
Abstract
Background
Little is known on the characteristics of patients diagnosed with idiopathic pulmonary fibrosis (IPF) in Spain. We aimed to characterize the demographic and clinical profile of IPF patients included in the IPF National Registry of the Spanish Respiratory Society (SEPAR).
Methods
This is a prospective, observational, multicentre and nationwide study that involved 608 IPF patients included in the SEPAR IPF Registry up to June 27th, 2017, and who received any treatment for their disease. IPF patients were predominantly males, ex-smokers, and aged in their 70s, similar to other registries.
Results
Upon inclusion, mean ± SD predicted forced vital capacity was 77.6% ± 19.4, diffusing capacity for carbon monoxide was 48.5% ± 17.7, and the 6-min walk distance was 423.5 m ± 110.4. The diagnosis was mainly established on results from the high-resolution computed tomography in the proper clinical context (55.0% of patients), while 21.2% of patients required invasive procedures (surgical lung biopsy) for definitive diagnosis. Anti-fibrotic treatment was prescribed in 69.4% of cases, 51.5% pirfenidone and 17.9% nintedanib, overall with a good safety profile.
Conclusions
The SEPAR IPF Registry should help to further characterize current characteristics and future trends of IPF patients in Spain and compare/pool them with other registries and cohorts.
Similar content being viewed by others


Background
Idiopathic pulmonary fibrosis (IPF) is a fatal, chronic fibrosing interstitial pneumonia, of unknown aetiology, which affects primarily adults older than 50 [1,2,3]. Although there is a great variability in the occurrence of IPF, possibly due to geographic and demographic differences, the most reliable data estimate a prevalence ranging approximately 13–20 per 100,000 inhabitants in women and men, respectively [4]. The IPF mean survival ranges between 2 and 4 years from diagnosis for patients not receiving anti-fibrotic treatment [5]. Some factors have been identified to be associated with poorer prognosis and shorter survival time, such as older age, smoking status (smokers and ex-smokers), lower body mass index, more impaired pulmonary function (mainly on forced vital capacity, FVC, total lung capacity, TLC, and diffusing capacity for carbon monoxide, DLCO), radiological findings (usual interstitial pneumonia, UIP), a pattern or greater extent of fibrosis, and the development of acute exacerbations or comorbidities, especially pulmonary hypertension and emphysema [6,7,8,9,10]. The diagnosis of IPF requires the collaboration of a multidisciplinary team of specialists to integrate and interpret complex clinical information [11, 12]. Anti-fibrotic treatments for IPF aim to slow down the disease progression and increase the survival time [13, 14]. To date, there are two effective disease-modifying therapies, pirfenidone and nintedanib [2]. Besides the performance of clinical trials for investigating the efficacy and safety of novel drugs, observational studies from routine clinical practice are also required for understanding the natural course of the disease, and identifying differential patterns of diagnosis and treatments [15,16,17]. Several national IPF registries have been created worldwide; however published results are still scarce [18,19,20,21,22,23,24]. The Spanish Society of Pneumology and Thoracic Surgery (‘Sociedad Española de Neumología y Cirugía Torácica’, SEPAR) started in 2012 a National IPF Registry aimed to know the clinical characteristics of IPF patients, procedures for diagnosis, and the evolution of patients in Spain. The primary objective of the present study was to characterize the demographic and clinical profile of IPF patients included in the SEPAR IPF National Registry, regardless of any received treatment.
Methods
Study design
This prospective, observational, multicentre and nationwide study involved patients with IPF who were included in the SEPAR IPF National Registry and received any treatment for their disease. A total of 28 public hospitals, widely distributed through Spain, participated in the study by including patients in the Registry. Patients were eligible if confirmed diagnosis of IPF. The diagnosis of IPF was based on criteria from international clinical guidelines [10]. Those cases receiving pirfenidone for at least 12 months were analysed to evaluate treatment effects in the real-world clinical practice. Procedures were in accordance with guidelines established in the Declaration of Helsinki, and with the principles of Good Clinical Practices. We have followed and endorsed the Strengthening the Reporting of Observational studies in Epidemiology (STROBE) guidance for reporting observational evidence [24]. Each participating hospital obtained the ethic approval from the Human Research Ethics Committee.
Data collection and statistical analysis
All pulmonologists from SEPAR were invited to participate in this IPF Registry. They collected the information during routine visits, and uploaded data to the SEPAR website, up to June 27th 2017 [25]. The first patient included was in January 10th, 2012. Database lock occurred in October 5th, 2017. Continuous variables are expressed as the mean, standard deviation (SD), whereas categorical variables as absolute and relative frequencies (%). Median survival time was determined including the corresponding 95% confidence interval (95% CI). Significant prognostic factors associated with mortality were identified by using a backward Cox regression analysis (Hazard ratio, HR). Survival was analysed by Kaplan-Meier methodology. Variables included in the analysis were: age, FVC (% of predicted) at diagnosis, DLCO at diagnosis, anti-fibrotic treatment (yes/no), proton-pump inhibitors (yes/no), reported comorbidities such as pulmonary emphysema (yes/no) or pulmonary hypertension (yes/no), and smoking habits. The patient comorbidities were reported by each participant and the Charlson comorbidity index was calculated after including the data in the Registry. All statistical procedures were performed by using SAS 9.4 software.
Results
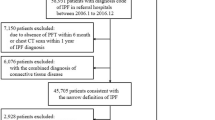
From 713 patients included in the SEPAR IPF National Registry, 105 were finally excluded (Fig. 1). Therefore, the number of patients evaluable for the primary endpoint was 608. Regarding participating centres, 18 were interstitial lung disease (ILD) academic centres, and 10 non-ILD academic centres [26].

Flowchart of patients. 608 patients were evaluable for the main objective of the study (IPF Spanish patient characterization). 231 patients that received pirfenidone for at least 12 months were analysed to evaluate safety of treatment and clinical features of this subgroup of cases
Registry patients
Demographics
Patients were predominantly male (80.8%), with a mean age of 70.2 years (SD 9.2), a mean body mass index of 28.2 kg/m2 (SD 4.2), and ex-smokers (63.7%). Demographic and clinical characteristics of patients are shown in Table 1. Time from the onset of symptoms to diagnosis was 20.4 months (SD 21.4).
Comorbidities
The Charlson comorbidity index was 3.5 (SD 1.7). Diabetes mellitus with no target organ damage (15.8% of patients), chronic respiratory disease (15.6%), arterial hypertension (14%), gastroesophageal reflux (12.8%), pulmonary emphysema (12.1%), and coronary artery disease (8.6%) were the most frequent comorbidities.
IPF characteristics
Regarding symptoms or signs indicative of IPF at the time of diagnosis, 89.6% of patients had inspiratory bibasilar crackles, 84.7% dyspnoea (mainly grade 2 or 1), 62.8% non-productive cough, and 29.4% digital clubbing. The mean FVC was 77.6% of predicted (SD 19.4), mean DLCO was 48.5% of predicted (SD 17.7), mean TLC was 72.5% of predicted (SD 16.5), and the 6-min walk distance (6MWD) was 423.5 m (SD 110.4; Table 2).
Diagnostic procedures
The high-resolution computed tomography (HRCT) was the most frequent procedure performed at diagnosis (99.2% of patients), followed by pulmonary function tests (98.3%), autoimmune serology (91.8%), chest radiography (81.7%), and the 6-min walking test (71.2%). The confident radiological UIP pattern was found in 65.4% of cases (Table 3). The definitive diagnosis of IPF was established in the 55% of cases by the results of the HRCT in the proper clinical context (after evaluation by ILD clinical and radiological experts). A multidisciplinary discussion with the whole ILD committee was required for definitive IPF diagnosis in 45% of cases; 21.2% of patients that underwent surgical lung biopsy (surgical or transbronchial cryobiopsy), and 23.8% without biopsy.
Treatment approach
Patients were receiving pirfenidone (51.5%) or nintedanib (17.9%) as disease-modifying therapies for IPF at inclusion in the Registry. Main concurrent treatments were: proton-pump inhibitors (68.9%), oxygen therapy (21.5%), or oral corticosteroids (17.8%). From 30.6% of cases not anti-fibrotic treatment: 8.5% were > 85 years, 43.5% presented an FVC > 80%. Of those treated patients, 24.7% experienced at least one adverse event, such as gastrointestinal discomfort (14.0% of patients), anorexia/weight loss (5.9%), alteration of liver enzymes (3.3%), and photosensitivity (2.6%). The adverse event (AE) was the reason for discontinuing the treatment in 27 patients (4.4% of total): pirfenidone (n = 15), nintedanib (n = 11), or oral corticosteroids (n = 1). Recommended non-pharmacological treatment such as rehabilitation and lung transplant were performed in 10.1 and 3.1%, respectively.
Survival
A total of 108 patients (17.8%) died during the follow-up, 88 male (81.48%) and 20 females (18.51%) (HR 1.5; 95% 0.94–2.3, p = 0.092) (Fig. 2a). The causes of death were: disease progression (45.4%), disease exacerbation (15.7%), lung cancer (5.6%), post-lung transplantation (3.7%), and others /unknown (29.6%). The median survival time was 5.8 years (95 CI 4.8–6.6) since diagnosis. The DLCO at diagnosis was the only prognostic factor associated with mortality (HR 0.609; 95%CI 0.525–0.706). A patient had 39.1% lower risk of death per 10 units of DLCO (%) increased.

Mortality and survival analyze depending on gender and pirfenidone treatment. a No statistically significant higher mortality was observed in males (HR 1.5; 95% 0.94–2.3, p = 0.092). b There was no statistically significant difference in mortality among pirfenidone treated patients depending on gender (HR 1.6; 95% 0.86–3, p = 0.139. c Survival time (weeks) analyzed by gender and pirfenidone treatment was not statistically different
Patients receiving pirfenidone
A total of 231 patients received pirfenidone for at least 12 months. Patients were predominantly male (79.7%), with a mean age of 68.2 years (SD 9.2), and ex-smokers (68.4%; Table 4). In these patients, the definitive diagnosis was established by results of the HRCT (52.2%), after undergoing the surgical lung biopsy (27.6%), or by multidisciplinary discussion (20.2%).
Changes in IPF characteristics
Patients receiving pirfenidone showed a stable lung function in FVC (71.5% of predicted, SD 16.7) and DLCO (47.2% of predicted, SD 17.6) after 12 months of treatment (compared with baseline, 74.1% of predicted, SD 15.5 for FVC; and 47.4% of predicted, SD 16.9 for DLCO). The mean 6MWT distance was similar after 12 months of treatment (429.9 m, SD 117.4) than at baseline (425.5 m, SD 114.7).
Safety profile
Of patients receiving pirfenidone, 23.4% experienced at least one adverse event (Table 4). Of 231 patients receiving pirfenidone, 15.2% had to modify the treatment during the follow-up period. Reasons of treatment modification (dose reduction n = 10, discontinuing the treatment n = 9) were as follows: clinical worsening of disease (3.5%), AEs (2.2%) and requiring concomitant medications (1.3%).
Survival
Eight patients receiving pirfenidone (3.5%) died during the first 12-month period of treatment. A total of 55 patients (23.8%) died during the follow-up. 87.3% were male and 12.7% female (Fig. 2b). The median survival time was 5.8 years (95 CI 4.2–9.2) since diagnosis (Fig. 2c). Causes of death were: disease progression (32 patients, 58.2%), disease exacerbation (8 patients, 14.5%), lung cancer (5 patients, 9.1%), and others /unknown (10 patients, 18.2%).
Discussion
Limited information is available about the demographic and clinical profile of IPF patients in Spain, the diagnostic decision-making, and treatments for IPF in real-life setting. To our knowledge, data from 7 national IPF registries have published so far [18,19,20,21,22,23,24]. While some IPF features are common in all countries such as male predominance, mean age, and smoking history, other demographic and clinical data differ from other registries, especially mean FVC, DLCO and 6MWD at inclusion, or the basis for the final diagnosis (Table 5). Probably, the heterogeneity of data among registries would depend on the different methodology and type of centres. Some authors have thus suggested creating a global IPF registry, or connecting current IPF networks, such as the ARIANE-IPF pan-European IPF registry and biobank [28]. The goal of the present study is to publish for the first time results from the Spanish IPF national registry on the profile of patients with IPF in routine clinical practice.
In agreement with international consensus, in most cases the diagnosis was based on typical HRCT images in the clinical context [11, 12, 29,30,31,32]. 45% of cases required a case-discussion by the whole ILD multidisciplinary committee. Walsh and colleagues showed a good agreement for the IPF diagnosis between pulmonologists, independently of the type of centre (academic or non-academic centres), with higher concordance in those cases with ILD MDT availability (32). The Fleischner Society recently stated that a confident IPF diagnosis can be achieved when HRCT shows a typical or probable UIP pattern [12]. On the other hand, the MDT discussion of each potential IPF case with probable, possible or inconsistent UIP pattern is recommended in the updated IPF guideline (11). Once made the diagnosis of IPF, the treatment with anti-fibrotics should start as soon as possible [33]. In our study 51.5 and 17.9% of the participants were receiving pirfenidone or nintedanib, respectively. Besides this, some pulmonologists seem reluctant to treat patients with “mild” or “stable” disease, and thus they perform a wait and see approach, probably for avoiding potential AEs or due to misunderstanding by pulmonologists [34, 35]. An international survey revealed that only 40% of patients with a confirmed diagnosis will receive anti-fibrotic treatment; and among untreated patients, 45% receive no treatment at all [34]. Another survey has recently shown that pulmonologists who initiated the anti-fibrotic treatment after more than 4 months of patient’s diagnosis (46% of total) saw fewer patients and had less confidence in the treatment than those who initiated it in ≤4 months [36]. On the other hand, patients with IPF have a higher risk of developing comorbidities [37]. In our study, 12.8% of our patients had gastro-oesophageal reflux, 12.1% pulmonary emphysema, 8.6% coronary artery disease, and 6.2% pulmonary hypertension. It is interesting to note the low number of cases of gastro-oesophageal reflux or cardiovascular disease, compared with literature. Previous studies have shown a prevalence of 87 and 66% for gastro-oesophageal reflux and coronary artery disease in IPF, respectively [38, 39]. Some studies have demonstrated an association between decreased disease progression and longer survival time and the treatment of gastro-oesophageal reflux with antacid [40]; whereas other have not so [41]. In our study, the high percentage of patients receiving proton-pump inhibitors (65.3%) does contrast with the low percentage of patients diagnosed with symptomatic gastro-oesophageal reflux. One explanation is that these treatments were prescribed at the time of IPF diagnosis, before the beginning of the “anti-fibrotic” era. Furthermore, in our study, approximately one in ten patients had family history of IPF, and 55.9% of patients experienced occupational exposures (inorganic, organic particles, or potentially harmful aerosols). In this line, diverse studies have reported an increased risk of UIP in workers exposed to fumes, metal or organic dust [42, 43].
We aimed to describe pirfenidone in clinical practice because it was the first anti-fibrotic available in Spain (more than 2 years before nintedanib). In our Registry, up to 313 patients (51.5%) received treatment with pirfenidone. Despite the proven effectiveness of pirfenidone [34], when receiving treatment, there is always a subgroup of patients who experience inadequate response to therapy. Patients who continue treatment with pirfenidone after having disease progression by month 6 of treatment have a lower risk of FVC decline or death during the subsequent 6 months of treatment [44]. For this reason, it seems recommendable to maintain the treatment with pirfenidone for, at least, 12 months. In our study 8 patients (3.5%) experienced clinical worsening during the treatment with pirfenidone, and 9 patients (3.9%) discontinued treatment. This percentage of discontinuation is slightly lower than previous studies, such as CAPACITY (7.5 and 5.8% of patients), or ASCEND (16%) [45, 46]. It is interesting to note that in our study the median survival rate of total patients and those receiving pirfenidone was similar (5.8 years). This result is in disagreement with other studies, such as the European Registry (EurIPFreg) which reported a significant improvement in survival rate in patients receiving anti-fibrotic treatment (mean 123.1 months; 83% of cases with pirfenidone and 17% with nintedanib) after 7 years of follow-up, comparing with patients not receiving it (mean 68.3 months) [24]. Although no definitive explanation can be provided, we suppose it is because pirfenidone has been only available to patients with FVC < 80% for a long time in most of Spanish hospitals. This fact might have limited the rate of survival in patients receiving pirfenidone. Finally, DLCO at diagnosis was the only factor significantly associated with mortality. In our study, a patient had 39.1% lower risk of death per 10 units of DLCO (%) increased. The impact of DLCO on survival has already been described in previous studies [47, 48]. In fact, some indices combine DLCO together with FVC (the Gender Age Physiology score) and with forced expiratory volume in 1 s (the Composite Physiological Index) for predicting mortality [49, 50].
Main limitations of our study were intrinsically related to the retrospective nature of data collection in the first participants in the Registry (those diagnosed before 2011). Presumably, no available data would improve the knowledge in management of IPF in clinical practice. For example, we only collected information of treatments at the time of inclusion. There is thus a lack of information regarding when they actually received the treatment or whether or not the patient received a new treatment during the follow-up. Another limitation derived from the heterogeneity of patients (including mild, moderate and severe disease) and some uncertainty associated with the diagnostic process, i.e. integrating information from different healthcare professionals, such as clinicians, thoracic radiologists, and pathologists; with varying degrees of experience; and different sites (university or non-university facilities, with or without access to multidisciplinary team meetings) [32]. Furthermore, we couldn’t identify those cases diagnosed based on disease behaviour (working diagnosis), which probably could be part of the IPF cases without lung biopsy that required multidisciplinary discussion. In this regard, SEPAR has recently created a registry of Spanish hospitals according to level of ILD expertise [27]. Differences in the access to medications among Spanish regions may also contribute to heterogeneity. Another limitation was that the type of centre of recruitment (ILD specialist or non- ILD specialist academic centres) may have biased the results as ILD specialist centres could preferentially enrol patients in clinical trials (an exclusion factor for the present study) or prescribe antifibrotic medication. Regarding the 5.6% (19 male/9 female) of cases with low titter of non-specific positive auto-antibodies, all of them had been evaluated by an expert rheumatologist, excluding the association with connective tissue diseases. Albeit, only pulmonologists from ILD and non- ILD specialist centres recruited the patients. Furthermore, this database was not established to evaluate the safety profile or effectiveness of pirfenidone, thus conclusions given with reference to this should be made carefully. Although a higher number of centres would strength results and conclusions, our cohort of patients is representative of the whole population of patients with IPF in Spain. This valuable information can be used in subsequent studies to build prediction models for Spanish patients with IPF. Another goal of the study is that all patients derived from public hospitals, having the same (free) access to procedures and medications.
Conclusions
Demographic characteristics of patients from the SEPAR IPF National Registry are in accordance with other national registries. In agreement with international guidelines, the diagnosis is mainly based on HRCT in the proper clinical context. A low percentage of patients require invasive procedures for the definitive diagnosis. The treatment with pirfenidone is generally safe and well tolerated, and most cases do not present disease progression after 12 months. Additional studies, including more patients and centres to the Registry, are required to corroborate these results. This SEPAR IPF Registry should help to further characterize current characteristics and future trends of IPF patients in Spain, and compare/pool them with other registries and cohorts.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- 95% CI:
-
95% confidence interval
- AE:
-
Adverse Event
- DLCO :
-
Diffusing Capacity for Carbon Monoxide
- FVC:
-
Forced Vital Capacity
- HR:
-
Hazard ratio
- HRCT:
-
The high-resolution computed tomography
- ILD:
-
Interstitial Lung Disease
- IPF:
-
Idiopathic pulmonary fibrosis
- SD:
-
Standard Deviation
- SEPAR:
-
National Registry of the Spanish Respiratory Society
- STROBE:
-
The Strengthening the Reporting of Observational studies in Epidemiology
- TLC:
-
Total lung capacity
- UIP:
-
Usual Interstitial Pneumonia
References
Martinez FJ, Safrin S, Weycker D, Starko KM, Bradford WZ, King TE Jr, Flaherty KR, Schwartz DA, Noble PW, Raghu G, et al. The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med. 2005;142:963–7.
Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389:1941–52.
Sgalla G, Iovene B, Calvello M, Ori M, Varone F, Richeldi L. Idiopathic pulmonary fibrosis: pathogenesis and management. Respir Res. 2018;19:32.
Ley B, Collard HR. Epidemiology of idiopathic pulmonary fibrosis. Clin Epidemiol. 2013;5:483–92.
Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011;183:431–440.
Erbes R, Schaberg T, Loddenkemper R. Lung function tests in patients with idiopathic pulmonary fibrosis. Are they helpful for predicting outcome? Chest. 1997;111:51–7.
King TE Jr, Tooze JA, Schwarz MI, Brown KR, Cherniack RM. Predicting survival in idiopathic pulmonary fibrosis. Scoring system and survival model. Am J Respir Crit Care Med. 2001;164:1171–81.
Alakhras M, Decker PA, Nadrous HF, Collazo-Clavell M, Ryu JH. Body mass index and mortality in patients with idiopathic pulmonary fibrosis. Chest. 2007;131:1448–53.
Sumikawa H, Johkoh T, Colby TV, Ichikado K, Suga M, Taniguchi H, Kondoh Y, Ogura T, Arakawa H, Fujimoto K, et al. Computed tomography findings in pathological usual interstitial pneumonia: relationship to survival. Am J Respir Crit Care Med. 2008;177:433–9.
Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824.
Raghu G, Remy-Jardin M, Myers J, Richeldi L, Ryerson C, Lederer D, Behr J, Cottin V, Danoff S, Morell F, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198:e44–68.
Lynch DA, Sverzellati N, Travis WD, Brown KK, Colby TV, Galvin JR, Goldin JG, Hansell DM, Inoue Y, Johkoh T, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner society white paper. Lancet Respir Med. 2018;6:138–53.
Center for Drug Evaluation and Research (CDER), U.S. Food and Drug Administration (FDA). The voice of the patient: a series of reports from the U.S. Food and Drug Administration's (FDA's) patient-focused drug development initiative. [Accessed 03 June 2018]. Available from URL: http://www.fda.gov/downloads/ForIndustry/UserFees/PrescriptionDrugUserFee/UCM440829.pdf.
European Medicines Agency. Esbriet (Pirfenidone). CHMP assessment report. [Accessed 03 June 2018]. Available from URL: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002154/WC500103073.pdf.
Antoniou KM, Margaritopoulos GA, Siafakas NM. Pharmacological treatment of idiopathic pulmonary fibrosis: from the past to the future. Eur Respir Rev. 2013;22:281–91.
Wuyts WA, Agostini C, Antoniou KM, Bouros D, Chambers RC, Cottin V, Egan JJ, Lambrecht BN, Lories R, Parfrey H, et al. The pathogenesis of pulmonary fibrosis: a moving target. Eur Respir J. 2013;41:1207–18.
Cordier JF, Cottin V. Neglected evidence in idiopathic pulmonary fibrosis: from history to earlier diagnosis. Eur Respir J. 2013;42:916–23.
Behr J, Kreuter M, Hoeper MM, Wirtz H, Klotsche J, Koschel D, Andreas S, Claussen M, Grohé C, Wilkens H, et al. Management of patients with idiopathic pulmonary fibrosis in clinical practice: the INSIGHTS-IPF registry. Eur Respir J. 2015;46:186–96.
Kaunisto J, Kelloniemi K, Sutinen E, Hodgson U, Piilonen A, Kaarteenaho R, Mäkitaro R, Purokivi M, Lappi-Blanco E, Saarelainen S, et al. Re-evaluation of diagnostic parameters is crucial for obtaining accurate data on idiopathic pulmonary fibrosis. BMC Pulm Med. 2015;15:92.
Ferrara G, Carlson L, Palm A, Einarsson J, Olivesten C, Sköld M. Idiopathic pulmonary fibrosis in Sweden: report from the first year of activity of the Swedish IPF-registry. Eur Clin Respir J. 2016;3:31090.
Singh S, Collins BF, Sharma BB, Joshi JM, Talwar D, Katiyar S, Singh N, Ho L, Samaria JK, Bhattacharya P, et al. Interstitial lung disease in India. Results of a prospective registry. Am J Respir Crit Care Med. 2017;195:801–13.
Jo HE, Glaspole I, Grainge C, Goh N, Hopkins PM, Moodley Y, Reynolds PN, Chapman S, Walters EH, Zappala C, et al. Baseline characteristics of idiopathic pulmonary fibrosis: analysis from the Australian idiopathic pulmonary fibrosis registry. Eur Respir J. 2017;49(2). https://doi.org/10.1183/13993003.01592-2016.
Doubková M, Švancara J, Svoboda M, Šterclová M, Bartoš V, Plačková M, Lacina L, Žurková M, Binková I, Bittenglová R, et al. EMPIRE registry, Czech part: impact of demographics, pulmonary function and HRCT on survival and clinical course in idiopathic pulmonary fibrosis. Clin Respir J. 2018;12:1526–35.
Guenther A, Krauss E, Tello S, Wagner J, Paul B, Kuhn S, Maurer O, Heinemann S, Costabel U, Barbero MAN, et al. The European IPF registry (eurIPFreg): baseline characteristics and survival of patients with idiopathic pulmonary fibrosis. Respir Res. 2018;19:141.
STrengthening the reporting of OBservational studies in epidemiology (STROBE) guidance for reporting observational research. [Accessed 03 June 2018]. Available from URL: http://strobe-statement.org/index.php?id=strobe-home.
SEPAR Website. [accessed 03 June 2018]. Available from URL: https://www.separ.es/?q=node/233.
Spanish Society of Pneumology and Thoracic Surgery (SEPAR). [Unidades acreditadas de enfermedades pulmonares intersticiales difusas]. [Accessed 03 June 2018]. Available from URL: https://www.separ.es/?q=node/826.
Ryerson CJ, Corte TJ, Collard HR, Richeldi L. A global registry for idiopathic pulmonary fibrosis: the time is now. Eur Respir J. 2014;44:273–6.
Xaubet A, Ancochea J, Bollo E, Fernández-Fabrellas E, Franquet T, Molina-Molina M, Montero MA, Serrano-Mollar A. Sociedad Española de Neumología y Cirugía Torácica (SEPAR) research group on diffuse pulmonary diseases. Guidelines for the diagnosis and treatment of idiopathic pulmonary fibrosis. Sociedad Española de Neumología y Cirugía Torácica (SEPAR) research group on diffuse pulmonary diseases. Arch Bronconeumol. 2013;49:343–53.
Raghu G, Lynch D, Godwin JD, Webb R, Colby TV, Leslie KO, Behr J, Brown KK, Egan JJ, Flaherty KR, et al. Diagnosis of idiopathic pulmonary fibrosis with high-resolution CT in patients with little or no radiological evidence of honeycombing: secondary analysis of a randomised, controlled trial. Lancet Respir Med. 2014;2:277–84.
Chung JH, Chawla A, Peljto AL, Cool CD, Groshong SD, Talbert JL, McKean DF, Brown KK, Fingerlin TE, Schwarz MI, et al. CT scan findings of probable usual interstitial pneumonitis have a high predictive value for histologic usual interstitial pneumonitis. Chest. 2015;147:450–9.
Walsh SLF, Maher TM, Kolb M, Poletti V, Nusser R, Richeldi L, Vancheri C, Wilsher ML, Antoniou KM, Behr J, et al. Diagnostic accuracy of a clinical diagnosis of idiopathic pulmonary fibrosis: an international case-cohort study. Eur Respir J. 2017;50(2). https://doi.org/10.1183/13993003.00936-2017.
Antoniou KM, Symvoulakis EK, Anyfantakis D, Wells AU. New treatments for idiopathic pulmonary fibrosis: ‘die another day’ if diagnosed early? Respiration. 2015;90:352.
Maher TM, Molina-Molina M, Russell AM, Bonella F, Jouneau S, Ripamonti E, Axmann J, Vancheri C. Unmet needs in the treatment of idiopathic pulmonary fibrosis-insights from patient chart review in five European countries. BMC Pulm Med. 2017;17:124.
Torrisi SE, Pavone M, Vancheri A, Vancheri C. When to start and when to stop antifibrotic therapies. Eur Respir Rev. 2017;26(145). https://doi.org/10.1183/16000617.0053-2017.
Maher TM, Swigris JJ, Kreuter M, Wijsenbeek M, Cassidy N, Ireland L, Axmann J, Nathan SD. Identifying barriers to idiopathic pulmonary fibrosis treatment: a survey of patient and physician views. Respiration. 2018;96:514–24.
Raghu G, Amatto VC, Behr J, Stowasser S. Comorbidities in idiopathic pulmonary fibrosis patients: a systematic literature review. Eur Respir J. 2015;46:1113–30.
Raghu G, Freudenberger TD, Yang S, Curtis JR, Spada C, Hayes J, Sillery JK, Pope CE 2nd, Pellegrini CA. High prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis. Eur Respir J. 2006;27:136–42.
Nathan SD, Basavaraj A, Reichner C, Shlobin OA, Ahmad S, Kiernan J, Burton N, Barnett SD. Prevalence and impact of coronary artery disease in idiopathic pulmonary fibrosis. Respir Med. 2010;104:1035–41.
Lee JS, Collard HR, Anstrom KJ, Martinez FJ, Noth I, Roberts RS, Yow E, Raghu G, Investigators IPF. Anti-acid treatment and disease progression in idiopathic pulmonary fibrosis: an analysis of data from three randomised controlled trials. Lancet Respir Med. 2013;1:369–76.
Kreuter M, Wuyts W, Renzoni E, Koschel D, Maher TM, Kolb M, Weycker D, Spagnolo P, Kirchgaessler KU, Herth FJ, et al. Antacid therapy and disease outcomes in idiopathic pulmonary fibrosis: a pooled analysis. Lancet Respir Med. 2016;4:381–9.
Barber CM, Fishwick D. Importance of past occupational exposures in the rising incidence of idiopathic pulmonary fibrosis in the U.K. Thorax. 2012;67:264–5.
Paolocci G, Folletti I, Torén K, Ekström M, Dell'Omo M, Muzi G, Murgia N. Occupational risk factors for idiopathic pulmonary fibrosis in southern Europe: a case-control study. BMC Pulm Med. 2018;18:75.
Nathan SD, Albera C, Bradford WZ, Costabel U, du Bois RM, Fagan EA, Fishman RS, Glaspole I, Glassberg MK, Glasscock KF, et al. Effect of continued treatment with pirfenidone following clinically meaningful declines in forced vital capacity: analysis of data from three phase 3 trials in patients with idiopathic pulmonary fibrosis. Thorax. 2016;71:429–35.
Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, King TE Jr, Lancaster L, Sahn SA, Szwarcberg J, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377:1760–9.
King TE Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, Gorina E, Hopkins PM, Kardatzke D, Lancaster L, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–92.
Kim HJ, Perlman D, Tomic R. Natural history of idiopathic pulmonary fibrosis. Respir Med. 2015;109(6):661-70.
du Bois RM, Weycker D, Albera C, Bradford WZ, Costabel U, Kartashov A, King TE Jr, Lancaster L, Noble PW, Sahn SA, et al. Forced vital capacity in patients with idiopathic pulmonary fibrosis: test properties and minimal clinically important difference. Am J Respir Crit Care Med. 2011;184:1382–9.
Wells AU, Desai SR, Rubens MB, Goh NS, Cramer D, Nicholson AG, Colby TV, du Bois RM, Hansell DM. Idiopathic pulmonary fibrosis: a composite physiologic index derived from disease extent observed by computed tomography. Am J Respir Crit Care Med. 2003;167:962–9.
Ley B, Ryerson CJ, Vittinghoff E, Ryu JH, Tomassetti S, Lee JS, Poletti V, Buccioli M, Elicker BM, Jones KD, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156:684–91.
Acknowledgements
Authors would like to thank to A. Xaubet for his contribution to the IPF Registry initiation. Authors would also like to express their gratitude to Roche Farma and Meisys for assisting in the preparation of the manuscript. The list of additional researchers participating in the present SEPAR National Registry is as follows (in alphabetical order): Aburto Barrenetxea, M. (Hospital Galdakao-Usansolo, Bizkaia); Alfageme Michavila, I. (Hospital Universitario Virgen de Valme, Sevilla); Bollo de Miguel, E. (Complejo Asistencial Universitario de León, León); Cano, E. (Hospital Universitario Lucus Augusti, Lugo); Casanova Espinosa, A. (Hospital Universitario del Henares, Madrid); Castillo Villegas, D. (Hospital de la Santa Creu i Sant Pau, Barcelona); Figuerola Mendal, J.A. (Hospital Clínico Universitario Lozano Blesa, Zaragoza); García Sevila, R. (Hospital General Universitario de Alicante, Alicante); Gaudó Navarro, J.I. (Hospital Universitario Ramón y Cajal, Madrid); Gómez Carrera, L. (Hospital Universitario La Paz, Madrid); González, J.M. (Hospital Universitario de Salamanca, Salamanca); Herrera Lara, S. (Hospital Universitario Doctor Peset, Valencia); Laporta Hernández, R. (Hospital Universitario Puerta de Hierro Majadahonda, Madrid); Marín González, M. (Hospital Clínico Universitario de Valencia, Valencia); Nieto Barbero, M.A. (Hospital Clínico San Carlos, Madrid); Portillo, K. (Hospital Universitari Germans Trias i Pujol, Barcelona); Romero, A.D. (Hospital Universitario Virgen de las Nieves, Granada); Sánchez Simón-Talero, R. (Hospital Universitario Nuestra Señora del Perpetuo Socorro, Albacete); Sancho Chust, J.N. (Hospital Universitari Sant Joan d’Alacant, Alicante); Sellarés Torres, J. (Hospital Clínic de Barcelona. Instituto del Tórax, Barcelona); Soler Sempere, M.J. (Hospital General Universitario de Elche, Alicante); Sauleda Roig, J. (Hospital Universitari Son Espases, Palma, Illes Balears); Tomás López, L. (Hospital Universitario de Araba. Txagorritxu. Álaba); Villanueva Montes, M. (Hospital San Agustin, Asturias).
Funding
This study was funded by Roche Farma SA Spain.
Author information
Authors and Affiliations
Author notes
Antoni Xaubet is deceased. This paper is dedicated to his memory.
- Antoni Xaubet
Consortia
Contributions
EF-F, MM-M and JB participated in the study design. All authors contributed in the data acquisition. JB, JAR, JA and CV analysed the data. EFF and JB did the manuscript drafting; and EFF and MMM performed the critical manuscript revision. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Each participating hospital obtained the ethic approval from the Human Research Ethics Committee.
Consent for publication
Not applicable.
Competing interests
EF-F received funding for research grants and advisory boards from Hoffman-La Roche, Boeringher Ing., GSK, Chiesi, Esteve Teijin Healthcare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fernández-Fabrellas, E., Molina-Molina, M., Soriano, J.B. et al. Demographic and clinical profile of idiopathic pulmonary fibrosis patients in Spain: the SEPAR National Registry. Respir Res 20, 127 (2019). https://doi.org/10.1186/s12931-019-1084-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-019-1084-0