
Abstract
Introduction
Bacteria have been extensively implicated in the development of smoking related diseases, such as COPD, by either direct infection or bacteria-mediated inflammation. In response to the health risks associated with tobacco exposure, the use of electronic cigarettes (e-cigs) has increased. This study compared the effect of e-cig vapour (ECV) and cigarette smoke (CSE) on the virulence and inflammatory potential of key lung pathogens (Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus and Pseudomonas aeruginosa).
Methods
Biofilm formation, virulence in the Galleria mellonella infection model, antibiotic susceptibility and IL-8/TNF-α production in A549 cells, were compared between bacteria exposed to ECV, CSE and non-exposed bacteria.
Results
Statistically significant increases in biofilm and cytokine secretion were observed following bacterial exposure to either ECV or CSE, compared to non-exposed bacteria; the effect of exposure to ECV on bacterial phenotype and virulence was comparable, and in some cases greater, than that observed following CSE exposure. Treatment of A549 cells with cell signaling pathway inhibitors prior to infection, did not suggest that alternative signaling pathways were being activated following exposure of bacteria to either ECV or CSE.
Conclusions
These findings therefore suggest that ECV and CSE can induce changes in phenotype and virulence of key lung pathogens, which may increase bacterial persistence and inflammatory potential.
Similar content being viewed by others


Background
Smoking is a risk factor for the development and progression of chronic lung diseases, such as chronic obstructive pulmonary disease (COPD) and asthma [1, 2]. Exposure to cigarette smoke initiates a cascade of tissue inflammatory responses and protease imbalances, which contribute to lung inflammation and aid establishment of chronic lung infection [3,4,5]. Electronic cigarettes (e-cigs) are widely perceived by the public as a safer alternative to tobacco smoking and their use has increased dramatically in recent years [6, 7]. Significant controversy exists around their use, dividing opinion amongst public health specialists [8, 9]. Since e-cigs contain fewer toxic chemicals, and in lower concentrations, than conventional cigarettes, they are viewed by some as a “lesser evil”. However, insufficient evidence regarding either their value as a smoking cessation tool or their safety compared to conventional cigarettes is currently available [10,11,12]. Of concern, recent reports have identified clusters of acute pulmonary disease associated with use of nicotine containing electronic cigarettes [13].
Bacteria, particularly Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus and Pseudomonas aeruginosa have all been implicated in the development of smoking-related chronic lung disease, through both direct infection and bacteria-mediated inflammation [14]. Sequencing based studies have shown that these bacteria are associated with the development of a lung community skewed towards loss of diversity, and associated with declining lung function [15, 16] Although many studies have focused on the interaction between bacteria and host lung tissues, it is unclear how this complex interplay is affected by bacterial exposure to either conventional cigarette smoke or e-cigarette vapour. We hypothesize that such exposure may act as an environmental pressure on the respiratory pathogens, driving establishment of chronic lung infection through changes in bacterial phenotype and virulence, subsequent development of inflammation, and ultimately result in poorer clinical outcomes. Therefore, in this study we compared the effect of cigarette smoke extract (CSE) and e-cig vapour extract (ECVE) on the phenotype and virulence of respiratory pathogens.
Methods
Bacterial isolates
Isolates used in this study were obtained from the American Type Culture Collection (ATCC): H. influenzae (ATCC 49766), S. aureus, (ATCC 29213), S. pneumoniae (ATCC 49619) and P. aeruginosa (ATCC 27853). All isolates were stored at -80 °C prior to inoculation onto chocolate blood agar (H. influenzae: Oxoid, Basingstoke, UK) or blood agar (S. aureus, S. pneumoniae, P. aeruginosa: Oxoid, Basingstoke, UK) and incubated at 37 °C in 5% CO2 (H. influenzae, S. pneumoniae), or in air (S. aureus, P. aeruginosa).
Preparation of cigarette smoke and electronic cigarette vapour
Preparation of cigarette smoke extract (CSE)
CSE was prepared from Marlboro Red™ cigarettes (0.8 mg nicotine, 10 mg Tar; 10 mg carbon monoxide /cigarette), as previously described with minor modifications [17]. Cigarette smoke (35 ml) was drawn, using a sterile syringe, through 100 ml of appropriate culture medium every 15 s for 5 min. This action was repeated with either four, three, two or one cigarette per 100 ml of culture medium (termed 100, 75, 50 and 25% CSE, respectively). Following sterilisation by filtration through both 0.45 μm and 0.2 μm filters, the optical density550nm was determined for all CSE solutions to ensure between batch consistency. All CSE exposed media was inoculated onto Mueller Hinton agar and incubated at 37 °C overnight to ensure sterility of the media prior to bacterial inoculation.
Preparation of E-cigarette vapor extract (ECVE)
ECVE was generated in identical fashion to CSE, except with a commercially available e-cigarette [Vapourlites™ (VL-EGO 650, (http://www.vapourlites.com/)] and using unflavoured e-liquid containing 10 mg/ml nicotine. Given the wide variety of e-cig devices currently available on the market, we chose one that at the time of study was a best –seller and widely available. Four, three, twice or once × 5 min vaping/100 ml of culture medium (termed 100, 75, 50 and 25%, ECVE respectively) was used. The resulting ECVE was then sterilised by filtration, and sterility of ECVE exposed media checked, as described above.
Determination of total viable count (TVC) of bacteria following growth in CSE or ECVE
A suspension of 1 x 107cfu of each bacteria (H. influenzae, S. pneumoniae, S. aureus and P. aeruginosa) was inoculated into 10mls culture media +/− 100, 75, 50 or 25% CSE/ECVE. Total viable counts were determined in triplicate at t = 0, 2, 4, 6, 24 and 48 h post inoculation as described previously and expressed as cfu/ml [18]. Bacterial growth in media, which had not been exposed to CSE/ECVE, was tested in parallel. Transmission electron micrograph (TEM) images were kindly prepared by Dr. Kathryn Whyte, EM Research Services, Newcastle University. Briefly, samples were fixed in 2% glutaraldehyde in Sorenson’s phosphate buffer, post-fixed in osmium tetroxide and dehydrated in graded acetone. They were then embedded in epoxy resin (TAAB premix medium) and polymerised for 24 h at 60 °C. Ultrathin sections (70 nm) were picked up on copper grids, stained with uranyl acetate and lead citrate before being imaged on a Hitachi HT7800 TEM with EMSIS camera.
Growth of bacterial biofilm in CSE and ECVE
Biofilm formation of each isolate grown in media alone, or media exposed to either either 100% CSE or ECVE was determined by crystal violet staining of adherent cells after 24 h, as described previously [19].
Effect of exposure to CSE/ECVE on bacterial virulence in the Galleria mellonella infection model
Changes in virulence of isolates in response to growth in media alone, or to media exposed to CSE/ECVE was determined using the G. mellonella infection model as described previously [20]. Following overnight growth in media +/− CSE/ECVE, the inoculum was washed by centrifugation and adjusted to 1 × 108 cfu/ml in broth, to obtain a sub-lethal inoculum concentration, which both avoided immediate larval kill and allowed a change in % survival to be observed (Additional file 1: Table S1). Inoculation of larvae was carried out as previously described [21]. Briefly, for each test condition, batches of 10 larvae were inoculated with bacteria grown in the presence or absence of CSE or ECVE, or PBS, into the left, last set of pro-legs on each larvae prior to incubation at 37 °C in air for 24 h. Experiments were carried out in triplicate and % survival recorded.
Development of resistance to antibiotics commonly used in the treatment of chronic lung infection
All isolates were inoculated in media alone, or media exposed to 100 or 50% CSE or ECVE. Following overnight incubation, each culture was adjusted to approximately 5 x106cfu and inoculated into 10mls of fresh culture medium +/− CSE or ECVE. This serial passage was repeated daily for 12 days, with the MIC determined at 0, 3, 6, 9 and 12 days post inoculation by E-test® (BioMerieux, BioMerieux UK Ltd., Basingstoke, UK) in accordance with manufacturers instructions. Antibiotics tested were amoxicillin, co-amoxiclav, tetracycline, doxycycline, erythromycin, azithromycin and ciprofloxacin. At day 12, isolates in which resistance development had been observed were cultured in CSE/ECVE-free media for a further 12 days and MICs determined once more.
Immune response to bacteria following exposure to CSE/ECVE
Human airway epithelial A549 cells (ATCC CCL-158) were passaged in complete medium [RPMI 1640, 10 μl/ml (v/v) penicillin/streptomycin solution, 10 μl/ml (v/v) HEPES buffer, 10% v/v foetal calf serum (Life Technologies, UK)] and incubated in 5% v/v CO2 at 37 °C. Bacterial infection of A549 cells was carried out by seeding cells into 24-well plates at a density of 2.5 x 105cells/ml and overnight incubation until 70–90% confluency was achieved. Bacteria which had been grown for 24 h in media alone or media + 100% CSE or ECVE were added to serum-starved cells at a multiplicity of infection of 100 cfu/cell. Negative controls of PBS only were also included in each experiment. The viability of A549 cells under each treatment condition was determined at 2, 4 and 6 h post infection, by staining with Alamar Blue® (ThermoFisher UK Ltd., Paisley, UK) in accordance with the manufacturers instructions. Viability was determined by measurement of fluorescence at 600nm and percentage viability calculated by fluorescencesample/fluorescencecontrol × 100.
At 0, 4 and 6 h post infection an aliquot of cell supernatant was removed and stored for cytokine analysis. All experiments were carried out in triplicate. Levels of IL-8, TNF-α and IL-1β were determined by ELISA (Peprotech, UK) in accordance with the manufacturers instructions, and standard curves generated using GraphPad Prism (version 5.00 for Windows, GraphPad Software, San Diego California USA). The above cell infection experiments were repeated, but with the addition of cell signaling inhibitors (BAY117085, SB203580, U0126 and SP600125, Tocris U.K.) which were added 1 h prior to bacterial infection of the cells, and levels of IL-8 and TNF-α in supernatants determined by ELISA (Additional file 1: Table S2).
Statistical analyses
Differences in the growth of bacterial biofilm in CSE and ECVE were analysed using the Wilcoxon signed-rank test with Bonferroni’s adjustment for multiple comparisons [GraphPad Prism (version 6, GraphPad Software, San Diego California USA]. A one-way ANOVA test with Tukeys test for multiple comparisons was used to compare changes in G.mellonella following bacterial infection +/− CSE/ECVE exposure [R Environment version 3.3.1 (http://www.r-project.org)]. Changes in IL-8 and TNF-α +/− CSE/ECVE were analyzed by the Mann Whitney test, and the effect of pathway inhibitors, by pairwise comparison using Kruskal-Wallace test and Dunn’s test [R Environment version 3.3.1 (http://www.r-project.org)].
Results
Determination of TVC of bacteria following growth in CSE or ECVE
CSE or ECVE had no observable effect on the growth of any isolate tested, at any concentration, compared to growth of the isolate in media without CSE/ECVE. (Additional file 1: Figure S1). With higher concentrations of CSE, a slight lag in initial growth rate was observed, particularly with H. influenzae, but this was not evident at 24 h. Comparison of TEM images following exposure to either CSE or ECVE showed no gross physiological changes compared to bacteria grown in media alone, with the exception of P.aeruginosa. Exposure of P.aeruginosa to either CSE or ECVE resulted in increased numbers of cells in which the cytoplasm appeared to be partly detached from the cell wall (Additional file 1: Figure S2). However, this was not associated with any change in P. aeruginosa viability.
Effect of CSE/ECVE on bacterial growth in biofilm
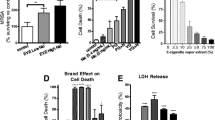
Growth of isolates in culture medium containing CSE resulted in an increase in biofilm formation for all species compared to isolates grown in media alone, with statistically significant increases apparent for S. pneumoniae (p = 0.0047) and P. aeruginosa (p = 0.0043) (Fig. 1). A significant increase in biofilm formation was also observed for S. aureus cultured in media + ECVE (p < 0.001) compared to that in media alone. No difference was observed in biofilm formation in isolates cultured in CSE vs. ECVE, with the exception of S. aureus (p = 0.001) in which biofilm formation was higher in ECVE compared to CSE.

Effect of CSE and ECVE on biofilm formation. A trend towards increased biofilm formation was observed in all isolates, with statistically significant differences observed with (a) S. aureus + CSE/ECVE, S. pneumoniae + CSE and (b) P. aeruginosa + CSE/ECVE. The mean OD was calculated based on values from 4 replicates, repeated twice
Effect of bacterial exposure to CSE/ECVE on survival of G. mellonella
We observed a statistically significant decrease in survival of G. mellonella infected with bacteria exposed to CSE or ECVE compared to larvae infected with bacteria not exposed to either CSE/ECVE (Fig. 2). The observed decrease was greater following bacterial exposure to CSE, compared to ECSE.

Effect of CSE and ECVE exposure on bacterial virulence in the G. mellonella infection model (n = 10). Larval survival decreased significantly in all isolates following exposure of isolates to both CSE and ECVE, compared to controls
Development of resistance to antibiotics commonly used in the treatment of chronic lung infection
The MIC of P. aeruginosa exposed to CSE to both tetracycline and doxycycline increased from 24 mg/ml and 48 mg/ml respectively, to > 256 mg/ml, within three days of exposure to CSE. This increase in MIC returned to original levels when isolates were cultured in the absence of CSE for 24 h, and the observed stability remained for the remaining 12 days of the experiment. No change in MIC of any other antibiotic was observed with the remaining isolates passaged in CSE or ECVE (Additional file 1: Table S3).
Immune response to bacteria +/− CSE/ECV
Exposure of A549 cells to bacteria exposed vs. bacteria not exposed to CSE resulted in a statistically significant increase in IL-8 secretion, with the exception of S. pneumoniae [H. influenzae (p = 0.0002); P. aeruginosa (p = 0.0022); S. aureus (p = 0.0372)] [Fig. 3(a)]. Exposure of bacteria to ECVE prior to A549 infection resulted in a statistically significant increase in IL-8 secretion with all bacteria + ECVE vs. bacteria not exposed to ECVE [H. influenzae (p = 0.0002); P. aeruginosa (p = 0.0019); S. aureus (p = 0.0372); S. pneumoniae (p = 0.0343)]. Levels of TNF-α were significantly increased in H. influenzae in response to CSE exposure (p = 0.0028) and in all bacteria exposed to ECVE with the exception of P. aeruginosa [H. influenzae (p = 0.0006); S. pneumoniae (p = 0.0017); S. aureus (p = 0.0104)] [Fig. 3(b)]. Viability of A549 cells remained at approximately 100% under each treatment condition and over the duration of the experiment, as determined by Alamar Blue® staining (Additional file 1: Figure S3).

The effect of cigarette smoke extract (CSE) and electronic cigarette vapour (ECVE) exposure on the capacity of key lung pathogens (i) H. influenzae ATCC 49766 (HI), (ii) P.aeruginosa ATCC 27853 (PA), (iii) S. pneumoniae ATCC 49619 (SP) and (iv) S.aureus ATCC 29213 (SA), to stimulate (a) IL-8 (n = 9) and (b)TNF-α (n = 9) production from A549 cells
The activation of NF-kB and MAP kinases, p38, ERK and JNK, is associated with the expression of inflammatory cytokines. To determine which one of these signaling pathways governed the increase in inflammation observed with ECVE-treated bacteria, infections were carried out in the presence of well-characterized pharmacological inhibitors. Use of pathway inhibitors resulted in a decrease in both IL-8 and TNF-α secretion by A549 cells following bacterial infection either alone, or following bacterial exposure to ECVE or CSE (Fig. 4, Table 1 and Additional file 1: Table S4). In general, the overall findings from these pathway inhibitor experiments show that the inflammatory pathway employed following bacterial exposure to ECVE was similar to that activated following infection with bacteria alone, or bacteria exposed to CSE.

The effect of cigarette smoke extract (CSE) and electronic cigarette vapour (ECVE) exposure on the capacity of key lung pathogens to stimulate IL-8 [(a)-(d) (i)] and TNF-α [(a)-(d)(ii)]production from A549 cells (n = 8). Cell pathway signaling inhibitors were added to determine the contribution of each pathway to the cytokine production observed and the subsequent reduction in secretion of IL-8 or TNF- α measured. P-values are shown in (Additional file 1: Table S3)
Discussion
In this study, changes in bacterial phenotype associated with virulence were observed following exposure to ECVE. In some cases the observed phenotypic changes were less than those observed with CSE-exposed bacteria (e.g. with virulence in the G. mellonella model). However, in general, there was little difference in the effect on exposure of bacteria to CSE or ECVE, with exposure to either resulting in increased virulence and inflammatory potential of the bacterial isolates.
Several studies have suggested an effect of ECVE on cultured lung cells, ranging from increased inflammation, measured by increased cytokine production, to changes in the microvasculature [22,23,24]. Increased cytokine production and evidence of lung injury has also been observed following exposure of mice to e-cig vapour and nicotine, together with a reduced capacity to clear either bacterial (S. pneumoniae) or viral (H1N1 Influenza) infection [23, 25, 26]. These findings suggest an inflammatory lung environment similar to that observed following cigarette smoking. Many e-cig users have previously been cigarette smokers; therefore, it is difficult to attribute any changes in lung function to e-cigs alone. However, perhaps driven by concerns over cigarette safety, many adolescents who have never smoked, are now taking up vaping [27], resulting in evidence of an association between e-cigarette use or exposure, and increased asthma exacerbations [28, 29]. There is therefore a need to understand the long-term impact of e-cigarette use and second hand ECV exposure, particularly on the lung health of vulnerable populations [12].
Bacterial colonization and infection of the airways is a contributing factor to lung function decline across a range of chronic lung diseases and a recognized risk of tobacco smoke exposure [30]. However, the extent to which cigarette smoke, or ECVE drives the establishment of bacterial colonization and aids persistence of these bacteria has not been extensively studied in all key pathogens implicated in chronic lung disease. H. influenzae, S. pneumoniae, P. aeruginosa and S. aureus are consistently associated with lung function decline, increased severity of disease and increased rate of exacerbation in chronic lung diseases in which smoking also plays an important role [31, 32]. Establishment of biofilm by these pathogens is a significant virulence determinant in the pathophysiology of chronic lung disease, and is associated with establishment and persistence of infection, resistance to antibiotics and evasion of the host immune system. In this study, biofilm formation increased in all isolates in response to both CSE and ECVE. Furthermore, the degree of biofilm formation observed following exposure of bacterial isolates to either CSE or ECVE, was similar and suggests that bacterial exposure to either CSE or ECVE may promote bacterial adhesion, biofilm formation and thus establishment of persistent infection. This reflects previous studies, which demonstrated similar findings following CSE exposure of lung (S. aureus, P.aeruginosa and S. pneumoniae) [33,34,35,35,35,35,39] and oral pathogens (Streptococcus gordonii, Porphyromonas gingivalis and Candida albicans) [40,41,42]. In all cases, genes associated with biofilm formation were found to be up-regulated, and this was linked to oxidative stress resultant from CSE exposure. Changes were also observed in expression of genes encoding for bacterial cell surface structures, resulting in increased bacterial adhesion to epithelial cells. MRSA exposed to CSE had increased hydrophobicity and altered surface charge, which resulted in increased adherence to epithelial cells and decreased bacterial susceptibility to antimicrobial peptides, respectively [35]. In the case of P. gingivalis, increased expression of fimbrial proteins induced TLR2 hyposensitivity and hence altered immune responses [41]. The effect of ECVE was not investigated in these studies, and further work will be required to determine if the observed increases in biofilm following ECVE exposure, occur by similar mechanisms. In this study, there was limited evidence of structural change by electron microscopy, following exposure of bacteria to either CSE or ECVE. Future work will therefore more fully investigate changes in bacterial transciptomes following exposure to vape or tobacco smoke.
Increased biofilm formation subsequent to CSE/ECVE bacterial exposure is suggestive of increased isolate virulence, and this hypothesis was further explored in the G. mellonella model. Numerous studies have shown that microbial pathogenesis and bacterial virulence are comparable in humans, mice and G. mellonella [21]. For the purposes of this study, it provided a high-throughput and cost-effective means by which changes in bacterial virulence could be assessed [43,44,45]. Statistically significant decreases in larvae survival (assumed to be consistent with increased bacterial virulence), were observed for all bacteria exposed to CSE, and for all bacteria exposed to ECVE, except H. influenzae. Mammalian models of lung infection will be required to more fully assess changes in host pathology following infection with CSE/ECVE exposed bacteria; however, our aim in this study was to assess gross changes in bacterial virulence.
A particularly striking finding of this study was the change in lung inflammation observed following infection of A549 cells with bacteria exposed to either CSE or ECVE. Dysregulation of the lung inflammatory response is a hallmark of chronic lung disease, such as COPD, where it is persistent, observed long after exposure to cigarette smoke has ceased, and attributed to bacterial colonization [46]. With the exception of S. pneumoniae, IL-8 secretion from A549 cells was significantly increased in all isolates following infection with bacteria exposed to CSE and ECVE, compared to infection with non-CSE/ECVE exposed bacteria. Of particular note, was that there was no difference observed between levels of IL-8 produced following infection with bacteria + CSE vs. bacteria + ECVE, with the exception of S.aureus. In this case, exposure to ECVE resulted in increased IL-8 levels compared to CSE. Levels of TNF-α were similarly increased following ECVE exposure with H. influenzae, S. pneumoniae and S. aureus. These data indicate that bacteria exposed to CSE promote a greater inflammatory response in A549 cells than in non-exposed bacteria, but that this is closely matched and in some cases exceeded by the level of inflammation observed following exposure to ECVE. Altered immune responses, which promote bacterial persistence, have previously been observed with S. pneumoniae, following airway cell-CSE exposure [47, 48] and with CSE- exposed MRSA [33]. MRSA exposure to ECVE has also been described as altering immunomodulatory cytokines in the airways of mice [49]. Our findings expand on this work to show that exposure of other key respiratory pathogens to both CSE and, in particular, ECVE, has the potential to modulate host response to infection and we speculate that this could contribute to the increased inflammation and bacterial persistence characteristic of smoking-related chronic lung disease. The epithelial cell-line A549 were considered to be suitable for this study since the epithelium is the major source of lung immunomodulatory factors and is hence critical in the modulation of inflammatory diseases such as COPD and bronchiectasis [50]. Furthermore, they are well characterized and standardized, allowing for rigorous comparison of bacterial infections. Future studies will more fully analyse the host response to CSE/ECVE exposed bacteria in a range of primary cell cultures, but this is outside the scope of the present study.
Addition of a range of immune pathway inhibitors suggested that the cell-signalling pathway utilized in response to infection is dependant on the bacterial species involved. Furthermore, the results did not indicate that increased cytokine production in response to bacterial exposure to ECVE was occurring via an alternative cell-signaling pathway, compared to bacterial infection alone or CSE-exposed bacteria. Moreover, bacterial CSE/ECVE exposure enhanced the immunomodulatory effect observed. Increased activation of both NFκB and MAPK signaling pathways have been implicated in the pathogenesis of COPD and asthma, with NFκB upregulation further associated with steroid insensitivity [51], but the potential contribution of bacterial infection to this pathway is still poorly understood. Our findings clearly indicate that these pathways may be further up-regulated by exposure of key lung pathogens to CSE or ECVE. The bacterial lung community is complex and increased airway inflammation subsequent to bacterial exposure to CSE/ECVE is likely to be mediated via a range of signaling pathways. Understanding each of these, and their respective contribution to inflammation in vivo may provide insight into potential therapies to reduce the effects of persistent bacterial-induced inflammation.
A recurring theme of this study is the similarity observed in the effect of exposure to CSE compared to ECVE on bacterial phenotype and virulence. CSE was generated in accordance with previously published and accepted protocols: however, this is a potential limitation of this study. In order to ensure comparability, CSE and ECVE were prepared using a similar method. This may not represent a true reflection of differences between smoking and vaping: e.g. it fails to take account of the differences in puffing topography (puff duration and flow rate) between conventional and electronic cigarettes, and between individuals [52]. E-cigarette users take larger and longer puffs, compared to conventional cigarette users, which may increase nicotine delivery. Our model may therefore underestimate the exposure of respiratory pathogens to ECVE [53]. Our current protocol is also based on a one-off exposure to CSE/ECVE, and used a brand of e-cigarettes with no added flavour: however, flavourings and e-cigarettes additives (such as PG/VG) have been associated with changes in the bronchial epithelia and impairment in respiratory innate immunity [54, 55]. Further studies are therefore required to investigate the effect of both common e-cigarette flavourings and long-term exposure of bacteria to CSE/ECVE. Furthermore, only reference isolates were used in this study and further work investigating a wider range of clinical isolates is required.
Conclusions
Exposure of respiratory pathogens to e-cigarette vapour induced changes in phenotype and virulence, which may increase bacterial persistence and inflammatory potential. These changes were similar, and in some cases exceeded, those observed following bacterial exposure to cigarette smoke and suggest that there is little difference between the effect of CSE and ECVE. There is therefore an urgent need for further robust clinical studies investigating and clarifying the long-term effect of e-cigarette use on both airway cells and respiratory pathogens to enable a better informed judgment to be made regarding their safety.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information files.
References
Mannino DM, Buist AS. Global burden of COPD: risk factors, prevalence, and future trends. Lancet. 2007;370(9589):765–73.
Thomson NC. Asthma and smoking-induced airway disease without spirometric COPD. Eur Respir J. 2017;49(5):1602061.
Comer DM, Kidney JC, Ennis M, Elborn JS. Airway epithelial cell apoptosis and inflammation in COPD, smokers and nonsmokers. Eur Respir J. 2013;41(5):1058–67.
Garmendia J, Morey P, Bengoechea JA. Impact of cigarette smoke exposure on host-bacterial pathogen interactions. Eur Respir J. 2012;39(2):467–77.
Sopori M. Effects of cigarette smoke on the immune system. Nat Rev Immunol. 2002;2(5):372–7.
Regan AK, Promoff G, Dube SR, Arrazola R. Electronic nicotine delivery systems: adult use and awareness of the 'e-cigarette' in the USA. Tob Control. 2013;22(1):19–23.
Kalkhoran S, Glantz SA. E-cigarettes and smoking cessation in real-world and clinical settings: a systematic review and meta-analysis. Lancet Respir Med. 2016;4(2):116–28.
Rowell TR, Tarran R. Will chronic e-cigarette use cause lung disease? Am J Physiol Lung Cell Mol Physiol. 2015;309(12):L1398–409.
Orellana-Barrios MA, Payne D, Mulkey Z, Nugent K. Electronic cigarettes-a narrative review for clinicians. Am J Med. 2015;128(7):674–81.
Jankowski M, Broek G, Lawson J, Skoczyaki S, Zejda JE. E-smoking: emerging public health problem? Int J Occup Med Environ Health. 2017;30(3):329–44.
Hartmann-Boyce J, McRobbie H, Bullen C, Begh R, Stead LF, Hajek P. Electronic cigarettes for smoking cessation. Cochrane Database Syst Rev. 2016;9:CD010216.
Bals R, Boyd J, Esposito S, Foronjy R, Hiemstra PS, Jiménez-Ruiz CA, et al. Electronic Cigarettes – Task Force report from the European Respiratory Society. Eur Respir J. 2019;53:1801151
Layden JE, Ghinai I, Pray I, Kimball A, Layer M, Tenforde M, et al. Pulmonary Illness Related to E-Cigarette Use in Illinois and Wisconsin — Preliminary Report. N Engl J Med. 2019; Epub ahead of print.
Faner R, Sibila O, Agusti A, Bernasconi E, Chalmers JD, Huffnagle GB, et al. The microbiome in respiratory medicine: current challenges and future perspectives. Eur Respir J. 2017;49(4). https://doi.org/10.1183/13993003.02086-2016.
Einarsson GG, Comer DM, McIlreavey L, Parkhill J, Ennis M, Tunney MM, et al. Community dynamics and the lower airway microbiota in stable chronic obstructive pulmonary disease, smokers and healthy non-smokers. Thorax. 2016;71(9):795–803.
Erb-Downward JR, Thompson DL, Han MK, Freeman CM, McCloskey L, Schmidt LA, et al. Analysis of the lung microbiome in the "healthy" smoker and in COPD. PLoS One. 2011;6(2):e16384.
Comer DM, Elborn JS, Ennis M. Comparison of nasal and bronchial epithelial cells obtained from patients with COPD. PLoS One. 2012;7(3):e32924.
Miles AA, Misra SS, Irwin JO. The estimation of the bactericidal power of the blood. J Hyg (Lond). 1938;38(6):732–49.
Stepanovic S, Vukovic D, Hola V, Di Bonaventura G, Djukic S, Cirkovic I, et al. Quantification of biofilm in microtiter plates: overview of testing conditions and practical recommendations for assessment of biofilm production by staphylococci. APMIS. 2007;115(8):891–9.
Insua JL, Llobet E, Moranta D, Pérez-Gutiérrez C, Tomás A, Garmendia J, et al. Modeling Klebsiella pneumoniae pathogenesis by infection of the wax moth galleria mellonella. Infect Immun. 2013;81(10):3552–65.
Harding CR, Schroeder GN, Collins JW, Frankel G. Use of Galleria mellonella as a model organism to study Legionella pneumophila infection. J Vis Exp. 2013;81:e50964.
Cervellati F, Muresan XM, Sticozzi C, Gambari R, Montagner G, Forman HJ, et al. Comparative effects between electronic and cigarette smoke in human keratinocytes and epithelial lung cells. Toxicol in Vitro. 2014;28(5):999–1005.
Lerner CA, Sundar IK, Yao H, Gerloff J, Ossip DJ, McIntosh S, et al. Vapors produced by electronic cigarettes and e-juices with flavorings induce toxicity, oxidative stress, and inflammatory response in lung epithelial cells and in mouse lung. PLoS One. 2015;10(2):e0116732.
Schweitzer KS, Chen SX, Law S, Van Demark M, Poirier C, Justice MJ, et al. Endothelial disruptive proinflammatory effects of nicotine and e-cigarette vapor exposures. Am J Physiol Lung Cell Mol Physiol. 2015;309(2):L175–87.
Sussan TE, Gajghate S, Thimmulappa RK, Ma J, Kim JH, Sudini K, et al. Exposure to electronic cigarettes impairs pulmonary anti-bacterial and anti-viral defenses in a mouse model. PLoS One. 2015;10(2):e0116861.
Ahmad S, Zafar I, Mariappan N, Husain M, Wei C-C, Vetal N, et al. Acute pulmonary effects of aerosolized nicotine. Am J Physiol Lung Cell Mol Physiol. 2019;316(1):L94–L104.
McCarthy M. “Alarming” rise in popularity of e-cigarettes is seen among US teenagers as use triples in a year. BMJ. 2015;350:h2083
Eaton DL, Kwan LY, Stratton K. National Academies of sciences, engineering, and medicine; health and medicine division; board on population health and public health practice; committee on the review of the health effects of electronic nicotine delivery systems: public health consequences of E-cigarettes. 2018.
Bayly JE, Bernat D, Porter L, Choi K. Secondhand exposure to aerosols from electronic nicotine delivery systems and asthma exacerbations among youth with asthma. Chest. 2019;155(1):88–93.
Dickson RP, Erb-Downward J, Huffnagle GB. The role of the bacterial microbiome in lung disease. Expert Rev Respir Med. 2013;7(3):245–57.
Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med. 2008;22:2355–65.
Ishak A, Everard ML. Persistent and recurrent bacterial bronchitis-a paradigm shift in our understanding of chronic respiratory disease. Front Pediatr. 2017;5:19.
Kulkarni R, Caskey J, Singh SK, Paudel S, Baral P, Schexnayder M, et al. Cigarette smoke extract–exposed methicillin-resistant Staphylococcus aureus regulates leukocyte function for pulmonary persistence. Am J Respir Cell Mol Biol. 2016;55(4):586–601.
Antunes MB, Chi JJ, Liu Z, Goldstein-Daruech N, Palmer JN, Zhu J, et al. Molecular basis of tobacco-induced bacterial biofilms. Otolaryngol Head Neck Surg. 2012;147(5):876–84.
McEachern EK, Hwang JH, Sladewski KM, Nicatia S, Dewitz C, Mathew DP, et al. Analysis of the effects of cigarette smoke on staphylococcal virulence phenotypes. Infect Immun. 2015;83(6):2443–52.
Kulkarni R, Antala S, Wang A, Amaral FE, Rampersaud R, Larussa SJ, et al. Cigarette smoke increases Staphylococcus aureus biofilm formation via oxidative stress. Infect Immun. 2012;80(11):3804–11.
Mutepe ND, Cockeran R, Steel HC, Theron AJ, Mitchell TJ, Feldman C, et al. Effects of cigarette smoke condensate on pneumococcal biofilm formation and pneumolysin. Eur Respir J. 2013;41(2):392.
Cockeran R, Herbert JA, Mitchell TJ, Dix-Peek T, Dickens C, Anderson R, et al. Exposure of a 23F serotype strain of Streptococcus pneumoniae to cigarette smoke condensate is associated with selective upregulation of genes encoding the two-component regulatory system 11 (TCS11). Biomed Res Int. 2014;4:976347.
Goldstein-Daruech N, Cope EK, Zhao K, Vukovic K, Kofonow JM, Doghramji L, et al. Tobacco Smoke Mediated Induction of Sinonasal Microbial Biofilms. PLoS One. 2011;6(1):e15700.
Semlali A, Killer K, Alanazi H, Chmielewski W, Rouabhia M. Cigarette smoke condensate increases C. albicans adhesion, growth, biofilm formation, and EAP1, HWP1 and SAP2 gene expression. BMC Microbiol. 2014;14:61 2180-14-61.
Bagaitkar J, Demuth DR, Daep CA, Renaud DE, Pierce DL, Scott DA. Tobacco upregulates P. gingivalis fimbrial proteins which induce TLR2 hyposensitivity. PLoS One. 2010;5(5):e9323.
Huang R, Li M, Ye M, Yang K, Xu X, Gregory RL. Effects of nicotine on Streptococcus gordonii growth, biofilm formation, and cell aggregation. Appl Environ Microbiol. 2014;80(23):7212–8.
Fuchs BB, O'Brien E, Khoury JB, Mylonakis E. Methods for using Galleria mellonella as a model host to study fungal pathogenesis. Virulence. 2010;1(6):475–82.
Brennan M, Thomas DY, Whiteway M, Kavanagh K. Correlation between virulence of Candida albicans mutants in mice and Galleria mellonella larvae. FEMS Immunol Med Microbiol. 2002;34(2):153–7.
Jander G, Rahme LG, Ausubel FM. Positive correlation between virulence of Pseudomonas aeruginosa mutants in mice and insects. J Bacteriol. 2000;182(13):3843–5.
Shapiro SD. End-stage chronic obstructive pulmonary disease: the cigarette is burned out but inflammation rages on. Am J Respir Crit Care Med. 2001;164(3):339–40.
Le Rouzic O, Konak B, Kluza J, Marchetti P, Hennegrave F, Olivier C, et al. Cigarette smoke alters the ability of human dendritic cells to promote anti-Streptococcus pneumoniae Th17 response. Respir Res. 2016;17(1):94.
Pichavant M, Sharan R, Le Rouzic O, Olivier C, Hennegrave F, Rémy G, et al. IL-22 Defect During Streptococcus pneumoniae Infection Triggers Exacerbation of Chronic Obstructive Pulmonary Disease. EBioMedicine. 2015;2(11):1686–96.
Hwang JH, Lyes M, Sladewski K, Enany S, McEachern E, Mathew DP, et al. Electronic cigarette inhalation alters innate immunity and airway cytokines while increasing the virulence of colonizing bacteria. J Mol Med. 2016;94(6):667–79.
Holgate ST, Lackie PM, Davies DE, Roche WR, Walls AF. The bronchial epithelium as a key regulator of airway inflammation and remodelling in asthma. Clin Exp Allergy. 1999;29:90–5.
Adcock IM, Barnes PJ. Molecular mechanisms of corticosteroid resistance. Chest. 2008;134(2):394–401.
Robinson RJ, Hensel EC, Roundtree KA, Difrancesco AG, Nonnemaker JM, Lee YO. Week long topography study of young adults using electronic cigarettes in their natural environment. PLoS One. 2016;11(10):e0164038.
Spindle TR, Breland AB, Karaoghlanian NV, Shihadeh AL, Eissenberg T. Preliminary Results of an Examination of Electronic Cigarette User Puff Topography: The Effect of a Mouthpiece-Based Topography Measurement Device on Plasma Nicotine and Subjective Effects. Nicotine Tob Res. 2014;17(2):142–9.
Ghosh A, Coakley RC, Mascenik T, Rowell TR, Davis ES, Rogers K, et al. Chronic E-cigarette exposure alters the human bronchial epithelial proteome. Am J Respir Crit Care Med. 2018;198(1):67–76.
Clapp PW, Lavrich KS, van Heusden CA, Lazarowski ER, Carson JL, Jaspers I. Cinnamaldehyde in flavored E-cigarette liquids temporarily suppresses bronchial epithelial cell Ciliary motility by Dysregulation of mitochondrial function. Am J Phys Lung Cell Mol Phys. 2019;316(3):L47–L486.
Acknowledgments
Not applicable.
Funding
KM and KG were supported by studentships from the Department for Education and Learning in Northern Ireland.
Author information
Authors and Affiliations
Contributions
DG was the major contributor in writing this manuscript; DG and KM analysed and interpreted the data; KM, KG and AD carried out the experimental work; GE provided statistical oversight; JB, JSE and MT provided critical revisions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Supplementary figures and tables.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Gilpin, D.F., McGown, KA., Gallagher, K. et al. Electronic cigarette vapour increases virulence and inflammatory potential of respiratory pathogens. Respir Res 20, 267 (2019). https://doi.org/10.1186/s12931-019-1206-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-019-1206-8