
Abstract
Extensive studies of the tumor microenvironment (TME) in the last decade have reformed the view of cancer as a tumor cell-centric disease. The tumor microenvironment, especially termed the "seed and soil" theory, has emerged as the key determinant in cancer development and therapeutic resistance. The TME mainly consists of tumor cells, stromal cells such as fibroblasts, immune cells, and other noncellular components. Within the TME, intimate communications among these components largely determine the fate of the tumor. The pivotal roles of the stroma, especially cancer-associated fibroblasts (CAFs), the most common component within the TME, have been revealed in tumorigenesis, tumor progression, therapeutic response, and tumor immunity. A better understanding of the function of the TME sheds light on tumor therapy. In this review, we summarize the emerging understanding of stromal factors, especially CAFs, in cancer progression, drug resistance, and tumor immunity with an emphasis on their functions in epigenetic regulation. Moreover, the importance of epigenetic regulation in reshaping the TME and the basic biological principles underpinning the synergy between epigenetic therapy and immunotherapy will be further discussed.
Similar content being viewed by others

Background
Cancer is one of the main life-threatening diseases worldwide. Although substantial improvements have been achieved in early detection and treatment paradigms, tumor recurrence, metastasis, and therapeutic resistance remain the major challenges in almost all kinds of cancers. A better understanding of the underlying mechanism of tumor development and progression may provide opportunities for cancer treatment.
Genomic instability and mutation in cancer cells have been considered the fundamental driving characteristics during cancer progression; therefore, substantial studies have largely been restricted to the epithelial component of tumors. However, a tumor is not an island originating from cancer cells but rather a complex cellular ecosystem. The “seed and soil” theory was first proposed by Stephen Paget in 1889 to interpret the preferences of cancer cells when metastasizing to organs [1, 2]. For the first time, this theory emphasizes the importance of the host environment to the appearance of tumor metastasis in addition to the intrinsic properties of cancer cells. It also has important reference significance for understanding tumorigenesis, tumor progression, and drug resistance in cancer. The maintenance and progression of tumors are highly supported by the tumor microenvironment (TME), also termed the tumor stroma, which mainly includes fibroblasts, immune cells, the basement membrane, the extracellular matrix, and the vasculature [3]. As the most abundant cell type in the TME, cancer-associated fibroblasts, which transdifferentiate from their main precursors, normal fibroblasts, play pivotal roles in tumor progression. The interplay between cancer cells and CAFs, which affects tumorigenesis and tumor development from almost every aspect, has become increasingly unveiled in recent years.
Epigenetic regulations, including DNA methylation, histone modification, chromatin remodeling, and noncoding RNA regulation, change gene expression without affecting germline DNA sequences. In addition to genetic mutations, epigenetic dysfunction is recognized as an important hallmark of cancer. It has been widely recognized that epigenetic alterations can drive oncogenesis and promote cancer progression by extensively regulating the aberrant expression of cancer-associated and immune-related genes. In cancer cells, the transcription of proto-oncogenes is commonly boosted as a result of promoter hyperacetylation, while tumor suppressor genes are repressed by promoter hypoacetylation and DNA hypermethylation. Many cancers show a global loss of DNA methylation across the genome, with gains of DNA methylation primarily at CpG islands, suppressing genes that control cancer progression [4]. As one of the most important mechanisms, epigenetic dysfunction fundamentally reshapes the tumor microenvironment by altering the phenotypes of cancer cells, tumor-associated stromal cells, and immune cells. The reversibility of epigenetic modifications has enabled pharmacological interventions with potential therapeutic strategies as either monotherapy or in combination with other therapies. In particular, epigenetic agents have shown great potential for synergizing with cancer immunotherapy, such as immune checkpoint blockade (ICB), due to their potent ability to convert the TME into a more immunopermissive type.
In this review, we summarize the latest understanding of stromal factors, especially CAFs, in cancer progression, therapeutic resistance, and tumor immunity with a particular emphasis on their functions in epigenetic regulation. In addition, the importance of epigenetic regulation in reshaping the TME and the basic biological principles underpinning the synergy between epigenetic therapy and immunotherapy will be further discussed.
CAF-tumor cell interaction regulates tumor progression and therapeutic response
As a principal constituent of the tumor stroma, CAFs play a central role in cross-communication between cells in tumors. In this section, we will mainly focus on colorectal cancer representing solid tumors, in which the transcriptional signatures of CAFs rather than tumor cells were robustly associated with poor disease prognosis and relapse across the various classifications [5, 6]. In the consensus molecular subtype (CMS) classification, which described a thoroughly studied and robust stratification strategy for CRC in large patient cohorts, CAFs were closely associated with CRC development [7, 8]. Subtype 4 (CMS4), the mesenchymal subtype, with overrepresentation of the stromal signature, predicted more aggressive tumor stages and worse prognosis.
Secreted effectors and oncogenic signaling
The protumorigenic function of CAFs in CRC can be mainly exerted via secreted effectors such as growth factors, cytokines, chemokines, or exosomes, including the transforming growth factor β (TGF-β) family, interleukin (IL)-6, IL-8, IL-11, Wnt, hepatocyte growth factor (HGF), IL-17A, Netrin-1, leukemia inhibitory factor (LIF), secreted glycoprotein stanniocalcin-1 (STC1), fibroblast growth factor 1 (FGF1), stromal cell-derived factor-1 (SDF-1), and bone morphogenetic proteins (BMPs) [9,10,11,12,13,14,15].
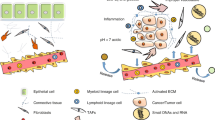
In a noncontact coculture system, conditioned medium from CAFs rather than normal fibroblasts was found to promote the proliferation [16], migration, and invasion [17, 18] of CRC cells. TGF-β is one of the most important cytokines secreted mainly by CAFs and is highly expressed at tumor invasive margins. Interestingly, prominent TGF-β activation was also found in CRC subtype CMS4 [7]. The activation of TGF-β/Smad2 signaling in CAFs stimulated by CRC cells enhances the expression of α-SMA and differentiation of CAFs into a myofibroblastic phenotype, resulting in the expression of invasion-related proteins such as matrix metalloproteinases (MMPs) [19]. By developing patient-derived models to dissect the microenvironmental interaction between CAFs and tumor cells, we also described CAF-secreted TGF-β2, a member of the TGF-β family, inducing the expression of GLI Family Zinc Finger 2 (GLI2), an important effector of Hedgehog signaling, as a predominant pathway to promote CRC stemness and chemoresistance [20] (Fig. 1). Endoglin, a transmembrane accessory receptor of TGF-β that is expressed in CAFs in CRC and its metastatic specimens, is implicated in CAF-mediated invasion and metastasis via TGF-β signaling activation [21]. Additionally, integrin β6 expressed by CRCs was able to increase CAF activation through active TGF-β, and activated CAFs were found to promote CRC cell invasion [11].

CAFs regulate tumor progression and therapeutic response. Our recent work demonstrated that CAFs promote tumor progression and therapeutic resistance through diverse mechanisms. CAFs secrete IL6 and IL8, which activate the JAK2-STAT3 pathway. JAK2-dependent phosphorylated BRD4 interacts with STAT3 to modulate chromatin remodeling (enhancer/super-enhancer reprogramming) and promote oncogene expression, leading to BETi resistance and tumor progression. IL6 also induces LRG1 expression through STAT3-dependent transactivation, which promotes EMT and metastasis. CAF-secreted TGF-β2 induced the expression of GLI2, which synergizes with hypoxia-induced HIF1α to promote CRC stemness and chemoresistance
Interleukin-6/signal transducer and activator of transcription 3 (IL-6/STAT3) signaling is a crucial and well-known pathway mediating malignant progression in CRC [22, 23]. CAFs in the tumor microenvironment play an active role in maintaining STAT3 activation in CRC. Heichler et al. found that the level of p-STAT3 activated by CAF-secreted IL-6/IL-11 was closely correlated with CRC patient survival [24]. TGF-β-stimulated CAFs activate STAT3 signaling in cancer cells, mediating tumor metastasis through the secretion of IL-11 [25]. Furthermore, STAT3 activation in fibroblasts promotes the expression of periostin, a multifunctional extracellular matrix protein, which ultimately facilitates CRC development by Integrin-FAK-Src pathway-mediated YAP/TAZ activation [26]. More recently, our work demonstrated that IL-6 could promote epithelial-mesenchymal transition (EMT), migration, and invasion of CRC cells through upregulation of leucine-rich α-2 glycoprotein 1 (LRG-1), which was found to be a direct transcriptional target of STAT3 [18]. Blocking the IL-6/LRG-1 axis remarkably attenuates metastasis in the xenograft CRC mouse model. IL-6-activated STAT3 in CAFs also regulates transcriptional patterns associated with angiogenesis. In a genetically engineered mouse model, the constitutive activation of STAT3 in fibroblasts promotes CRC growth, which is blocked by the inhibition of proangiogenic signaling [24]. It was also reported that the secretion of IL-6 from CAFs promotes angiogenesis by enhancing the production of the key angiogenic factor, vascular endothelial growth factor A (VEGFA) from endothelial cells [27]. IL-6 was also reported to promote colorectal cancer stem-like properties via induction of fos-related antigen 1 (FRA1) deacetylation [28]. Thus, targeting the microenvironment through STAT3 and its related signaling may provide a promising therapeutic opportunity for CRC treatment. Sanchez-Lopez et al. reported that targeting insulin-like growth factor-1 receptor (IGF-1R) and STAT3 decreased CRC proliferation and increased apoptosis by inhibiting CAF activation and inflammation [29].
The Wnt/beta-catenin signaling, one of the most activated pathways in CRC, was observed preferentially in tumor cells located close to stromal myofibroblasts. Myofibroblast-secreted factors, specifically hepatocyte growth factor (HGF), activate beta-catenin-dependent transcription and subsequently restore the cancer stem cell phenotype in more differentiated tumor cells both in vitro and in vivo [30]. In addition, Straussman et al. found that CAF-secreted HGF activates the MAPK and PI3K-AKT signaling pathways, resulting in resistance to RAF inhibitors in BRAF-mutant cancer cells [31]. In addition, CAF-derived Wnt2 can increase tumor angiogenesis [32] through the upregulation of some proangiogenic proteins and promote cell invasion and migration in CRC [33].
Epigenetic regulation
Accompanying extensive regulation of signaling transduction and the tumor cell transcriptome, rewiring of the epigenetic landscape in tumor cells or CAFs can also modulate the extent and quality of the antitumor treatment response and overall disease outcome. Agrawal et al. discovered that fibroblasts promote cell proliferation and variably affect the expression of DNA methylation-regulating enzymes, hence improving decitabine-induced demethylation in CRC cells and increasing their stemness [34]. Bromodomain-containing protein 4 (BRD4), an epigenetic reader of histone acetylation, is highly expressed in different types of tumor cells, and it can protect these tumor cells against targeted therapy [35,36,37,38] and immunogenic cell death [39,40,41]. Our recent work demonstrated a tumor microenvironment mechanism by which CAF-associated inflammatory paracrine IL6/IL8-JAK2 signaling induces BRD4 activation by phosphorylation in CRC, leading to chromatin reprogramming through increased enhancer and super-enhancer activity. Interestingly, the chromatin remodeling induced by CAF-derived IL6/IL8 was established through the convergence of p-BRD4 and STAT3 co-occupancy on a set of crucial oncogenes associated with tumor growth and metastasis, such as MYC, C-X-C motif chemokine ligand (CXCL)1, and CXCL2. Additionally, noncoding RNAs are also involved in CAF-mediated tumor progression and therapeutic resistance. Several studies have reported that CAFs exert their roles by directly transferring exosomes to CRC cells, leading to a significant increase in noncoding RNA levels in CRC cells [42,43,44]. It was found that all CAF-derived lncRNA CCAL (colorectal cancer-associated lncRNA) [44], lncRNA H19 [43], and miR-92a-3p [42] transferred via exosomes can activate the Wnt/β-catenin pathway in CRC, then suppress cell apoptosis, promote cell stemness, and/or confer resistance to chemotherapy.
In addition to cancer cells, CAFs are also extensively regulated by epigenetic mechanisms in the TME. Leukemia inhibitory factor (LIF) is a cytokine highly expressed in CRC cells that can inhibit cell apoptosis and promote chemoresistance via the activation of STAT3 signaling [45]. Interestingly, LIF was also reported to activate quiescent CAFs by an epigenetic switch. Mechanistically, DNA methyltransferase 3 beta (DNMT3B), a de novo DNA methyltransferase that is activated in a LIF/STAT3-dependent manner, can methylate CpG sites and silence the expression of SHP-1 phosphatase, leading to the activation of CAFs [46]. Adenosine-to-inosine (A-to-I) RNA editing is a newly described epigenetic modification that is thought to represent a crucial carcinogenic mechanism in human malignancies. A study with a large cohort of 627 colorectal cancer (CRC) specimens by Takeda et al. revealed that adenosine deaminase acting on RNA (ADAR), the key enzyme involved in A-to-I RNA editing, was upregulated in both cancer cells and cancer-associated fibroblasts, which increased the RNA edition level of antizyme inhibitor 1 (AZIN1). Interestingly, conditioned medium from cancer cells led to both induction of ADAR1 expression and activation of AZIN1 RNA editing in CAFs, resulting in the increased invasive potential of CAFs within the TME in the colon [47].
These studies clearly showed that the tumor microenvironment is a comprehensive ecosystem in which intimate communication between cancer cells and CAFs fundamentally regulates tumor development and progression. These studies also strengthened that the therapeutic strategies mainly targeting tumor cells might be insufficient to achieve a curative outcome in cancer, which has been repeatedly observed in clinical practice. The tumor stroma supports cancer cell survival and drug resistance after exposure to these tumor-targeting therapies, leading to fatal progression. Interestingly, Lotti et al. discovered a considerable increase in the number of CAFs in CRC patients when chemotherapy was given. These CAFs maintain cancer-initiating cell self-renewal and lead to increased resistance to chemotherapy [48]. Thus, targeting CAFs must be considered to eliminate cancer. Notably, multiple strategies have been developed in preclinical and clinical models to target CAFs and their related pathways [49, 50]. Nevertheless, targeting CAFs alone is unlikely to be efficient in eliminating the whole tumor. Combination strategies that co-target tumor cells and CAFs may elicit tumor eradication. This relies on both mechanistic studies dissecting the complex interplay among cells in the tumor and the discovery of reliable biomarkers to stratify patients who may benefit from the treatment.
It is worth noting that the abovementioned stromal mechanism to regulate tumor progression and therapeutic response also deeply modulates tumor immunity within the TME, which will be discussed below. For instance, in addition to our finding that CAF-secreted TGF-β signaling and hypoxic environment-induced HIF-1α synergistically induce GLI2 expression to regulate tumor stemness and chemoresistance, it is well known that TGF-β signaling plays a vital role in tumor immunity in the TME by repressing the antitumor functions of various immune cell populations, including T cells and natural killer cells [51]. Interestingly, GLI2 and HIF-1α have also been found to regulate T cell and NK cell infiltration and activity in tumors [52,53,54,55,56,57,58,59]. Again, intriguingly, in addition to directly regulating angiogenesis and metastasis [18, 60], LRG1 has been shown to promote the extravasation and activation of neutrophils as well as to regulate NETosis [61], which is implicated in tumor immune suppression and neutrophil extracellular traps (NETs)-dependent metastasis [62, 63]. Thus, a more comprehensive understanding of the communications within the tumor microenvironment is needed for cancer therapies.
Stromal mechanisms reshaping the TME and tumor immune response
Immunotherapy, specifically immune checkpoint blockade (ICB), has been the most promising paradigm in cancer therapies in the past decade. According to the abundance of tumor-infiltrating lymphocytes, tumors were arbitrarily classified as "hot tumors" or "cold tumors" with high infiltrated or low infiltrated lymphocytes, respectively [64]. While ICB has shown effectiveness in multiple cancers, such as melanoma and lung cancer, most patients cannot benefit from the treatment, especially those with cold tumors, such as CRC and breast cancer. In these “cold tumors”, due to the lack or paucity of tumor T cell infiltration, ICB treatment rarely triggers a strong immune response, leading to ICB failure [65]. Based on the underlying mechanism of ICB action, several potential features are proposed to be related to immunotherapy response, including programmed death-ligand 1 (PD-L1) expression level, immune composition within the TME (immune score), neoantigens and tumor mutation burden [66]. These features of tumors are determined not only by the genetic status of tumor cells (such as genetic mutations related to tumor antigens and mutation burden) but also by the stromal mechanisms by which CAFs reshape the TME through interplay with immune cells. Meanwhile, the epigenetic mechanism in the TME controlling these events has also been extensively documented, implying that certain epigenetic alterations could be used as potential targets for immunotherapy.
The interplay between CAFs and immune cells to modulate tumor immunity
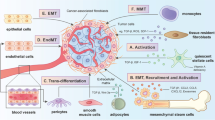
Recent studies have suggested that CAFs in the TME are linked to immunotherapy response by diverse mechanisms. For instance, CAFs and secreted ECM serve as a physical barrier to prevent drug delivery and infiltration of immune cells, thus restraining the effectiveness of immunotherapy [67, 68]. Moreover, the induction of immune checkpoint molecules such as PD-L1, PD-L2, and B7-H3 by CAF-secreted factors, exosomes in cancer cells or CAFs themselves substantially induce T cell exhaustion and deactivation, leading to intrinsic resistance to immunotherapy [69]. Additionally, cytokines such as IL-1β, IL-6, and TGF-β that can be produced by activated immune cells have been broadly implicated in CAF activation [19, 70, 71]. By interacting with immune cells such as T lymphocytes, myeloid-derived suppressor cells, dendritic cells, and others within the TME, CAFs can establish the so-called immunosuppressive microenvironment (Fig. 2).

CAFs modulate the immunosuppressive microenvironment. CAFs promote immune suppression and abolish immune surveillance in the TME. CAFs secrete TGFβ and CCL5 to differentiate naïve T cells into Tregs and to recruit Tregs. CCL2, IL6, and IL33 secreted by CAFs help to recruit MDSCs and strengthen their immunosuppressive function. CAFs promote NETosis and M2 polarization of TMAs in the TME by releasing amyloid β or IL8. However, TGF-β secreted by CAFs suppresses Th cell function and reduces CTL infiltration. The expression of PD-L2 and FasL induces AICD in CTLs. CAFs can suppress the DC-mediated antitumor T cell response and inactivate NK cell-mediated tumor killing by PGE2 and IDO secretion. TME: tumor microenvironment; Th: T helper cell; Treg: regulatory T cell; MDSC: myeloid-derived suppressor cell; TAM: tumor-associated macrophage; NK cell: natural killer cell; AICD: activation-induced cell death
CAFs and T lymphocytes
T lymphocytes function as essential modulators mediating the immune response, which comprises distinct subtypes such as cytotoxic CD8+ T lymphocytes (CTLs), Fox3p + regulatory T cells (Tregs), and CD4+ T helper (Th) cells. CTLs, the most critical immune cells of antitumor immunity, are substantially modulated by CAFs to reduce their infiltration, growth, and antitumor activity. CAF-secreted TGF-β inhibits the expression of cytolytic genes in CTLs, which are responsible for CTL-mediated tumor cytotoxicity [72]. Surprisingly, Lakins et al. found that CAFs isolated from murine melanoma and lung tumors can directly participate in antigen presentation, leading to antigen-mediated activation-induced cell death (AICD) of tumor-reactive CD8+ T lymphocytes via engagement of PD-L2 and Fas ligand to promote cancer immune evasion [73]. Furthermore, CAFs were reported to markedly stimulate Treg cell migration and increase their infiltration into tumor sites in CRC [74]. CAF-derived secreted factors such as TGF-β or CCL5 are also responsible for the recruitment of Tregs and differentiation of naïve T cells to Tregs, ultimately inducing immune suppression [75,76,77].
Several studies have indicated the significant influence of CAFs on Th cell polarization. For example, lactate release from CAFs reduced the percentage of antitumoral Th1 cells and concomitantly increased Tregs, thus driving immunosuppression in prostate cancer [78]. As one of the most frequently secreted cytokines by CAFs, TGF-β can suppress type 2 immunity by repressing Th2 cell responses in cancer [79].
CAFs and MDSCs
Myeloid-derived suppressor cells (MDSCs) are well documented for their immunosuppressive role in the TME. C–C motif chemokine ligand 2 (CCL2) released from CAFs in liver tumors was reported to promote the recruitment of MDSCs through the activation of STAT3 [80]. Similarly, CAF-produced IL-6 and IL-33 were able to educate MDSCs in the TME via hyperactivation of 5-lipoxygenase (5-LO), thus potentiating the capability of MDSCs to enhance cancer stemness [81]. Whereas, Yang et al. found that nonalcoholic fatty liver disease (NAFLD)-associated hepatocellular carcinoma (HCC) expressed low levels of CCL2 as well as other cytokines, such as CCL4, CXCL2, and CXCL6, compared with nontumor tissues [82]. Although somehow contradictory to the immunosuppressive function of CCL2, this study demonstrated that CCL4, a crucial chemokine for T cell migration, is more responsible under this circumstance. Interestingly, pharmacological inhibition of histone deacetylase 8 (HDAC8), a histone H3 lysine 27 (H3K27)-specific isozyme overexpressed in a variety of human cancers, increased global and enhancer acetylation of H3K27 to reactivate the production of CCL4 by HCC cells, thus dampening HCC tumorigenicity in a T cell-dependent manner.
CAFs and other immune cells
Many reports have also uncovered the importance of CAFs in mediating tumor immune evasion by regulating innate immune cells, such as dendritic cells (DCs), tumor-associated macrophages (TAMs), neutrophils, natural killer (NK) cells, and myeloid cells. In CRC, CAF-secreted Wnt2 led to evasion of immune surveillance by suppressing the DC-mediated antitumor T cell response through the SOCS3/p-JAK2/p-STAT3 signaling cascades [83]. Moreover, a comprehensive map to elaborate the interaction between diverse types of cells in the TME of CRC has been depicted recently by taking advantage of scRNA-seq using clinical samples [84]. Of note, SPP1+ TAMs displayed direct interaction with CAFs, which might be due to the binding of MMP2 produced from CAFs and SDC2 that was preferentially expressed in SPP1+ TAMs [84]. In line with this, another work by Zhang et al. also confirmed that CAFs promoted TAM infiltration and subsequent M2 polarization in CRC via IL-8 [85]. Furthermore, TAMs could synergize with CAFs to suppress NK cell killing ability, therefore promoting CRC progression. Neutrophils release histone-bound nuclear DNA and cytotoxic granules as extracellular traps (NETs). A novel finding demonstrated that CAF-secreted amyloid β drives the formation of tumor-associated NETs (t-NETs), thus supporting tumor progression [86]. More interestingly, it was also observed that t-NETs could reciprocally activate CAFs by promoting their expansion, contractility, and deposition of matrix components [86]. CAFs inhibit NK cells through a variety of mechanisms. CAFs, for example, reduce the expression of NK cell-activating receptors, including NKp30 and NKp44, and transition NK cells into an inactivated state by secreting prostaglandin E2 (PGE2) and indoleamine 2,3-dioxygenase (IDO) [87, 88]. Surprisingly, NK cells can enhance this suppressive loop by boosting the release of PGE2 by CAFs [87]. It has also been reported that CAFs indirectly decrease NKG2D-dependent cytotoxic activity and interferon (IFN) secretion of NK cells by reducing the ligands of NK-activating receptors on melanoma cells [89]. Previous research has shown that various myeloid cell subsets expand in CRC cancers. However, these tumor-infiltrating myeloid cells have both pro- and anti-tumor roles in CRC progression. Salman et al. discovered that CD33+ myeloid cells from patients with advanced stages expressed more pro-angiogenic and hypoxia-related genes but fewer immune and inflammatory response genes compared to those with early-stage diseases [90]. This study implies that immune cell recruitment and activation could be compromised under the TME, which dynamically evolves along with tumor progression.
These works highlighted that CAFs and immune cells formed an intimate connection within the TME, implying a promising potential strategy to reshape the immune microenvironment by perturbing crosstalk between the two cell populations.
Epigenetic mechanisms in the TME modulate immunotherapy efficacy
The complex interplay between stroma, immune, and cancer cells alters the epigenome of each other, which is important for antitumor immunity. The idea of converting noninflamed cold tumors into hot tumors using epigenetic intervention may help to achieve a better response to immunotherapy [91]. Early testing of the therapeutic potential of combining epigenetic agents and immunotherapies showed elevated immune-related gene expression and a durable response to anti-CTLA4 or anti-PD1 treatment [92,93,94]. Epigenetic modifications of immune-related genes may strengthen immune surveillance and increase the efficacy of immunotherapy by three key mechanisms (Fig. 3): (1) activating immune pathways or reprogramming the tumor microenvironment to counteract immunosuppression. (2) Increasing the tumor antigenicity by enhancing the processing and presentation of tumor antigens (3) reversing the exhaustion of infiltrated cytotoxic immune cells in the tumor.

Epigenetic regulation of the immune response in the tumor microenvironment. DNA methylation and histone modification regulate the tumor immune response in the TME. The epigenetic mechanisms of DNA methylation induced by DNMT, transcriptional suppression by EZH2, and HDAC play crucial roles in immune-related signal inactivation, immune cell recruitment, antigen processing and presentation, and immune cell exhaustion by suppressing the expression of ERVs, MHC I genes, antigen processing machinery, and cancer testis antigens in the TME. TME: tumor microenvironment; IFNs: interferons
Modulation of key immune signaling pathways in the TME
As evidenced by the existence of an IFN-responsive gene profile in some tumors, an inflamed "hot" TME is compatible with effective IFN-mediated antitumor immune responses. IFN signaling, including type I IFN (IFNα and IFNβ) and type II IFN (IFN-γ), is a well-controlled molecular network that plays pivotal roles in tumor immunity.
Type I interferons control the development of innate and adaptive immune responses by activating intracellular viral defense pathways. Viral double-stranded DNA (dsDNA) or dsRNA can activate the production of type I interferons when captured by their sensors. It is worth noting that the cytosolic dsDNA sensing pathway, especially the cyclic GMP-AMP synthase and stimulator of interferon genes (cGAS-STING) pathway, is usually epigenetically silenced in human cancers through DNA hypermethylation at their promoter regions [95,96,97,98]. The reactivation of ancient endogenous retroviruses (ERVs) and retrotransposons in our genome that are typically silenced (so-called viral mimicry) has been emerging as a robust strategy to boost the immune response in cancer [99, 100] by inducing type I IFN activation after being recognized by sensors of dsRNA, such as RIG-I and MDA5. Recent studies have shown that ERVs can be reinvigorated by drugs targeting epigenetic modulators, including DNMTs, HDACs, or HMTs. In many cancers, including CRC, DNA methyltransferase inhibitors (DNMTis) can induce dsRNA expression mainly derived from ERVs and subsequently trigger cytosolic sensing of dsRNA, causing a type I interferon response and apoptosis [93, 101]. Interestingly, similar to DNMT1 inhibition, ablating the histone demethylase LSD1, which is elevated in diverse cancers, improves tumor immunogenicity by simultaneously activating the dsRNA-IFN pathway by stimulating ERV expression and downregulating the RNA-induced silencing complex (RISC) [102]. These findings may provide an opportunity to reactivate the pathway and promote the immune response by targeting specific epigenetic regulators.
Moreover, Morel et al. demonstrated that EZH2 represses the production of dsRNA and genes implicated in the IFN response, antigen presentation, and T-cell attraction through its catalytic function in prostate cancer [103]. As a histone methyltransferase, SETDB1 was first found to keep silencing the transposable elements (TEs) that lead to the production of dsRNAs in acute myeloid leukemia (AML) [104]. SETDB1 is located at a frequently amplified chromosome area in many other solid tumors, chromosome 1q21.3, which was also implicated in worse tumor prognosis in breast cancer [105]. The amplification of SETDB1 (1q21.3) in tumors is associated with immune exclusion and resistance to immune checkpoint blockade [106]. SETDB1 loss derepresses latent TE-derived regulatory elements, immunostimulatory genes, and TE-encoded retroviral antigens in these regions and triggers TE-specific cytotoxic T cell responses in vivo. Using melanoma and colon cancer as models, Zhang et al. uncovered that KDM5B—an H3K4 demethylase—recruits the H3K9 methyltransferase SETDB1 to repress endogenous retroelements in a demethylase-independent manner [107]. Although it remains to be further determined whether these epigenetic regulations commonly occur in colon cancer, the viral mimicry induced by epigenetic intervention provides an apparent strategy to trigger a robust IFN response and antitumor immunity within the TME. Strikingly, ERV regulation also determines T helper cell lineage integrity. In immune T helper cells, SETDB1 controls the deposition of the restrictive H3K9me3 mark at a restricted and cell-type-specific set of endogenous retroviruses positioned in the vicinity of genes implicated in immunological processes [108]. These retrotransposons operate as Th1 gene enhancers or influence Th1 gene cis-regulatory elements. By suppressing a range of ERVs to shape and govern the Th1 gene network, H3K9me3 deposition by SETDB1 ensures Th cell lineage fidelity.
IFN-γ binds to interferon gamma receptors (IFNGRs) and activates the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) signaling pathway, which modulates the immune response by activating an IFN-stimulated gene (ISG) transcriptional program. The presence of an IFN-γ-responsive gene signature predicts a better response to immunotherapy compared with tumors lacking the IFN-γ signature [109].
Epigenetic histone modifications and DNA methylation are closely involved in the regulation of the IFN-γ signaling pathway in colorectal cancer. Tumor production of CXCL9 and CXCL10, which are Th1-type chemokines, can be repressed by either enhancer of zeste homolog 2 (EZH2, a core of the PRC2 complex)-mediated histone H3 lysine 27 trimethylation or DNA methyltransferase 1 (DNMT1)-induced DNA methylation, subsequently resulting in less recruitment of IFN-γ-producing immune cells [110]. Conversely, ARID1A, a core member of the SWItch/Sucrose Non-Fermentable (SWI/SNF) complex, promotes tumor expression of CXCL9 and CXCL10 [111]. Genetic deficiency in ARID1A has been reported to result in a reduction in chromatin accessibility at these chemokine loci in colorectal cancer cells. ARID1A interacts with EZH2 through its carboxyl terminus, preventing EZH2 from inhibiting IFN signaling-mediated gene expression. Moreover, our previous work discovered that EZH2 can suppress the IFN-γ signaling pathway by directly silencing the expression of interferon-γ receptor 1 (IFNGR1) [112] and ISG activation [113], which led to cancer cells being insensitive to IFN-γ treatment or resistant to trastuzumab treatment, respectively.
Improvements to tumor antigenicity
Aberrant epigenetic mechanisms driving the dysregulation of genes involved in the processing or presentation of tumor antigens, essential for T cell activation, are a recurring characteristic of cancer cells escaping from immune surveillance. In addition to activating IFN signaling, DNMTis such as 5-azacytidine, decitabine, and guadecitabine, which induce global hypomethylation, significantly boost the expression of MHC class I genes and PD-L1 [114, 115]. Additionally, DNMTi can also increase the expression of cancer-testis antigens (CTAs), promising immunotherapy targets such as MAGE-11 and NY-ESO-1 that are expressed in early embryonic cells but suppressed in mature somatic cells due to promoter CpG island DNA methylation [116, 117]. In cancer cells, deacetylation of histone lysine residues is frequently linked to hypermethylated and repressed genes. Histone deacetylase inhibitors (HDACis), such as trichostatin A (TSA), restore gene expression by targeting these regions. HDACis have been shown to boost the expression of various antigen processing machinery components, such as TAP-1, TAP-2, LMP-2, and tapasin. Treatment of metastatic cancer cells with TSA increases MHC class I expression on the cell surface, which functionally translates to increased vulnerability to killing by antigen-specific CTLs [118]. PRC2 was also reported to silence the MHC-I antigen processing and presentation pathway and evade immune surveillance. Pharmacological inhibition of EED or EZH2 and EZH1 reverses the silencing of these pathways, leading to the reestablishment of effective T cell-mediated antitumor immunity.
Reversed immune exhaustion
Tumor-infiltrating lymphocytes, particularly cytotoxic CD8+ T cells (CTLs), often display dysfunction and exhaustion due to the persistent existence of antigen stimulation and other factors in the TME, such as hypoxia and metabolic stress [119]. They frequently lose the capacity to produce cytokines such as tumor necrosis factor-α, IFN-γ, and interleukin (IL)-2 but retain the expression of inhibitory receptors such as programmed cell death protein (PD)-1, lymphocyte-activation gene (LAG)-3, or T cell immunoglobulin and mucin-domain containing (TIM)-3 [120, 121]. Specific chromatin-accessible areas linked with an altered transcriptional profile are found in CD8+ T cell exhaustion, including enrichment for genes in interferon signaling, PD-1 signaling and the cytokine IL-10 response [122]. Immune checkpoint blockade, such as anti-PD-1 antibody treatment, has been shown to partially reverse CD8+ T cell exhaustion; however, extensive epigenetic reprogramming during T cell exhaustion, which differs substantially from that of effector and memory T cells, limits the durable success of immunotherapies [123]. By characterizing the critical epigenetic reprogramming mechanisms of T cell exhaustion, the exhaustion status may be reversible [124,125,126,127]. Ghoneim et al. demonstrated that epigenetic changes introduced by the DNA methyltransferase DNMT3A are needed to acquire an exhausted phenotype[126]. DNMT3A methylates thousands of genes de novo, many of which are critical for effector CD8+ T cell function. A study of exhausted CD8+ T cells in humans and a chronic viral infection mouse model by Sen et al. revealed that a state-specific epigenetic landscape organized into functional modules of enhancers is required for exhaustion [124]. Using an in vitro system that models human T cell exhaustion, our data recently reported that hypoxia in the TME induces transcriptional suppression of the immune effectors IFN-γ, tumor necrosis factor α (TNFα), and granzyme B, resulting in immune effector cell dysfunction and resistance to immunotherapy [128]. Furthermore, the chromatin remodeling enforced by HIF1α interaction with HDAC1 and subsequent dependence on PRC is identified as a crucial epigenetic mechanism conferring immune effector suppression. In addition, under continuous stimulation with tumor antigen, hypoxia further induces TIM-3 and ITGIT to potentiate T cell exhaustion in a HIF-1α-independent manner. In addition, microenvironmental stressors coordinated with T cell receptor stimulation, and PD-1 signaling can promote terminal exhaustion of T cells through epigenetic reprogramming as a result of mitochondrial dysfunction [129].
Implications of epigenetic modulators in cancer intervention
Many studies have focused on evaluating combinations of immunotherapies with various therapies, including chemotherapy, radiation therapy, and targeted therapy, to increase the infiltration of CTLs [130]. With the idea of converting "cold tumors" to "hot tumors", epigenetic therapy offers a unique opportunity to remodel the TME from immunosuppressive to immunopermissive by regulating stromal and immune cells via multiple mechanisms [91]. Multiple preclinical studies have discovered that epigenetic agents can reinvigorate the immune response in various tumor types. As discussed in the previous sections, DNA hypomethylating agents such as DNMTi (5-AZA), EZH2 inhibitors, or HDACi (TSA) can improve the efficacy of ICB by reducing immune suppression through the initiation of the type I IFN response via dsRNA production. 5-AZA increased the infiltration of both CD8+ T and natural killer (NK) cells and reduced the percentages of macrophages and MDSCs in the TME. Interestingly, Zhou et al. recently revealed that p53 activation by MDM2 inhibitors induced the type I IFN response, abolishing tumor immune evasion and promoting antitumor immunity in an LSD1- and DNMT1-dependent manner [131]. The importance of p53 during cancer progression is unequivocal since more than half of all sporadic cancers show p53 dysfunction. Furthermore, the MDM2 inhibitor ALRN-6924 induced a viral mimicry response and tumor inflammation signature genes in melanoma patients, which provided a rationale for the synergistic strategy of MDM2 inhibitors and immunotherapy. Additionally, in mouse mammary tumor models (MMTV-rtTA/tetO-HER2, MMTV-PyMT) and patients with breast and colon carcinoma, treatment with CDK4/6 inhibitors reduced DNMT1 expression, resulting in hypomethylation of immune-related genes, enhancing antitumor immunity by both promoting antigen presentation and reducing Treg cell expansion [132]. These events ultimately promoted clearance of tumor cells by cytotoxic T cells, which could be further improved by the addition of immune checkpoint blockade (anti-PD-L1), thus opening a new avenue to treat cancer by combination regimens comprising CDK4/6 inhibitors and immunotherapies.
Interestingly, many epigenetic modulation agents play roles in different aspects of immune modulations. For example, DNMTi can initiate the type I IFN response and has functions in regulating tumor antigen presentation. HDACis could restore tumor antigen expression and reverse T cell exhaustion. Although these functions may be played under different contexts, it is interesting to determine how to leverage them to augment antitumor immunity. In some circumstances, the combination of different epigenetic agents plus ICB may confer the best antitumor effect. For example, a triple combination of DNMTi/HDACi plus the immune checkpoint inhibitor α-PD-1 provides the most prolonged overall survival in an ovarian cancer model [133]. Similarly, histone deacetylase 6 (HDAC6) inhibitors with enhanced antitumor immunity of anti-PL-L1 immunotherapy were recently developed for melanoma treatment [134]. A concern is that many epigenetic inhibitors have been shown to limit T cell growth, which could compromise the long-term effectiveness of immunotherapy that relies on a persistent T cell population. Inhibition of EZH2, for example, has been shown to impair T cell function [135]. EZH2 is required to generate and maintain memory T cells, which are responsible for effector T cell production and antitumor activity. In conclusion, more research is needed to determine whether the benefit of combining epigenetic therapy and immunotherapy is dependent on the type of cancer or other circumstances. Recently, many strategies combining epigenetic therapy and immunotherapy are being evaluated in numerous clinical trials (summarized in Table 1), which may improve clinical practice in the future.
Concluding remarks
In summary, this review broadly discusses recent studies exploring the complex interaction networks across key cell components within the TME, which consist of CAFs, tumor cells, and immune cells. Reciprocal crosstalk between different cell populations ultimately determines tumor progression via diverse "intermediate massagers". Epigenetic dysfunction has emerged as a novel hallmark of cancer. Although in-depth research has indicated the critical influence of epigenetic regulation on cancer cells, rising evidence has pointed to the other appealing property of epigenetic modulators in reshaping the TME, especially from the perspective of creating a tumor-favor immunosuppressive condition. As comprehensively stated above, various epigenetic modulators contribute to immune evasion, and hence, targeting them with small molecules could boost the immune response. Thus, these findings present a promising strategy to combine epi-drugs with other therapies, such as immune checkpoint blockade (ICB) therapy, which requires an immune-permissive TME as the prerequisite for successful treatment. Moreover, while ICB therapy undoubtedly became one of the most powerful tools to treat multiple cancers with a durable response and acceptable toxicity, up to approximately 85% of patients displayed intrinsic or acquired resistance to ICB, which profoundly limits its utility in the clinic. Therefore, the identification of epigenetic markers that can predict patients benefiting from ICB treatment merits further investigation in the future.
Availability of data and materials
Not applicable.
References
Paget S. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev. 1889;8(2):98–101.
Fidler IJ, Poste G. The, “seed and soil” hypothesis revisited. Lancet Oncol. 2008;9(8):808.
Anderson NM, Simon MC. The tumor microenvironment. Curr Biol. 2020;30(16):R921–5.
Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462(7271):315–22.
Isella C, Terrasi A, Bellomo SE, Petti C, Galatola G, Muratore A, et al. Stromal contribution to the colorectal cancer transcriptome. Nat Genet. 2015;47(4):312–9.
Calon A, Lonardo E, Berenguer-Llergo A, Espinet E, Hernando-Momblona X, Iglesias M, et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat Genet. 2015;47(4):320–9.
Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21(11):1350–6.
Becht E, de Reynies A, Giraldo NA, Pilati C, Buttard B, Lacroix L, et al. Immune and stromal classification of colorectal cancer is associated with molecular subtypes and relevant for precision immunotherapy. Clin Cancer Res. 2016;22(16):4057–66.
Peña C, Céspedes MV, Lindh MB, Kiflemariam S, Mezheyeuski A, Edqvist PH, et al. STC1 expression by cancer-associated fibroblasts drives metastasis of colorectal cancer. Cancer Res. 2013;73(4):1287–97.
Henriksson ML, Edin S, Dahlin AM, Oldenborg PA, Öberg Å, Van Guelpen B, et al. Colorectal cancer cells activate adjacent fibroblasts resulting in FGF1/FGFR3 signaling and increased invasion. Am J Pathol. 2011;178(3):1387–94.
Peng C, Zou X, Xia W, Gao H, Li Z, Liu N, et al. Integrin αvβ6 plays a bi-directional regulation role between colon cancer cells and cancer-associated fibroblasts. Biosci Rep. 2018;38(6):BSR20180243.
De Wever O, Nguyen QD, Van Hoorde L, Bracke M, Bruyneel E, Gespach C, et al. Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent pro-invasive signals to human colon cancer cells through RhoA and Rac. FASEB J. 2004;18(9):1016–8.
Kobayashi H, Gieniec KA, Wright JA, Wang T, Asai N, Mizutani Y, et al. The balance of stromal BMP signaling mediated by GREM1 and ISLR drives colorectal carcinogenesis. Gastroenterology. 2021;160(4):1224–39.
Deng L, Jiang N, Zeng J, Wang Y, Cui H. The versatile roles of cancer-associated fibroblasts in colorectal cancer and therapeutic implications. Front Cell Dev Biol. 2021;9: 733270.
Sung PJ, Rama N, Imbach J, Fiore S, Ducarouge B, Neves D, et al. Cancer-associated fibroblasts produce Netrin-1 to control cancer cell plasticity. Cancer Res. 2019;79(14):3651–61.
Nakagawa H, Liyanarachchi S, Davuluri RV, Auer H, Martin EW Jr, de la Chapelle A, et al. Role of cancer-associated stromal fibroblasts in metastatic colon cancer to the liver and their expression profiles. Oncogene. 2004;23(44):7366–77.
Berdiel-Acer M, Bohem ME, López-Doriga A, Vidal A, Salazar R, Martínez-Iniesta M, et al. Hepatic carcinoma-associated fibroblasts promote an adaptative response in colorectal cancer cells that inhibit proliferation and apoptosis: nonresistant cells die by nonapoptotic cell death. Neoplasia. 2011;13(10):931–46.
Zhong B, Cheng B, Huang X, Xiao Q, Niu Z, Chen Y-F, et al. Colorectal cancer-associated fibroblasts promote metastasis by up-regulating LRG1 through stromal IL-6/STAT3 signaling. Cell Death Dis. 2021;13(1):16.
Hawinkels LJ, Paauwe M, Verspaget HW, Wiercinska E, van der Zon JM, van der Ploeg K, et al. Interaction with colon cancer cells hyperactivates TGF-β signaling in cancer-associated fibroblasts. Oncogene. 2014;33(1):97–107.
Tang YA, Chen YF, Bao Y, Mahara S, Yatim S, Oguz G, et al. Hypoxic tumor microenvironment activates GLI2 via HIF-1alpha and TGF-beta2 to promote chemoresistance in colorectal cancer. Proc Natl Acad Sci USA. 2018;115(26):e5990–9.
Paauwe M, Schoonderwoerd MJA, Helderman R, Harryvan TJ, Groenewoud A, van Pelt GW, et al. Endoglin expression on cancer-associated fibroblasts regulates invasion and stimulates colorectal cancer metastasis. Clin Cancer Res. 2018;24(24):6331–44.
Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15(2):103–13.
Putoczki TL, Thiem S, Loving A, Busuttil RA, Wilson NJ, Ziegler PK, et al. Interleukin-11 is the dominant IL-6 family cytokine during gastrointestinal tumorigenesis and can be targeted therapeutically. Cancer Cell. 2013;24(2):257–71.
Heichler C, Scheibe K, Schmied A, Geppert CI, Schmid B, Wirtz S, et al. STAT3 activation through IL-6/IL-11 in cancer-associated fibroblasts promotes colorectal tumour development and correlates with poor prognosis. Gut. 2020;69(7):1269–82.
Calon A, Espinet E, Palomo-Ponce S, Tauriello DV, Iglesias M, Céspedes MV, et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell. 2012;22(5):571–84.
Ma H, Wang J, Zhao X, Wu T, Huang Z, Chen D, et al. Periostin promotes colorectal tumorigenesis through Integrin-FAK-Src pathway-mediated YAP/TAZ activation. Cell Rep. 2020;30(3):793–806.
Nagasaki T, Hara M, Nakanishi H, Takahashi H, Sato M, Takeyama H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour-stroma interaction. Br J Cancer. 2014;110(2):469–78.
Wang T, Song P, Zhong T, Wang X, Xiang X, Liu Q, et al. The inflammatory cytokine IL-6 induces FRA1 deacetylation promoting colorectal cancer stem-like properties. Oncogene. 2019;38(25):4932–47.
Sanchez-Lopez E, Flashner-Abramson E, Shalapour S, Zhong Z, Taniguchi K, Levitzki A, et al. Targeting colorectal cancer via its microenvironment by inhibiting IGF-1 receptor-insulin receptor substrate and STAT3 signaling. Oncogene. 2016;35(20):2634–44.
Vermeulen L, De Sousa EMF, van der Heijden M, Cameron K, de Jong JH, Borovski T, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12(5):468–76.
Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487(7408):500–4.
Unterleuthner D, Neuhold P, Schwarz K, Janker L, Neuditschko B, Nivarthi H, et al. Cancer-associated fibroblast-derived WNT2 increases tumor angiogenesis in colon cancer. Angiogenesis. 2020;23(2):159–77.
Aizawa T, Karasawa H, Funayama R, Shirota M, Suzuki T, Maeda S, et al. Cancer-associated fibroblasts secrete Wnt2 to promote cancer progression in colorectal cancer. Cancer Med. 2019;8(14):6370–82.
Agrawal K, Das V, Táborská N, Gurský J, Džubák P, Hajdúch M. Differential regulation of methylation-regulating enzymes by senescent stromal cells drives colorectal cancer cell response to DNA-demethylating epi-drugs. Stem Cells Int. 2018;2018:6013728.
Wang W, Tang Y-A, Xiao Q, Lee WC, Cheng B, Niu Z, et al. Stromal induction of BRD4 phosphorylation results in chromatin remodeling and BET inhibitor resistance in colorectal cancer. Nat Commun. 2021;12(1):4441–57.
Dai X, Gan W, Li X, Wang S, Zhang W, Huang L, et al. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat Med. 2017;23(9):1063–71.
Zhang P, Wang D, Zhao Y, Ren S, Gao K, Ye Z, et al. Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT-mTORC1 activation. Nat Med. 2017;23(9):1055–62.
Janouskova H, El Tekle G, Bellini E, Udeshi ND, Rinaldi A, Ulbricht A, et al. Opposing effects of cancer-type-specific SPOP mutants on BET protein degradation and sensitivity to BET inhibitors. Nat Med. 2017;23(9):1046–54.
Zhu H, Bengsch F, Svoronos N, Rutkowski MR, Bitler BG, Allegrezza MJ, et al. BET bromodomain inhibition promotes anti-tumor immunity by suppressing PD-L1 expression. Cell Rep. 2016;16(11):2829–37.
Kagoya Y, Nakatsugawa M, Yamashita Y, Ochi T, Guo T, Anczurowski M, et al. BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. J Clin Invest. 2016;126(9):3479–94.
Milner JJ, Toma C, Quon S, Omilusik K, Scharping NE, Dey A, et al. Bromodomain protein BRD4 directs and sustains CD8 T cell differentiation during infection. J Exp Med. 2021;218(8):e20202512.
Hu JL, Wang W, Lan XL, Zeng ZC, Liang YS, Yan YR, et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol Cancer. 2019;18(1):91.
Ren J, Ding L, Zhang D, Shi G, Xu Q, Shen S, et al. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Theranostics. 2018;8(14):3932–48.
Deng X, Ruan H, Zhang X, Xu X, Zhu Y, Peng H, et al. Long noncoding RNA CCAL transferred from fibroblasts by exosomes promotes chemoresistance of colorectal cancer cells. Int J Cancer. 2020;146(6):1700–16.
Yu H, Yue X, Zhao Y, Li X, Wu L, Zhang C, et al. LIF negatively regulates tumour-suppressor p53 through Stat3/ID1/MDM2 in colorectal cancers. Nat Commun. 2014;5:5218.
Albrengues J, Bertero T, Grasset E, Bonan S, Maiel M, Bourget I, et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat Commun. 2015;6:10204.
Takeda S, Shigeyasu K, Okugawa Y, Yoshida K, Mori Y, Yano S, et al. Activation of AZIN1 RNA editing is a novel mechanism that promotes invasive potential of cancer-associated fibroblasts in colorectal cancer. Cancer Lett. 2019;444:127–35.
Lotti F, Jarrar AM, Pai RK, Hitomi M, Lathia J, Mace A, et al. Chemotherapy activates cancer-associated fibroblasts to maintain colorectal cancer-initiating cells by IL-17A. J Exp Med. 2013;210(13):2851–72.
Saw PE, Chen J, Song E. Targeting CAFs to overcome anticancer therapeutic resistance. Trends Cancer. 2022;8(7):527–55.
Wang W, Cheng B, Yu Q. Cancer-associated fibroblasts as accomplices to confer therapeutic resistance in cancer. Cancer Drug Resist. 2022;5:889–901.
Derynck R, Turley SJ, Akhurst RJ. TGFβ biology in cancer progression and immunotherapy. Nat Rev Clin Oncol. 2021;18(1):9–34.
Furmanski AL, Barbarulo A, Solanki A, Lau C-I, Sahni H, Saldana JI, et al. The transcriptional activator Gli2 modulates T-cell receptor signalling through attenuation of AP-1 and NFκB activity. J Cell Sci. 2015;128(11):2085–95.
Chakrabarti J, Holokai L, Syu L, Steele NG, Chang J, Wang J, et al. Hedgehog signaling induces PD-L1 expression and tumor cell proliferation in gastric cancer. Oncotarget. 2018;9(100):37439.
Furler RL, Uittenbogaart CH. GLI2 regulates TGF-β1 in human CD4+ T cells: implications in cancer and HIV pathogenesis. PLoS ONE. 2012;7(7): e40874.
Grund-Gröschke S, Stockmaier G, Aberger F. Hedgehog/GLI signaling in tumor immunity—new therapeutic opportunities and clinical implications. Cell Commun Signal. 2019;17(1):172.
Jayaprakash P, Ai M, Liu A, Budhani P, Bartkowiak T, Sheng J, et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J Clin Invest. 2018;128(11):5137–49.
Ni J, Wang X, Stojanovic A, Zhang Q, Wincher M, Buhler L, et al. Single-cell RNA sequencing of tumor-infiltrating NK cells reveals that inhibition of transcription factor HIF-1alpha unleashes NK cell activity. Immunity. 2020;52(6):1075–87.
Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014;74(3):665–74.
Scharping NE, Rivadeneira DB, Menk AV, Vignali PDA, Ford BR, Rittenhouse NL, et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat Immunol. 2021;22(2):205–15.
Wang X, Abraham S, McKenzie JAG, Jeffs N, Swire M, Tripathi VB, et al. LRG1 promotes angiogenesis by modulating endothelial TGF-β signalling. Nature. 2013;499(7458):306–11.
Camilli C, Hoeh AE, De Rossi G, Moss SE, Greenwood J. LRG1: an emerging player in disease pathogenesis. J Biomed Sci. 2022;29(1):6.
Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature. 2020;583(7814):133–8.
Hedrick CC, Malanchi I. Neutrophils in cancer: heterogeneous and multifaceted. Nat Rev Immunol. 2022;22(3):173–87.
Derks S, de Klerk LK, Xu X, Fleitas T, Liu KX, Liu Y, et al. Characterizing diversity in the tumor-immune microenvironment of distinct subclasses of gastroesophageal adenocarcinomas. Ann Oncol. 2020;31(8):1011–20.
Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, et al. Cold tumors: a therapeutic challenge for immunotherapy. Front Immunol. 2019;10:168.
Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19(3):133–50.
Sorokin L. The impact of the extracellular matrix on inflammation. Nat Rev Immunol. 2010;10(10):712–23.
Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348(6230):74–80.
Barrett RL, Pure E. Cancer-associated fibroblasts and their influence on tumor immunity and immunotherapy. Elife. 2020;9:e57243.
Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell. 2010;17(2):135–47.
Sanz-Moreno V, Gaggioli C, Yeo M, Albrengues J, Wallberg F, Viros A, et al. ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell. 2011;20(2):229–45.
Thomas DA, Massague J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8(5):369–80.
Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD. Cancer-associated fibroblasts induce antigen-specific deletion of CD8+T Cells to protect tumour cells. Nat Commun. 2018;9(1):948.
Jacobs J, Deschoolmeester V, Zwaenepoel K, Flieswasser T, Deben C, Van den Bossche J, et al. Unveiling a CD70-positive subset of cancer-associated fibroblasts marked by pro-migratory activity and thriving regulatory T cell accumulation. Oncoimmunology. 2018;7(7): e1440167.
Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ, Hoffman RM, et al. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL–RANK signalling. Nature. 2011;470(7335):548–53.
Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449(7162):557–63.
Chen W, Jin W, Hardegen N, Lei K-J, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198(12):1875–86.
Comito G, Iscaro A, Bacci M, Morandi A, Ippolito L, Parri M, et al. Lactate modulates CD4(+) T-cell polarization and induces an immunosuppressive environment, which sustains prostate carcinoma progression via TLR8/miR21 axis. Oncogene. 2019;38(19):3681–95.
Liu M, Kuo F, Capistrano KJ, Kang D, Nixon BG, Shi W, et al. TGF-beta suppresses type 2 immunity to cancer. Nature. 2020;587(7832):115–20.
Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W, et al. FAP promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via STAT3–CCL2 signaling. Can Res. 2016;76(14):4124–35.
Lin Y, Cai Q, Chen Y, Shi T, Liu W, Mao L, et al. CAFs shape myeloid-derived suppressor cells to promote stemness of intrahepatic cholangiocarcinoma through 5-lipoxygenase. Hepatology. 2022;75(1):28–42.
Yang W, Feng Y, Zhou J, Cheung OK, Cao J, Wang J, et al. A selective HDAC8 inhibitor potentiates antitumor immunity and efficacy of immune checkpoint blockade in hepatocellular carcinoma. Sci Transl Med. 2021;13(588):eaaz6804.
Huang TX, Tan XY, Huang HS, Li YT, Liu BL, Liu KS, et al. Targeting cancer-associated fibroblast-secreted WNT2 restores dendritic cell-mediated antitumour immunity. Gut. 2022;71(2):333–44.
Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O’Brien SA, et al. Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell. 2020;181(2):442–59.
Zhang R, Qi F, Zhao F, Li G, Shao S, Zhang X, et al. Cancer-associated fibroblasts enhance tumor-associated macrophages enrichment and suppress NK cells function in colorectal cancer. Cell Death Dis. 2019;10(4):273.
Munir H, Jones JO, Janowitz T, Hoffmann M, Euler M, Martins CP, et al. Stromal-driven and Amyloid beta-dependent induction of neutrophil extracellular traps modulates tumor growth. Nat Commun. 2021;12(1):683.
Li T, Yi S, Liu W, Jia C, Wang G, Hua X, et al. Colorectal carcinoma-derived fibroblasts modulate natural killer cell phenotype and antitumor cytotoxicity. Med Oncol. 2013;30(3):663.
Li T, Yang Y, Hua X, Wang G, Liu W, Jia C, et al. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. 2012;318(2):154–61.
Balsamo M, Scordamaglia F, Pietra G, Manzini C, Cantoni C, Boitano M, et al. Melanoma-associated fibroblasts modulate NK cell phenotype and antitumor cytotoxicity. Proc Natl Acad Sci U S A. 2009;106(49):20847–52.
Toor SM, Taha RZ, Sasidharan NV, Saleh R, Murshed K, Abu NM, et al. Differential gene expression of tumor-infiltrating CD33(+) myeloid cells in advanced- versus early-stage colorectal cancer. Cancer Immunol Immunother. 2021;70(3):803–15.
Zhang J, Huang D, Saw PE, Song E. Turning cold tumors hot: from molecular mechanisms to clinical applications. Trends Immunol. 2022;43(7):523–45.
Tsai HC, Li H, Van Neste L, Cai Y, Robert C, Rassool FV, et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. 2012;21(3):430–46.
Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015;162(5):974–86.
Wrangle J, Wang W, Koch A, Easwaran H, Mohammad HP, Vendetti F, et al. Alterations of immune response of non-small cell lung cancer with azacytidine. Oncotarget. 2013;4(11):2067–79.
Konno H, Yamauchi S, Berglund A, Putney RM, Mulé JJ, Barber GN. Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene. 2018;37(15):2037–51.
Kitajima S, Ivanova E, Guo S, Yoshida R, Campisi M, Sundararaman SK, et al. Suppression of STING associated with LKB1 loss in KRAS-driven lung cancer. Cancer Discov. 2019;9(1):34–45.
Xia T, Konno H, Barber GN. Recurrent loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Can Res. 2016;76(22):6747–59.
Xia T, Konno H, Ahn J, Barber GN. Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. 2016;14(2):282–97.
Janin M, Esteller M. Epigenetic awakening of viral mimicry in cancer. Cancer Discov. 2020;10(9):1258–60.
Chen R, Ishak CA, De Carvalho DD. Endogenous retroelements and the viral mimicry response in cancer therapy and cellular homeostasis. Cancer Discov. 2021;11(11):2707–25.
Roulois D, Loo YH, Singhania R, Wang Y, Danesh A, Shen SY, et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell. 2015;162(5):961–73.
Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, et al. LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell. 2018;174(3):549–63.
Morel KL, Sheahan AV, Burkhart DL, Baca SC, Boufaied N, Liu Y, et al. EZH2 inhibition activates a dsRNA-STING-interferon stress axis that potentiates response to PD-1 checkpoint blockade in prostate cancer. Nat Cancer. 2021;2(4):444–56.
Cuellar TL, Herzner AM, Zhang X, Goyal Y, Watanabe C, Friedman BA, et al. Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J Cell Biol. 2017;216(11):3535–49.
Goh JY, Feng M, Wang W, Oguz G, Yatim S, Lee PL, et al. Chromosome 1q21.3 amplification is a trackable biomarker and actionable target for breast cancer recurrence. Nat Med. 2017;23(11):1319–30.
Griffin GK, Wu J, Iracheta-Vellve A, Patti JC, Hsu J, Davis T, et al. Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature. 2021;595(7866):309–14.
Zhang SM, Cai WL, Liu X, Thakral D, Luo J, Chan LH, et al. KDM5B promotes immune evasion by recruiting SETDB1 to silence retroelements. Nature. 2021;598(7882):682–7.
Adoue V, Binet B, Malbec A, Fourquet J, Romagnoli P, van Meerwijk JPM, et al. The histone methyltransferase SETDB1 controls T helper cell lineage integrity by repressing endogenous retroviruses. Immunity. 2019;50(3):629–44.
Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127(8):2930–40.
Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527(7577):249–53.
Li J, Wang W, Zhang Y, Cieślik M, Guo J, Tan M, et al. Epigenetic driver mutations in ARID1A shape cancer immune phenotype and immunotherapy. J Clin Investig. 2020;130(5):2712–26.
Wee ZN, Li Z, Lee PL, Lee ST, Lim YP, Yu Q. EZH2-mediated inactivation of IFN-gamma-JAK-STAT1 signaling is an effective therapeutic target in MYC-driven prostate cancer. Cell Rep. 2014;8(1):204–16.
Ong LT, Lee WC, Ma S, Oguz G, Niu Z, Bao Y, et al. IFI16-dependent STING signaling is a crucial regulator of anti-HER2 immune response in HER2+ breast cancer. Proc Natl Acad Sci U S A. 2022;119(31): e2201376119.
Serrano A, Tanzarella S, Lionello I, Mendez R, Traversari C, Ruiz-Cabello F, et al. Rexpression of HLA class I antigens and restoration of antigen-specific CTL response in melanoma cells following 5-aza-2’-deoxycytidine treatment. Int J Cancer. 2001;94(2):243–51.
Siebenkäs C, Chiappinelli KB, Guzzetta AA, Sharma A, Jeschke J, Vatapalli R, et al. Inhibiting DNA methylation activates cancer testis antigens and expression of the antigen processing and presentation machinery in colon and ovarian cancer cells. PLoS ONE. 2017;12:e0179501.
Karpf AR, Bai S, James SR, Mohler JL, Wilson EM. Increased expression of androgen receptor coregulator MAGE-11 in prostate cancer by DNA hypomethylation and cyclic AMP. Mol Cancer Res. 2009;7(4):523–35.
De Smet C, Loriot A, Boon T. Promoter-dependent mechanism leading to selective hypomethylation within the 5’ region of gene MAGE-A1 in tumor cells. Mol Cell Biol. 2004;24(11):4781–90.
Setiadi AF, Omilusik K, David MD, Seipp RP, Hartikainen J, Gopaul R, et al. Epigenetic enhancement of antigen processing and presentation promotes immune recognition of tumors. Can Res. 2008;68(23):9601–7.
Lim AR, Rathmell WK, Rathmell JC. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. Elife. 2020;9:e55185.
Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–99.
Speiser DE, Utzschneider DT, Oberle SG, Münz C, Romero P, Zehn D. T cell differentiation in chronic infection and cancer: functional adaptation or exhaustion? Nat Rev Immunol. 2014;14(11):768–74.
Miller BC, Sen DR, Al AR, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. 2019;20(3):326–36.
Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science. 2016;354(6316):1160–5.
Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, et al. The epigenetic landscape of T cell exhaustion. Science. 2016;354(6316):1165–9.
Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell. 2018;175(4):998–1013.
Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, et al. De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell. 2017;170(1):142–57.
Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature. 2017;545(7655):452–6.
Ma S, Zhao Y, Lee WC, Ong LT, Lee PL, Jiang Z, et al. Hypoxia induces HIF1alpha-dependent epigenetic vulnerability in triple negative breast cancer to confer immune effector dysfunction and resistance to anti-PD-1 immunotherapy. Nat Commun. 2022;13(1):4118.
Yu YR, Imrichova H, Wang H, Chao T, Xiao Z, Gao M, et al. Disturbed mitochondrial dynamics in CD8(+) TILs reinforce T cell exhaustion. Nat Immunol. 2020;21(12):1540–51.
Yan Y, Kumar AB, Finnes H, Markovic SN, Park S, Dronca RS, et al. Combining immune checkpoint inhibitors with conventional cancer therapy. Front Immunol. 2018;9:1739.
Zhou X, Singh M, Sanz SG, Guerlavais V, Carvajal LA, Aivado M, et al. Pharmacologic activation of p53 triggers viral mimicry response thereby abolishing tumor immune evasion and promoting antitumor immunity. Cancer Discov. 2021;11(12):3090–105.
Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature. 2017;548(7668):471–5.
Stone ML, Chiappinelli KB, Li H, Murphy LM, Travers ME, Topper MJ, et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc Natl Acad Sci USA. 2017;114(51):e10981–90.
Peng X, Li L, Chen J, Ren Y, Liu J, Yu Z, et al. Discovery of novel histone deacetylase 6 (HDAC6) inhibitors with enhanced antitumor immunity of anti-PD-L1 immunotherapy in melanoma. J Med Chem. 2022;65(3):2434–57.
Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L, et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat Immunol. 2016;17(1):95–103.
Center M.D.A.C. and National Cancer I. Nivolumab and azacitidine with or without ipilimumab in treating patients with refractory/relapsed or newly diagnosed acute myeloid leukemia. 2022.
Therapeutic Advances in Childhood Leukemia C. and Bristol-Myers S. Nivolumab in combination with 5-azacytidine in childhood relapsed/refractory aml. 2023.
Sidney Kimmel Comprehensive Cancer Center at Johns H., Rising Tide F., Stand Up To C., Bristol-Myers S., Celgene, Syndax Pharmaceuticals I.et al. Phase ii anti-pd1 epigenetic therapy study in nsclc. 2023.
Center H.L.M.C., Research I. and Bristol-Myers S. Nivolumab or nivolumab and azacitidine in patients with recurrent, resectable osteosarcoma. 2022.
Celgene. Safety and efficacy study of cc-486 with mk-3475 to treat locally advanced or metastatic non-small cell lung cancer. 2017.
Incyte C. Azacitidine combined with pembrolizumab and epacadostat in subjects with advanced solid tumors (echo-206). 2019.
Sidney Kimmel Comprehensive Cancer Center at Johns H., Merck S. and Dohme L.L.C. Study of azacitidine in combination with pembrolizumab in relapsed/refractory acute myeloid leukemia (aml) patients and in newly diagnosed older (≥65 years) aml patients. 2021.
Technische Universität D., Merck S. and Dohme L.L.C. Mrd-guided treatment in npm1mut aml patients. 2023.
Anuradha K., Merck S., Dohme L.L.C. and University of P. A phase 2 study of pembrolizumab (mk-3475) in combination with azacitidine in subjects with chemo-refractory metastatic colorectal cancer. 2016.
Sidney Kimmel Comprehensive Cancer Center at Johns H., Merck S., Dohme L.L.C. and Celgene C. A study of enhancing response to mk-3475 in advanced colorectal cancer. 2021.
Center M.D.A.C. and National Cancer I. Azacitidine and pembrolizumab in treating patients with myelodysplastic syndrome. 2022.
Center M.D.A.C., Merck S., Dohme L.L.C. and Celgene. Study of oral azacitidine (cc-486) in combination with pembrolizumab (mk-3475) in patients with metastatic melanoma. 2025.
Rachael A Safyan M.D. and Columbia U. Azacitidine and pembrolizumab in pancreatic cancer. 2021.
Translational Research in O. and Celgene. Study of pembrolizumab with or without cc-486 in patients with platinum-resistant ovarian cancer. 2021.
The First Affiliated Hospital of Soochow U. Pd-1 inhibitor combined with azacytidine and homoharringtonine,cytarabine, g-csf for refractory or relapsed aml. 2023.
Hoffmann-La R. A study of atezolizumab administered alone or in combination with azacitidine in participants with myelodysplastic syndromes. 2019.
Center M.D.A.C. and National Cancer I. Avelumab and azacitidine in treating patients with refractory or relapsed acute myeloid leukemia. 2019.
Center M.D.A.C. and National Cancer I. Ox40, venetoclax, avelumab, glasdegib, gemtuzumab ozogamicin, and azacitidine in treating patients with relapsed or refractory acute myeloid leukemia. 2023.
Celgene. An efficacy and safety study of azacitidine subcutaneous in combination with durvalumab (medi4736) in previously untreated adults with higher-risk myelodysplastic syndromes (mds) or in elderly patients with acute myeloid leukemia (aml). 2018.
MedImmune L.L.C. Phase 1 study to evaluate medi4736 in subjects with myelodysplastic syndrome. 2019.
Celgene. Safety and efficacy study of nab®-paclitaxel with cc-486 or nab®-paclitaxel with durvalumab, and nab®-paclitaxel monotherapy as second/third-line treatment for advanced non-small cell lung cancer. 2017.
Massachusetts General H., AstraZeneca and Astex Pharmaceuticals I. Oral decitabine (astx727) and durvalumab in recurrent and/or metastatic head and neck cancer patients. 2023.
University Health Network T. Study of azacitidine and durvalumab in advanced solid tumors. 2018.
Center M.D.A.C. and National Cancer I. Nivolumab and/or ipilimumab with or without azacitidine in treating patients with myelodysplastic syndrome. 2022.
Chinese P.L.A.G.H. The clinical trial of chidamide+decitabine+camrelizumab versus decitabine+camrelizumab in anti-pd-1 antibody resistant patients with classical hodgkin lymphoma. 2022.
Chinese P.L.A.G.H. Shr-1210 alone or in combination with decitabine in relapsed or refractory hodgkin lymphoma. 2022.
Chinese P.L.A.G.H. Two stage study of combination of chemotherapy, shr-1210 and/or decitabine for relapsed/refractory pmbcls. 2019.
Case Comprehensive Cancer C. and National Cancer I. Rational epigenetic immunotherapy for second line therapy in patients with nsclc: Precise trial. 2019.
National Heart L., Blood I. and National Institutes of Health Clinical C. Pembrolizumab and decitabine for refractory or relapsed acute myeloid leukemia. 2019.
City of Hope Medical C. and National Cancer I. Pembrolizumab and decitabine with or without venetoclax in treating patients with acute myeloid leukemia or myelodysplastic syndrome that is newly diagnosed, recurrent, or refractory. 2023.
Virginia Commonwealth U., Merck S., Dohme L.L.C. and National Cancer I. Neoadjuvant pembrolizumab + decitabine followed by std neoadj chemo for locally advanced her2- breast ca. 2025.
Children's Hospital Medical Center C. Pembrolizumab in combination with decitabine and hypofractionated index lesion radiation in pediatrics and young adults. 2023.
National Cancer I. and National Institutes of Health Clinical C. Oral decitabine and tetrahydrouridine as epigenetic priming for, pembrolizumab-mediated immune checkpoint blockade in patients with inoperable, or unresectable locally advanced or metastatic non-small cell lung cancers and esophageal carcinomas. 2026.
Milton S.H.M.C. and Pfizer. Avelumab with decitabine as first line for aml treatment of patients with aml, who are unfit for intensive chemotherapy. 2019.
National Cancer I. Ipilimumab and decitabine in treating patients with relapsed or refractory myelodysplastic syndrome or acute myeloid leukemia. 2023.
University of Southern C., National Cancer I., Bristol-Myers S. and Astex Pharmaceuticals I. Guadecitabine and nivolumab in treating refractory metastatic colorectal cancer. 2022.
Royal Marsden N.H.S.F.T., Astex Pharmaceuticals I., Merck S., Dohme L.L.C. and Institute of Cancer Research U.K. Combination study of guadecitabine and pembrolizumab. 2024.
Northwestern U., Merck S., Dohme L.L.C., Astex Pharmaceuticals I. and National Cancer I. Guadecitabine and pembrolizumab in treating patients with recurrent ovarian, primary peritoneal, or fallopian tube cancer. 2020.
Hoffmann-La R. A study evaluating the safety and pharmacology of atezolizumab administered in combination with immunomodulatory agents in participants with acute myeloid leukemia (aml). 2019.
University of Southern C., National Cancer I. and Van Andel Research I. Guadecitabine and atezolizumab in treating patients with advanced myelodysplastic syndrome or chronic myelomonocytic leukemia that is refractory or relapsed. 2023.
Fox Chase Cancer C., Stand Up To C. and Van Andel Research I. Atezolizumab + guadecitabine in patients with checkpoint inhibitor refractory or resistant urothelial carcinoma. 2020.
National Cancer I. Atezolizumab, guadecitabine, and cdx-1401 vaccine in treating patients with recurrent ovarian, fallopian tube, or primary peritoneal cancer. 2023.
Ajjai Alva M.D., AstraZeneca and Big Ten Cancer Research C. Study of durvalumab and guadecitabine in advanced kidney cancer. 2022.
University of Southern C., National Cancer I. and Van Andel Research I. Guadecitabine and durvalumab in treating patients with advanced liver, pancreatic, bile duct, or gallbladder cancer. 2021.
Italian Network for Tumor Biotherapy F. and Astex Pharmaceuticals I. A study investigating sgi-110 in combination with ipilimumab in unresectable or metastatic melanoma patients. 2016.
Memorial Sloan Kettering Cancer C., Merck S., Dohme L.L.C., Astex Pharmaceuticals I., Mirati Therapeutics I., Stand Up To C.et al. Pembrolizumab (immunotherapy drug) in combination with guadecitabine and mocetinostat (epigenetic drugs) for patients with advanced lung cancer. 2023.
National Cancer I. Testing the addition of tazemetostat to the immunotherapy drug, pembrolizumab (mk-3475), in advanced urothelial carcinoma. 2023.
Hoffmann-La R. A safety and pharmacology study of atezolizumab (mpdl3280a) administered with obinutuzumab or tazemetostat in participants with relapsed/refractory follicular lymphoma and diffuse large b-cell lymphoma. 2020.
Constellation P. Orion-e: a study evaluating cpi-1205 in patients with advanced solid tumors. 2019.
Bristol-Myers S. Study of bms-986158 in subjects with select advanced cancers. 2021.
Incyte C. An open-label, dose-escalation/dose-expansion safety study of incb059872 in subjects with advanced malignancies. 2022.
Chinese PLAGH Study of chidamide, decitabine and immune checkpoint inhibitors in r/r nhl and advanced solid tumors. 2024.
Sidney Kimmel Comprehensive Cancer Center at Johns H., Syndax P., Bristol-Myers S. and National Cancer I. A clinical trial of entinostat in combination with nivolumab for patients with previously treated unresectable or metastatic cholangiocarcinoma and pancreatic adenocarcinoma. 2020.
Memorial Sloan Kettering Cancer C, Merck S, Dohme LLC and Syndax P. Combination therapy with entinostat and pembrolizumab in relapsed and refractory lymphomas. 2023.
National Cancer I. Testing the safety and efficacy of the combination of the antibody pembrolizumab and entinostat in patients with myelodysplastic syndrome who are not responding to hypomethylating agents. 2023.
Center UNCLCz, Merck S., Dohme L.L.C. and Syndax Pharmaceuticals I. Window of opportunity study of pembrolizumab alone and in combinations in bladder cancer. 2023.
Syndax P, Merck S, Dohme L.L.C. Ph1b/2 dose-escalation study of entinostat with pembrolizumab in nsclc with expansion cohorts in nsclc, melanoma, and colorectal cancer. 2022.
Syndax P, Merck S, Dohme LLC. Continuation study of entinostat in combination with pembrolizumab in patients with advanced solid tumors. 2019.
Center U.N.C.L.C.C. and Syndax Pharmaceuticals I. An exploratory study of pembrolizumab plus entinostat in non-inflamed stage iii/iv melanoma. 2022.
National Cancer I. Entinostat, nivolumab, and ipilimumab in treating patients with solid tumors that are metastatic or cannot be removed by surgery or locally advanced or metastatic her2-negative breast cancer. 2022.
Roberto P., Bristol-Myers S., Syndax P., Indiana University School of M. and Hoosier Cancer Research N. Study of entinostat with nivolumab plus ipilimumab in previously treated renal cell carcinoma. 2022.
Syndax P, Merck KgaA DG. and Pfizer. Phase 1b/2 study of avelumab with or without entinostat in patients with advanced epithelial ovarian cancer. 2019.
Mirati Therapeutics I. Phase 2 study of glesatinib, sitravatinib or mocetinostat in combination with nivolumab in non-small cell lung cancer. 2021.
Mirati Therapeutics I. Phase 1/2 study of mocetinostat and durvalumab in patients with advanced solid tumors and nsclc. 2019.
Health N.Y.U.L. A phase 1b study of the selective hdac inhibitor mocetinostat in combination with ipilimumab and nivolumab in patients with unresectable stage iii or stage iv melanoma. 2020.
Center H.L.M.C., Research I., Merck S. and Dohme L.L.C. Pembro and vorinostat for patients with stage iv non-small cell lung cancer (nsclc). 2024.
Center H.L.M.C., Research I., Merck S. and Dohme L.L.C. Pembrolizumab and vorinostat combined with temozolomide for newly diagnosed glioblastoma. 2021.
City of Hope Medical C. and National Cancer I. Pembrolizumab and vorinostat in treating patients with relapsed or refractory diffuse large b-cell lymphoma, follicular lymphoma, or hodgkin lymphoma. 2022.
University of W. and National Cancer I. Pembrolizumab and vorinostat in treating patients with recurrent squamous cell head and neck cancer or salivary gland cancer that is metastatic and/or cannot be removed by surgery. 2019.
Nabil A. and Indiana U. Phase i/ib study of pembrolizumab with vorinostat for patients with advanced renal or urothelial cell carcinoma. 2023.
University of California S.F., Avon F., Merck S. and Dohme L.L.C. Reversing therapy resistance with epigenetic-immune modification. 2019.
Acknowledgements
We apologize to the researchers not cited in the manuscript due to limited space.
Funding
Wenyu Wang is supported by the National Natural Science Foundation of China (Nos. 31900515, 81972818). Bing Cheng is supported by the National Natural Science Foundation of China (82003163), and GuangDong Basic and Applied Basic Research Foundation (2019A1515110483).
Author information
Authors and Affiliations
Contributions
Writing, review, and/or revision of the manuscript: BC, QY, WW. Conception and design of the perspective: BC, QY, WW. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Cheng, B., Yu, Q. & Wang, W. Intimate communications within the tumor microenvironment: stromal factors function as an orchestra. J Biomed Sci 30, 1 (2023). https://doi.org/10.1186/s12929-022-00894-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-022-00894-z