patients with IPF. Overall, excluded patients were similar to patients in the study cohort. While the 302 patients with missing lung function data had generally similar patient characteristics, the 348 patients with incomplete lung function data were somewhat more likely on long-term oxygen therapy and seemed to have, slightly lower predicted lung function measures, such as TLCO and FVC. However, these values were based on a limited subset of patients with partial information and need to be interpreted with caution. The 305 patients excluded because of only one baseline visit were generally similar to patients included in the study cohort. These were mainly patients recently enrolled in the registry in 2018 and thus had not yet had another follow-up visit yet. Therefore, the exclusion of patients with incomplete data in this study could have led to decreased precision due to a smaller sample size but unlikely to have introduced selection bias.
Future studies evaluating IPF therapies need to consider confounding by indication and potential prevalent user biases as registries typically enrol prevalent and incident patients. This incident-prevalent phenomenon can lead to unexpected findings such as zero deaths in the first 6–12 months of follow-up [38]. The EMPIRE registry avoided the survival bias from incident-prevalent cohorts by enrolling about 75% of patients within 6 months of diagnosis and using the proper left truncation survival analyses [39].
Conclusions
The current enrollment of more than 2500 IPF patients in the EMPIRE registry since September 2014 is a promising cohort size for future clinical research in IPF, making it one of the largest ongoing multinational registries. Data from this registry will provide valuable longitudinal real-world data to describe regional characteristics of patients with IPF, including co-morbidities and complications of IPF, the quality of life of patients with IPF and management of the disease.
In all, with its large size of over 1600 patients with IPF with long follow-up, the EMPIRE registry provided some novelty by its more accurate confirmation of the prognostic factors and co-morbidities on longer term five-year mortality. Its results suggest that some fine-tuning of the commonly used classification indices of mortality may be possible to provide a more accurate long-term prognostic profile for these patients.
Availability of data and materials
The dataset analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- CI:
-
Confidence interval
- EMPIRE:
-
European Multipartner IPF Registry
- FEV1 :
-
Forced expiratory volume
- FVC:
-
Forced vital capacity
- HR:
-
Hazard ratio
- IPF:
-
Idiopathic pulmonary fibrosis
- TLco :
-
Carbon monoxide transfer factor
References
Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824.
Ley B, Collard HR, King TE. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183(4):431–40.
Martinez FJ, Safrin S, Weycker D, Starko KM, Bradford WZ, King TE Jr, et al. The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med. 2005;142(12 Pt 1):963–7.
Ley B, Collard HR. Epidemiology of idiopathic pulmonary fibrosis. Clin Epidemiol. 2013;5(1):483–92.
King TE Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083–92.
Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377(9779):1760–9.
Richeldi L, Costabel U, Selman M, Kim DS, Hansell DM, Nicholson AG, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365(12):1079–87.
Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–82.
Raghu G, Rochwerg B, Zhang Y, Garcia CA, Azuma A, Behr J, et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med. 2015;192(2):e3–19.
Belkin A, Swigris JJ. Health-related quality of life in idiopathic pulmonary fibrosis: where are we now? Curr Opin Pulm Med. 2013;19(5):474–9.
Swigris JJ, Stewart AL, Gould MK, Wilson SR. Patients’ perspectives on how idiopathic pulmonary fibrosis affects the quality of their lives. Health Qual Life Outcomes. 2005;3:61.
Behr J, Hoeper MM, Kreuter M, Klotsche J, Wirtz H, Pittrow D. Investigating significant health trends in idiopathic pulmonary fibrosis (INSIGHTS-IPF): rationale, aims and design of a nationwide prospective registry. BMJ Open Respir Res. 2014;1(1):e000010.
O'Brien EC, Durheim MT, Gamerman V, Garfinkel S, Anstrom KJ, Palmer SM, et al. Rationale for and design of the idiopathic pulmonary fibrosis-PRospective outcomes (IPF-PRO) registry. BMJ Open Respir Res. 2016;3(1):e000108.
Ferrara G, Carlson L, Palm A, Einarsson J, Olivesten C, Skold M. Idiopathic pulmonary fibrosis in Sweden: report from the first year of activity of the Swedish IPF-registry. Eur Clin Respir J. 2016;3:31090.
Moodley Y, Goh N, Glaspole I, Macansh S, Walters EH, Chapman S, et al. Australian idiopathic pulmonary fibrosis registry: vital lessons from a national prospective collaborative project. Respirology. 2014;19(7):1088–91.
BTS ILD Registry [Available from: https://www.brit-thoracic.org.uk/standards-of-care/lung-disease-registries/bts-ild-registry/. Accessed 20 Oct 2017.
Bouros D, Daniil Z, Papakosta D, Antoniou KM, Markopoulou K, Kolilekas L, et al. Design, rationale, methodology, and aims of a Greek prospective idiopathic pulmonary fibrosis registry: investigating idiopathic pulmonary fibrosis in Greece (INDULGE IPF). Respiration. 2018;96(1):41–7.
Doubkova M, Uher M, Bartos V, Sterclova M, Lacina L, Lostakova V, et al. Idiopathic pulmonary fibrosis prognostic factors - analysis of the Czech registry. Cas Lek Cesk. 2016;155(4):22–8.
Kim ES, Choi SM, Lee J, Park YS, Lee CH, Yim JJ, et al. Validation of the GAP score in Korean patients with idiopathic pulmonary fibrosis. Chest. 2015;147(2):430–7.
Kondoh S, Chiba H, Nishikiori H, Umeda Y, Kuronuma K, Otsuka M, et al. Validation of the Japanese disease severity classification and the GAP model in Japanese patients with idiopathic pulmonary fibrosis. Respir Investig. 2016;54(5):327–33.
Harari S, Caminati A, Confalonieri M, Poletti V, Vancheri C, Pesci A, et al. The prognostic role of gender-age-physiology system in idiopathic pulmonary fibrosis patients treated with pirfenidone. Clin Respir J. 2019;13(3):166–73.
Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122(2):156–63.
Ley B, Ryerson CJ, Vittinghoff E, Ryu JH, Tomassetti S, Lee JS, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156(10):684–91.
Coultas DB, Zumwalt RE, Black WC, Sobonya RE. The epidemiology of interstitial lung diseases. Am J Respir Crit Care Med. 1994;150(4):967–72.
Fernandez Perez ER, Daniels CE, Schroeder DR, St Sauver J, Hartman TE, Bartholmai BJ, et al. Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis: a population-based study. Chest. 2010;137(1):129–37.
American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med. 2000;161:646–64.
Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174(7):810–6.
Johnston I, Britton J, Kinnear W, Logan R. Rising mortality from cryptogenic fibrosing alveolitis. BMJ. 1990;301(6759):1017–21.
Schwartz DA, Helmers RA, Galvin JR, Van Fossen DS, Frees KL, Dayton CS, et al. Determinants of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1994;149(2 Pt 1):450–4.
Patel NM, Lederer DJ, Borczuk AC, Kawut SM. Pulmonary hypertension in idiopathic pulmonary fibrosis. Chest. 2007;132(3):998–1006.
Kreuter M, Ehlers-Tenenbaum S, Palmowski K, Bruhwyler J, Oltmanns U, Muley T, et al. Impact of comorbidities on mortality in patients with idiopathic pulmonary fibrosis. PLoS One. 2016;11(3):e0151425.
Raghu G, Amatto VC, Behr J, Stowasser S. Comorbidities in idiopathic pulmonary fibrosis patients: a systematic literature review. Eur Respir J. 2015;46(4):1113–30.
Douglas WW, Ryu JH, Schroeder DR. Idiopathic pulmonary fibrosis: impact of oxygen and colchicine, prednisone, or no therapy on survival. Am J Respir Crit Care Med. 2000;161(4 Pt 1):1172–8.
Kondoh Y, Taniguchi H, Kataoka K, Furukawa T, Ando M, Murotani K, et al. Disease severity staging system for idiopathic pulmonary fibrosis in Japan. Respirology. 2017;22(8):1609–14.
Lee SH, Park JS, Kim SY, Kim DS, Kim YW, Chung MP, et al. Comparison of CPI and GAP models in patients with idiopathic pulmonary fibrosis: a nationwide cohort study. Sci Rep. 2018;8(1):4784.
Torrisi SE, Ley B, Kreuter M, Wijsenbeek M, Vittinghoff E, Collard HR, et al. The added value of comorbidities in predicting survival in idiopathic pulmonary fibrosis: a multicentre observational study. Eur Respir J. 2019;53(3).
Lee SH, Kim SY, Kim DS, Kim YW, Chung MP, Uh ST, et al. Predicting survival of patients with idiopathic pulmonary fibrosis using GAP score: a nationwide cohort study. Respir Res. 2016;17(1):131.
Jo HE, Glaspole I, Grainge C, Goh N, Hopkins PM, Moodley Y, et al. Baseline characteristics of idiopathic pulmonary fibrosis: analysis from the Australian Idiopathic Pulmonary Fibrosis Registry. Eur Respir J. 2017;49(2).
Miller DP, Gomberg-Maitland M, Humbert M. Survivor bias and risk assessment. Eur Respir J. 2012;40(3):530–2.
Acknowledgements
EMPIRE Registry investigators: Beata Zolnowska: 1st Department of Pulmonary Diseases, Institute of Tuberculosis and Lung Diseases, Warsaw, Poland; Vladimír Bartoš: Pulmonary Department, University Hospital and Medical Faculty of Charles University in Hradec Kralove, Hradec Králové, Czech Republic; Martina Plačková; Department of Pneumology, Faculty Hospital Ostrava, Ostrava, Czech Republic; Robert Slivka: National Institute of Tuberculosis, Lung Disorders and Thoracic Surgery Vyšné Hagy, Slovakia;, Ladislav Lacina: Clinic of Pneumology and Thoracic Surgery, Hospital Na Bulovce, Prague, Czech Republic; Martina Doubková: Department of Pneumology, Faculty of Medicine and University Hospital, Brno, Czech Republic; Radka Bittenglová: Department of Respiratory Diseases, Charles University in Plzen, Pilsen, Czech Republic; Magdalena Martusewicz-Boros: 3rd Department of Pulmonary Diseases, Institute of Tuberculosis and Lung Diseases, Warsaw, Poland; Monika Žurková: Department of Respiratory Medicine, Faculty of Medicine and Dentistry, Palacky University and University Hospital, Olomouc, Czech Republic; Imrich Jonner: National Institute of Tuberculosis, Lung Disorders and Thoracic Surgery Vyšné Hagy, Slovakia; Amelia Szymanowska-Narloch: Department of Allergology and Pneumology, Medical University of Gdansk, Gdansk, Poland; Ilona Binková: Department of Pneumology, Faculty of Medicine and University Hospital, Brno, Czech Republic; Małgorzata Sobiecka: 1st Department of Pulmonary Diseases, Institute of Tuberculosis and Lung Diseases, Warsaw, Poland; Vladimíra Lošťáková: Department of Respiratory Medicine, Faculty of Medicine and Dentistry, Palacky University and University Hospital, Olomouc, Czech Republic; Marzena Trzaska-Sobczak: Department of Pneumology, Medical University of Silesia in Katowice, Poland; Richard Tyl: Department of Respiratory Diseases, Hospital in Novy Jicin, Novy Jicin, Czech Republic; Pawel Sliwinski: 4th Department of Pulmonary Diseases, Institute of Tuberculosis and Lung Diseases, Warsaw, Poland; Miklós Zsiray: National Korányi Tuberculosis and Pulmonology Institute, Budapest, Hungary; Pavlína Lisá: Department of Pneumology, 2nd Faculty of Medicine, Charles University in Prague and Motol University Hospital, Prague, Czech Republic; Anikó Bohács: Department of Pulmonology, Semmelweis University, Budapest, Hungary; Sebastian Majewski: Department of Pneumology and Allergy, Medical University of Lodz, Lodz, Poland; Hana Šuldová: Pulmonary Department, Hospital Ceske Budejovice, Ceske Budejovice, Czech Republic; Katarzyna Lewandowska: 1st Department of Pulmonary Diseases, Institute of Tuberculosis and Lung Diseases, Warsaw, Poland; Bohumil Matula: Pneumology and Phthisiology, Specialized Hospital of St. Zoerardus Zobor, Nitra, Slovakia; Lenka Šišková: Department of Respiratory Diseases, Thomas Bata Regional Hospital Zlin, Zlin, Czech Republic; Ján Plutinský: Pneumology and Phthisiology Outpatient Centre, ZAPA JJ Ltd., Levice, Slovakia; Ana Jakić: Institute for Pulmonary Diseases of Vojvodina, Clinic for Tuberculosis and Granulomatous Diseases, Sremska Kamenica, Serbia; Štefan Tóth: Clinic of Pneumology and Phthisiology, L. Pasteur University Hospital Kosice, Kosice, Slovakia; Zoltán Balikó: Department of Pulmonology, Faculty of Medicine, University of Pecs, Pecs, Hungary; Margita Bučeková: National Institute of Tuberculosis, Lung Disorders and Thoracic Surgery Vyšné Hagy, Slovakia; Jana Pšíkalová: Pneumology and Allergology Department, Kromeriz Hospital, Kromeriz, Czech Republic; Tomasz Stachura: Jagiellonian University, School of Medicine, Department of Pulmonology, Kraków, Poland; František Petřík: Department of Pneumology, 2nd Faculty of Medicine, Charles University in Prague and Motol University Hospital, Prague, Czech Republic; Jan Anton: Department of Respiratory Diseases of the First Faculty of Medicine Charles University, Thomayer Hospital, Prague, Czech Republic; Jaroslav Lněnička: Department of Pulmonary Diseases and Tuberculosis, Masaryk Hospital, Usti nad Labem, Czech Republic; Marina Roksandić Milenkovic: University Hospital of Pulmonology, Clinical Center of Serbia, Belgrade, Serbia; Imre Lajkó: Department of Pulmonology, University of Szeged, Szeged, Hungary; Vladimír Řihák: Department of Respiratory Diseases, Thomas Bata Regional Hospital Zlin, Zlin, Czech Republic; Zsuzsanna Szalai: Department of Pulmonology, Petz Aladár County Teaching Hospital, Gyor, Hungary; Paulina Jurek: Jagiellonian University, School of Medicine, Department of Pulmonology, Kraków, Poland; Aleksander Kania: Jagiellonian University, School of Medicine, Department of Pulmonology, Kraków, Poland; Štefan Laššán: Clinic of Pneumology and Phthisiology, University Hospital Bratislava, Bratislava, Slovakia; Tatjana Pejcic: Clinic for Lung Diseases Knes Selo, Clinical Centre of Nis, Niš, Serbia; Pavel Reiterer: Department of Pulmonary Diseases and Tuberculosis, Masaryk Hospital, Usti nad Labem, Czech Republic; Lukasz Borucki: Poznan University of Medical Sciences, Poznań, Poland; Renata Králová: Department of Pulmonology, Regional Hospital Pardubice, Pardubice, Czech Republic; Pavlína Musilová: Department of Respiratory Diseases, Hospital Jihlava, Jihlava, Czech Republic; Tomáš Snížek: Department of Respiratory Diseases, Hospital Jihlava, Jihlava, Czech Republic; Daniel Doležal: Department of Pulmonary Diseases and Tuberculosis, Masaryk Hospital, Usti nad Labem, Czech Republic; Jiří Homolka: 1st Clinic of Tuberculosis and Respiratory Diseases, 1st Medical School, Charles University, Prague, Czech Republic; Hana Hortvíková: Department of Pneumology, Faculty Hospital Ostrava, Ostrava, Czech Republic; Suzana Mladinov: Split University Hospital Center, Department of Pulmonology, Split, Croatia; Peter Palúch: Department of Respiratory Diseases of the First Faculty of Medicine Charles University, Thomayer Hospital, Prague, Czech Republic; Roman Hrdina: Department of Respiratory Diseases, Hospital Znojmo, Znojmo, Czech Republic; Maria Szilasi: Centre for Pulmonology, University of Debrecen, Clinical Centre, Debrecen, Hungary; Violeta Vučinić-Mihailović: University Hospital of Pulmonology, Clinical Center of Serbia, Belgrade, Serbia; Róbert Vyšehradský: Clinic of Pneumology and Phthisiology, University Hospital Martin, Martin, Slovakia; Radka Mokošová: Department of Pneumology, Faculty Hospital Ostrava, Ostrava, Czech Republic; Agata Nowicka: Poznan University of Medical Sciences, Poznań, Poland; Tatjana Radjenovic Petkovic: Clinic for Lung Diseases Knes Selo, Clinical Centre of Nis, Niš, Serbia.
Funding
The EMPIRE registry is supported by a grant from Boehringer Ingelheim (since 2014). The sponsor did not have any involvement in the collection, analysis, and interpretation of the data or in the writing of or commenting on this manuscript
Author information
Authors and Affiliations
Consortia
Contributions
MV acts as the guarantor of this entire manuscript. TT, SS drafted the manuscript; TT, SS participated in the data analysis; TT, SS, MV were involved in the design of the study and the interpretation of the data; NM, MSt, VM, JT-T, DJ, JK, MH, MSv, JS, MV, LD, KH were involved in the design of the registry and the acquisition of the data. All authors revised the work for important intellectual content, gave final approval of the version to be published, and agree to accountable for all aspects of the work.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was conducted in accordance with the ethical principles detailed in the Declaration of Helsinki and written informed consent was obtained from all patients. The study was approved by the Ethical Committee of the Thomayer Hospital, Prague, Czech Republic (approval no. 3195/14 (A 15–01-02)).
Consent for publication
Not applicable.
Competing interests
S.S. has received research grants from Boehringer Ingelheim and Novartis and has participated in advisory board meetings or as speaker for AstraZeneca, Boehringer-Ingelheim, and Novartis. L.D. and K.H. have received grants from Boehringer-Ingelheim and Roche. J.T.-T. has received support from Boehringer-Ingelheim and Roche. None of the other authors have any competing interests to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:
EMPIRE. Supplementary information: Detailed description of EMPIRE, supplementary Figures and Tables.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tran, T., Šterclová, M., Mogulkoc, N. et al. The European MultiPartner IPF registry (EMPIRE): validating long-term prognostic factors in idiopathic pulmonary fibrosis. Respir Res 21, 11 (2020). https://doi.org/10.1186/s12931-019-1271-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-019-1271-z