
Abstract
Post-translational modifications (PTMs) are crucial for the adaptation of various signalling pathways to ensure cellular homeostasis and proper adaptation to stress. PTM is a covalent addition of a small chemical functional group such as a phosphate group (phosphorylation), methyl group (methylation), or acetyl group (acetylation); lipids like hydrophobic isoprene polymers (isoprenylation); sugars such as a glycosyl group (glycosylation); or even small peptides such as ubiquitin (ubiquitination), SUMO (SUMOylation), NEDD8 (neddylation), etc. SUMO modification changes the function and/or fate of the protein especially under stress conditions, and the consequences of this conjugation can be appreciated from development to diverse disease processes. The impact of SUMOylation in disease has not been monotonous, rather SUMO is found playing a role on both sides of the coin either facilitating or impeding disease progression. Several recent studies have implicated SUMO proteins as key regulators in various cardiovascular disorders. The focus of this review is thus to summarize the current knowledge on the role of the SUMO family in the pathophysiology of cardiovascular diseases.
Similar content being viewed by others

Introduction
Every cell has countless biological processes occurring simultaneously, right from its genesis till its terminus, recalibrating its fate perpetually. Each of these processes such as growth, molecule and ion transport or cell division involves molecular pathways with an array of intermediates which ultimately converge on a gene or a group of genes getting modulated, which further regulate a cascade of more such pathways. Although the effect of each pathway is fairly pre-determined, a small molecular modification in one of the players can shift the entire direction of a signaling cascade, altering the fate of the intermediates and definitely changing the outcome. Such modifications, typically post-translational modifications (PTM), can serve as a ‘by-default setting’ in the pathway or could be brought about by the state at which the cell is at that instant, e.g. in a situation of cellular stress.
Small Ubiquitin like Modifier (SUMO) proteins have been recognized as one of the key PTMs modulating the function and half-life of many proteins, thereby serving as master switches in multiple molecular signalling pathways. In the following, we provide a detailed overview on the individual SUMO proteins and their (patho)physiological roles.
The SUMO family
SUMO is a conserved family of proteins that has one member in yeast, around eight in plants and four in mammals. In the late 90s, a single protein, now called SUMO1, was reported by several researchers with different names with differing functions. SUMO1 was found to be associated with DNA recombination and DNA damage response proteins RAD 51/52, and since it showed similarity to ubiquitin, it was termed as ubiquitin-like protein 1 (UBL1) [101]; in another report it prevented cell death by associating with the cytoplasmic tails or the “death domains” of FAS/APO 1 and tumour necrosis factor (TNF) receptor 1 and thus was given the name Sentrin (after ‘sentry’ for the protein’s function as a guardian) [85]; moreover, due to interaction with promyelocytic leukaemia (PML) protein,, it was also named as PIC1 (PML interacting clone 1) [10]; its posttranslational modification of RanGAP1 with subsequent translocation to RanBP2, a nuclear pore complex protein, led to the name GAP modifying protein 1 (GMP1) or SUMO1 [72, 74]; whereas, homology with the yeast protein Smt3 gave SUMO1 the name Smt3a [62]. To the contrary, SUMO2 and SUMO3 (SUMO2/3) were discovered using a database search [51]. SUMO2/3 share almost 95% homology with one other, but the two share only around 50% homology with SUMO1. SUMO4 was identified while studying the association of single nucleotide polymorphisms with type I diabetes, however, it is currently viewed as an intron-less pseudogene [12, 30]. The latest member of the family, a primate-specific SUMO5 with high tissue specificity, was found to facilitate promyelocytic leukaemia nuclear body growth [67].
Despite the similarity of SUMO with ubiquitin in sequence, size and even conjugation, their functions differ drastically. Ubiquitin is best known for its role in redirecting the proteins towards degradation via proteasomal or lysosomal pathways by binding to the target protein in a polymer fashion (also known as poly-ubiquitin tags), but it is also involved in regulating nuclear localization and DNA repair pathways [65]. SUMO, on the other hand, has been reported to play a role in a plethora of functions like DNA replication, transcriptional regulation, chromatin organization, cell cycle regulation, sub-cellular localization, protein–protein interactions, protein–DNA interactions, DNA damage response, and degradation [27, 33, 70, 74, 90, 100, 105, 108]. It is interesting and important to note that either ubiquitination or SUMOylation of a single substrate results in a different outcome. For example, ubiquitination of Histone deacetylase 1 (HDAC1), an epigenetic regulator, sends the protein to proteasome mediated degradation, whereas, SUMOylation promotes transcriptional repression of HDAC1 by enhancing its histone deacetylation activity [16]. The so called ‘Guardian of the genome’, p53 and the tumour suppressor promyelocytic leukaemia (PML) protein both get degraded upon ubiquitination. However, SUMOylation of PML is a prerequisite for nuclear body formation [124] and SUMO conjugation relegates the transcriptional activity of p53 [116].
The SUMO cycle
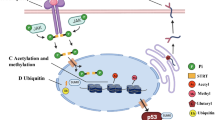
SUMO proteins are synthesized as propeptides and the maturation of these proteins requires the activity of proteins called the SENPs (sentrin/SUMO-specific proteases). SENPs chop off four amino acid residues from the SUMO propeptide, exposing the diglycine residue, thereby rendering the protein ready for substrate conjugation. The SUMO conjugation pathway is very similar to the ubiquitin conjugation pathway (Fig. 1). Post maturation, the SUMO molecule moves to the activation step by an E1 activating enzyme. In mammals, a SAE1–SAE2 heterodimer catalyses the adenylation of SUMO in the presence of Mg2+ ions [20, 84, 98]. This high energy bond between AMP and SUMO is broken and a new thioester linkage is formed between the active cysteine residue of SAE2 and the C terminal glycine of SUMO [49]. The next step is conjugation where SUMO is transferred to Ubc9, a SUMO conjugating E2 enzyme. The association between SUMO and the active cysteine of Ubc9 is another thioester bond [26, 48, 63, 99]. The penultimate step of the cycle is SUMO conjugation to a substrate. Ubc9 is potent enough to execute the conjugation process, but there have also been studies that suggest the involvement of E3 ligases in this process. Unlike ubiquitin E3 ligases, SUMO E3s act as a scaffold bringing the substrate and the SUMO bound E2 in proper orientation for the conjugation to materialize. SUMO E3 ligases have been classified into different families. PIAS (protein inhibitor of activated STAT) is a eukaryotic SUMO E3 ligase family with its counterparts called Siz proteins in the yeast. PIAS have two domains, one called the SAP domain for binding to AT rich DNA sequences while the other called the Siz/PIAS (SP) RING domain like the ubiquitin E3s, which allow for attachment with E2 [39]. PIAS also has the capability of binding to the substrate covalently via a PINIT domain [87]. However, its attachment to the SUMO molecule is non-covalent via a SUMO interacting motif (SIM) present on PIAS [75]. Ran binding protein 2 (RanBP2) is another SUMO E3 ligase family containing an internal repeat (IR) domain which facilitates the ligase activity. This family of E3s bind to Ubc9 promoting an optimal orientation but interestingly does not bind to the substrate [81, 97]. Polycomb 2 (Pc2) is a multimeric complex SUMO E3 ligase which does not possess the RING domain, yet binds to both Ubc9 and the substrate to promote the conjugation [50]. ZNF451 is the latest family added to the SUMO E3 ligase list. ZNF451 proteins have tandem SIM and inter SIM motifs which help perform the SUMO ligation [52]. Along with their ligation (E3) function, ZNF451 proteins also indulge in the SUMO elongation (E4) function [23]. Extensive research has revealed that SUMO substrates have a consensus sequence where the SUMO proteins bind. Interestingly, this sequence is also present in SUMO2/3 but not in SUMO1. This could be the reason why the former form polymeric chains while the latter does not [60]. In fact, this also corresponds to the presence of large pools of free SUMO2/3 but comparatively little SUMO1 [96]. Like deubiquitylation, the SUMO cycle concludes with deconjugation of SUMO by SENPs, setting the SUMO proteins free to enter the cycle again for changing the fate of another molecule (Fig. 1).

The SUMO cycle. SUMO is produced as a precursor which is processed by SENP to expose the C terminal diglycine residues (MATURATION). SUMO is then activated by heterodimer E1 enzyme in an ATP dependent manner (ACTIVATION). SUMO is then transferred to the SUMO E2 conjugating enzyme, Ubc9 (CONJUGATION). Further, SUMO is attached to the substrate either in the presence of E3 (with or without substrate binding) or directly by Ubc9 (LIGATION). Depending on the substrate and function, more than one SUMO molecules can be bound (POLYSUMOYLATION). SUMOylation is reversed by SENPs, releasing the SUMO molecule(s) for the next cycle
SUMO consensus motif v/s SUMO interacting motifs
Consensus motifs for SUMO conjugation are specific sequences in the SUMO substrates which are recognized by the E2 enzymes. This consensus motif is a highly conserved 4 amino acid sequence ψKxD/E, wherein ψ stands for a large hydrophobic amino acid and x is any amino acid [95]. Substrate recognition is assisted by the electrostatic interaction and the hydrogen bonds between E2 and the residues surrounding the lysine of this motif. A spike in the rate of reaction is brought about by the decrease in the pKa of the lysine residue via the residues in the E2, and further stabilization of this interaction is facilitated by co-factors and E3 enzymes. For example, SUMOylation of Peroxisome proliferator-activated receptor (PPAR)-γ2 at k107 is enhanced by the phosphorylation at S112; such motifs with a phosphorylatable serine residue at the C-terminal of the consensus motif, ψKX(D/E)XXSp, are called phosphorylation-dependent SUMO motifs (PDSMs) [119]. This phosphate group interacts with the basic patch in the Ubc9, aiding in recognition [76]. Another example of such a variant are negatively charged amino acid-dependent SUMO motifs (NDSMs), also present at the C-terminus of the consensus and important for the recognition of some proteins by Ubc9 [76, 120].
Around half of the SUMO substrates bind via the consensus motif, yet upon cellular stress conditions, interactions with non-consensus motifs seem to surge. In the early 2000s, a group of investigators noticed a few proteins interacting with an already SUMOylated p73, a tumour suppressor. Upon further investigation, they found that a SxS sequence (S = Serine) between a hydrophobic stretch and an acidic stretch in the protein is necessary for this interaction [75]. Further on, a few other groups showed that a (V/I)X(V/I)(V/I) motif, a hydrophobic core in between acidic stretches, is the obligatory sequence in this interaction [103]. Such sequences are called SIMs, another set of loose consensus motif bringing about a non-covalent interaction between the protein and a SUMO molecule. For this interaction, the beta sheets in the SIMs take on a parallel or an antiparallel orientation with the beta sheet of the SUMO molecule, where, the acidic residues around the SIMs and the SUMO basic residues form salt bridges [25, 53]. Like the PDSMs, SIMs also have a family which comprises of a phosphorylatable serine residue. Phosphorylation of this serine residue at the C terminal of the hydrophobic core by casein kinase 2 augments the SUMO-substrate interaction [22].
SUMOylation in cardiovascular diseases
Atherosclerosis
Atherosclerosis is an inflammatory disorder characterized by accumulation of lipids and immune cells, termed as plaques, on the intima of blood vessels ultimately triggering conditions like myocardial infarction and stroke. These plaques are a result of chronic activation of molecular signalling pathways in the inner endothelial lining of the blood vessels that mediate inflammation and apoptosis (Fig. 2). Signalling inside these cells depends on the flow dynamics of blood in the specific areas. A steady state “laminar” flow is regarded to be atheroprotective, while “turbulent” flow has been shown to be atherogenic [18]. NFκB is a transcription factor which upon activation enters the nucleus and initiates transcription of a number of genes responsible for stimulating the inflammatory signals in the cell. SUMOylation of IκBα, a component of NFκB inhibitor, via SUMO 1 rescues IκBα from ubiquitin mediated proteasomal degradation, in turn disabling the NFκB pathway [19]. On the contrary, SUMOylation of the same molecule via SUMO2/3 detaches it from NFκB, stimulating this pathway [19]. A component of the NFκB inhibitor kinase, NEMO, acts as a stimulant for NFκB upon SUMOylation [45].

Participation of SUMOylation in molecular signalling pathways involved in atherosclerosis. In the NFκB pathway, SUMOylation of IκBα via SUMO1 proves atheroprotective while IκBα SUMO2/3 conjugation proves atherogenic and so does NEMO SUMOylation. MK2 SUMOylation inhibits VCAM1/MCP1 mediated inflammation acting as a friend while p53 SUMOylation is apoptotic, hence SUMOylation acts as a foe here. SUMO when bound to MAGI activates p30RSK which further stabilizes ERK–SUMO interaction by retaining SENP2 in the cytoplasm, hence SUMOylation here in both the scenarios prove to be hazardous to the cells. Bcl2 B cell lymphoma 2, CO carbon monoxide, d flow distributed flow, ERK5 extracellular-signal-regulated kinase 5, HO hemeoxygenase, IκBα inhibitor of κB-α, IκK inhibitor of κB kinase, Klf Kruppel-like Factor, MAGI membrane-associated guanylate kinase with inverted domain structure-1, MCP1 monocyte chemoattractant protein 1, MK2 mitogen-activated protein kinase-activated protein kinase 2, NEMO NFκB essential modulator, P90RSK P90 Ribosomal s6 Kinase, PIASγ protein inhibitor of activated STAT γ, PKCζ protein kinase C ζ, PPAR peroxisome proliferator-activated receptor, s flow steady flow, SENP sentrin/SUMO-specific proteases, SMRT1 silencing mediator of retinoic acid and thyroid hormone receptor, VCAM vascular cell adhesion protein
Mitogen-activated protein kinase-activated protein kinase 2 (MAPKAPK2, or MK2) is another proinflammatory molecule which can further surge NFκB activation [47]. LDLR−/−/MK2−/− compound-mutant mice showed a reduction in TNFα induced inflammation via downregulation of VCAM1 and MCP1 expression and thus reduction in plaque formation compared to LDLR−/− mice, confirming the proinflammatory role of MK2 [47]. SUMOylation of MK2 inhibits the kinase activity of MK2 in turn inhibiting its inflammatory ability [14].
Extracellular-signal-regulated kinase 5-ERK5 (a member of the MAPK family) is a master molecule which inhibits atherosclerosis either by interacting with various proteins to inhibit inflammatory pathways or by controlling the expression of various anti-atherogenic targets [35]. The steady state flow activates ERK5, which in turn associates with peroxisome proliferator-activated receptor γ (PPARγ) [2]. Binding of ERK5 with the PPARγ-SMRT complex discharges SMRT (Silencing mediator of retinoic acid and thyroid hormone receptor) activating PPARγ for NFκB inhibition [2]. Carbon monoxide and heme oxygenase-1 mediated ERK5 activation also leads to association of ERK5 with PPARδ, rendering it active for NFκB inhibition [115]. ERK5 activation upregulates KLF2 and KLF4 which also help reduce inflammation [80, 83, 89]. SUMOylation of ERK5 occurs due to turbulent flow mediated ROS production, and impedes the atheroprotective role of ERK5 [37]. ROS mediated Protein kinase C ζ (PKCζ) activation brings about the association of activated PKCζ with protein inhibitor of activated STAT protein gamma (PIASγ)- an E3 SUMO ligase [37]. Once the E3 ligase is activated, it SUMOylates p53 resulting in its nuclear expulsion [37]. p53 associates with Bcl2 and inhibits its anti-apoptotic activity, thus activating the apoptotic signal [34]. Upon ER stress caused by distributed flow, the P90 Ribosomal s6 Kinase (p90RSK) and SUMOylated Membrane-associated guanylate kinase with inverted domain structure-1 (MAGI1) complex leads to phosphorylation of both MAGI1 and SENP2 [1, 36]. The phosphorylation of SENP2 enables its cytoplasmic retention, whereas, the phosphorylation of MAGI1 enables its deSUMOylation by SENP1 [1, 36]. The deSUMOylated MAGI1 facilitates the nuclear localization of p90RSK-MAGI1 and ER stress by product ATF6-MAGI1 complex. p30RSK further stabilizes the ERK5-SUMO in the nucleus by phosphorylating the nuclear SENP2 causing its export [1, 36]. Activating transcription factor 3 (ATF3) is a stress response transcription factor downstream of various environmental stresses. In Angiotensin II induced hypertensive mice, the colocalization of ATF3 and SUMO1 was observed in the thoracic aorta [122]. SUMOylation of ATF3 inhibits its ubiquitin mediated degradation and stabilizes the protein. SUMO deficient ATF3 K42R mutant on the other hand showed reduced nitric oxide production and knockdown of ATF3 or SUMO1 inhibited various inflammatory molecules [122]. These results indicate that the SUMOylation acts as a regulatory step in ATF3 mediated endothelial inflammation [122].
Reverse cholesterol transport (RCT) is a mechanism in which cholesterol from the peripheral tissues is delivered back to the liver [31]. A cross between an atherosclerosis-prone LDL receptor knockout mice (LDLR−/−) with a nuclear receptor liver receptor homolog 1 (LRH1) SUMOylation mutant LRH1 K289R, showed increased RCT in the mice [106]. The reason being, the wild type SUMO binds to prospero homeobox protein 1 (PROX1) and represses hepatic RCT genes, however, the SUMOylation deficient LRH1 fails to interact with PROX1, derepressing the RCT genes and reducing atherosclerosis in the mice [106].
Cardiomyopathy and heart failure
Cardiac hypertrophy
Cardiac hypertrophy is a condition in which cardiomyocytes grow and undergo cytoskeletal remodelling, thus increasing its size and sarcomere stability as a compensatory response to the increased biomechanical stress. However, sustained cardiac hypertrophy is usually maladaptive and results in contractile dysfunction and arrhythmias, clinically associated with heart failure and sudden death. Hypertrophy caused by pressure overload due to transverse aortic constriction is inhibited by SUMO1 gene transfer and left ventricular function is restored [107]. Likewise, oxidative stress mediated modifications in SERCA2a are inhibited by SUMO1 preventing the cardiomyocytes from undergoing hypertrophy [107]. Angiotensin II and insulin growth factor II receptor (IGF-IIR) have also been reported to be involved in cardiac hypertrophy [42]. Heat shock protein-2 (HSP2) SUMOylated at K82 by SUMO1 is unable to bind to the IGF-IIR promoter. A study revealed that angiotensin II upon binding to its receptor upregulates a polycomb protein MEL18, which then mediates deSUMOylation of HSP2 protein allowing it to activate IGF-IIR gene by direct interaction [42]. The increased protein expression of IGF-IIR subsequently leads to cardiac hypertrophy [42]. The possible mechanisms underlying MEL18 mediated deSUMOylation could be either interaction of MEL18 with UBC9 leading to its inhibition or MEL18 activating SENP1 rendering deSUMOylation, or both [64].
SUMO1 conjugation of myocardin (a smooth muscle and cardiac specific transcriptional coactivator) assisted by PIAS1 can cause hypertrophy in the heart, which is attenuated by p65 (a unit of NFκB heterodimer), as it inhibits both PIAS1 and SUMO1 [68]. p65 also increases the expression of microRNA miR-1, which in turn silences myocardin [44]. Activation of ZAK (Sterile alpha motif and leucine zipper containing kinase), a pro apoptotic protein from MAPKKK, activates MAPKs like p-JUN and P-P38, which further promotes c-JUN and GATA -induced hypertrophy [40]. ZAK translocate to the nucleus via SUMO1 conjugation, and this is attenuated by oestrogen receptor β (ERβ) overexpression which retains ZAK in the cytoplasm [86]. Overexpression of human myofibrillogenesis regulator 1 (MR1) in NRVCMs showed increased sarcomere organization followed by hypertrophy [113]. MR1 overexpression also showed an increased nuclear to cytoplasmic translocation of myomesin-1 mimicking SUMO1 overexpression [113]. Interestingly, SUMO1 overexpression along with a stealth siRNA for MR1 did not result in nuclear translocation of myomesin-1. These results indicate that MR1 SUMOylates myomesin-1, which then mediates increased sarcomere organization preceding hypertrophy [113]. In 2017, a group of investigators observed that treatment of primary cardiomyocytes with phenylephrine (PE), a hypertrophy causative agent, led to an increase in the expression of the SUMO E3 ligase Mitochondrial anchored protein ligase (MAPL), localized to the mitochondrial membrane [123]. This was accompanied by increased mitochondrial fission and decrease in mitochondrial fusion protein-2 (Mfn2), a protein necessary for mitochondrial fusion [123]. The authors suggested that the hypertrophic stimulus upregulates MAPL which promotes SUMOylation of mitochondrial fission protein Drp1 thus facilitating hypertrophy [123]. They also found a microRNA miR-485-5p to abrogate hypertrophy by silencing MAPL expression [123]. The calcineurin (Cn)-NFAT pathway is one of the most important signalling pathways associated with cardiac hypertrophy [77]. Bernt et al. used a luciferase mediated screening approach to discover SUMO2 as a novel candidate modulator associated with Cn-NFAT signalling [7]. We found that SUMO2 interacts with Cn and translocate Cn to the nucleus envelope whereby it activates NFAT signalling [7]. Overexpression of a mutant diglycine deficient SUMO2 (ΔGG) failed to alter Cn-NFAT signalling, pointing towards a SUMOylation independent activation of the pathway by SUMO2-Cn. These findings summarised in Fig. 3 signify the importance of SUMOylation in cardiac hypertrophy.

Participation of SUMOylation in molecular signalling pathways involved in cardiac hypertrophy. SUMO conjugation of Cn facilitated NFAT mediated hypertrophy, while SUMO binding to Myocardin could be a direct hypertrophic stimulus. Likewise, SUMOylated Myom1 can cause sarcomeric reorganization and SUMOylated Drp1 can cause mitochondrial disfunction, both leading to cardiac hypertrophy. These portray SUMO as a foe. But, ZAK, which mediates hypertrophy via c-JUN/GATA and HSP, which causes IGFIIR mediated hypertrophy, both upon SUMO conjugation obstructs their hypertrophic role, attesting SUMO as a friend here. Cn calcineurin, Drp1 dynamin related protein 1, ERβ oestrogen receptor β, HSP2 heat shock protein 2, IGFIIR Insulin growth factor 2 receptor, MAPL mitochondrial anchored protein ligase, Mfn2 mitochondrial fusion protein2, Myom1 myomesin 1, NFAT nuclear factor of activated T cells, PE phenylephrine, PIAS1 protein inhibitor of activated STAT; SENP1 sentrin/SUMO-specific proteases, SUMO2 small ubiquitin related modifier, ZAK sterile alpha motif and leucine zipper containing kinase
Dilated cardiomyopathy and heart failure
Dilated cardiomyopathy is a disease characterized by dilation in the left ventricular wall owing to reduction in contractile function. Anomalies in SUMO-modification of several substrates have been shown to contribute to dilated cardiomyopathy (Fig. 4). Cardiac specific overexpression of SENP5 results in mitochondrial dysfunction ultimately leading to dilated cardiomyopathy in aging mice. These adverse effects are caused by SENP5 mediated deSUMOylation of SUMO2/3 modified dynamin related protein (Drp1), a mitochondrial fission protein [58]. Results with SENP2 transgenic mice concurred with that of SENP5 overexpressing mice exhibiting cardiac fibrosis and cardiomyopathy with age [56]. Lamin A, an intermediate filament nuclear envelope protein, reportedly accounts for almost 10% of familial DCM due to autosomal dominant hereditary mutations [32]. SUMOylation of lamin A at K201 in the consensus sequence MKEE is necessary for normal cellular function. Mutants for K201R showed decreased SUMOylation of lamin A and exhibited abnormal lamin A localization and eventual cardiomyopathy, suggesting an important role of lamin A sumoylation in cardiomyopathy [121].

Participation of SUMOylation in molecular signalling pathways involved in dilated cardiomyopathy. SUMO plays a friendly role upon conjugating with Drp1 to inhibit mitochondrial disfunction and binding to Lamin A for its proper localization, but has a harmful effect when binding to CAST and Capn to induce apoptosis. Capn2 calpain small subunit 2, CAST calpastatin, Drp1 dynamin related protein 1, SENP sentrin/SUMO-specific proteases
The calpain–calpastatin pathway is an apoptotic pathway with its members found to be associated with SUMO2. Of note, cardiomyopathy is observed in transgenic mice overexpressing SUMO2. The calpain small subunit 2 (Capns2) inhibitor CAST (calpastatin), when conjugated to SUMO2, shows a reduced inhibition ability, whereas SUMO2 conjugation of Capns2 increases the enzymatic activity of Capns2. As a matter of fact, both SUMOylation events here prove to be detrimental in cardiomyopathy and subsequent heart failure [59].
Myocardial infarction/ischemic cardiomyopathy
Ischemia reperfusion injury is the damage caused by the reoxygenation of tissue after a short period of ischemia. Ischemic reperfusion leads to a decrease in the overall SUMO conjugation in cardiomyocytes. Like other forms of cardiomyopathies, SUMOylation defects in certain SUMO substrates causes ischemic cardiomyopathy (Fig. 5). Upon treatment with zinc, SUMO1 mediated SUMOylation of Drp1 increases zinc induced mitophagy protecting the cells against injury [9]. Ischemic reperfusion also increases SENP1 levels which further activates the hypoxia inducible factor 1α (HIF1α) pathway, thus supporting its cardioprotective role [29]. Moreover, there is a decrease in protein inhibitor of activated STAT 1 (PIAS 1) levels, a SUMO E3 ligase, upon ischemia reperfusion. An increased SUMOylation of PPARγ was observed upon ectopic expression of PIAS 1, inhibiting the NFκB pathway. It also revealed a decrease in the phosphorylation of IκBα, attenuating the NFκB signalling [117]. Luteonin is a plant flavonoid with antioxidant and immunomodulatory properties. A study shows that pre-treatment with luteonin stabilizes Sarco/endoplasmic reticulum Ca2+-ATPase (SERCA2a) by stabilizing SUMO1 protein binding and aids in the SUMOylation of SERCA2a specifically at K585 by SUMO1 [41]. SUMO conjugated SERCA2a enhances the mitochondrial membrane potential, rescuing the cell from ischemia reperfusion injury mediated apoptosis [21, 41, 54]. A group of investigators showed that myocardial ischemia reperfusion injury activates DJ1- an E3 ligase which inhibits the action of SUMO1 modified Drp1 [102]. In the absence of this event, DRP1 would be recruited to the mitochondria, activate mitochondrial fission followed by release of caspase 3, promoting infarction expansion [102]. A study with an adeno-associated vector type 1 gene transfer of SUMO1 in swine models with ischemic heart failure showed an improvement in left ventricular ejection fraction and a decrease in left ventricular volume compared to saline treated controls [107].

Participation of SUMOylation in molecular signalling pathways involved in ischemic cardiomyopathy. Zinc mediated Drp1 SUMOylation inhibits mitochondrial disfunction and SERCA2a SUMOylation increases mitochondrial membrane potential inhibiting apoptosis demonstrating the friendly nature of SUMO. Decreased levels of SUMOylated PPARγ activates the NFκB pathway, again displaying the beneficial role of SUMO conjugation. Drp1 dynamin related protein 1, HIF 1α hypoxia inducible factor 1α, NFκB nuclear factor κB, PIAS1 protein inhibitor of activated STAT 1, PPAR peroxisome proliferator-activated receptor, SENP sentrin/SUMO-specific proteases, SERCA2a Sarco/endoplasmic reticulum Ca2+-ATPase 2a, Zn zinc
Congenital heart defects
Gam1, an adenovirus protein, selectively leads to the degradation of SUMO E1 activating enzyme [11]. Microinjections of Gam1 mRNA in embryos at different developmental stages revealed sensitivity of non-canonical Wnt/PCP, Ets-1 and snail/twist to the loss of SUMOylation [8]. This loss of SUMOylation further manifested in defects of neural tube formation and heart development [8]. Importantly, Wang et al. demonstrated atrial and ventricular septal defects leading to premature death of SUMO1-deficient mice [109]. Furthermore, to substantiate these findings in humans, they reported a single 16 bp substitution in the regulatory sequence of SUMO1 gene sequence, probably causing loss of SUMOylation activity, in newborns with atrial septal defects and cleft lip [109].
A single point mutation in any transcription factor can largely disrupt the developmental orchestration, ultimately leading to congenital defects. Nkx2.5 SUMOylation deficient mutant added with haplosufficiency led to congenital heart defects in mouse hearts, indicating the importance of SUMOylation in development [57]. An increasing number of mutations in regulatory transcription factors like GATA, Myocardin, Mef2c, Nfatc1, tbx5, SRF, Smad4 have also been discovered which form the basis of numerous congenital defects [17, 43, 44, 69, 78, 82, 93]. Interestingly, these proteins have also been suggested to be modulated by SUMOylation being SUMO substrates [4, 5, 28, 71, 73, 79, 88, 110,111,112]. However, the relation between these mutations and altered SUMO conjugation is yet to be established.
Ion channel defects and cardiac arrhythmias
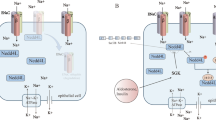
Ion channels are the key regulators of electrical conduction system of the heart, and impaired function typically causes cardiac arrhythmias. The channels producing the slowly activating K+ currents, called the IKs channels are crucial for ventricular repolarization. A recent study shows that all the four KCNQ1 subunits of the IKs can potentially be SUMOylated and addition of each SUMO molecule shifts the half maximum activation voltage to the positive side [118]. It is known that Kv1.5 mediates the ultra-rapid delayed rectifier potassium current in cardiomyocytes. Studies show that Kv1.5 are also SUMO conjugatable, and that SUMOylation of Kv1.5 can modulate the excitability of cardiomyocytes [6]. SUMO also works as the “gatekeeper” of K2P1 channels, channels that help maintaining the resting membrane potential of the cells [92]. It was shown in oocytes that non SUMOylable K274E and K274R mutant K2P1 channels were constitutively active while the SUMO modified wildtype is silent [92]. To further understand this phenomenon, another group tried to study both the mutations and how these mutations differ from each other [24]. To their surprise, K274E showed an increased current density in K21P1 but K274R failed to show the upsurge, suggesting the increment is actually because of the glutamate and not SUMO dependent in COS7 cells [24]. To further add insights to this notion, Kim and Kang induced oxidative stress to impel deSUMOylation in rat cardiomyocytes using buthionine sulfoximine and hydrogen peroxide, but failed to see any activation in the K+ channels [55]. This puts the role of SUMO in K2P1 regulation in question and demands in-depth research for answering the same. Transient receptor potential cation channel subfamily M member 4 gene (TRPM4) encodes a protein highly expressed in the purkinje fibres, which is a non-selective calcium activated ion channel which depolarizes the membrane by mediating the transport of monovalent cations [61]. A mutation from G to A transcribes a defective protein TRPM4 E7K which is constitutively SUMOylated, insensitive to deSUMOylation [61]. This constitutively SUMOylated mutant does not get endocytosed and remains on the cell membrane, thus impairing the action potential and causing arrhythmias [61].
Clinical and translational relevance of SUMO
As SUMOylation is intimately involved in various signalling pathways relevant for cardiovascular disease, an obvious question is how this knowledge could be exploited for translation to new diagnostics and therapies. Years of research have produced a plethora of molecules which can inhibit the entire SUMOylation cascade just by inhibiting one of the players [15, 125], for instance, by inhibition of SUMO E1 activating enzymes. Molecules inhibiting the E1 enzyme mostly act as AMP mimics and bind to the E1 enzymes instead of AMP hence further hampering the SUMO-E1 attachment [104]. These compounds are investigated for treating various cardiovascular, neurological, inflammatory, and proliferative disorders [104]. Ginkgolic acid is a well-known compound that inhibits the SUMO E1 enzyme via the same mechanism [91]. Studies show that delivery of Ginkgolic acid via osmotic pumps embedded in the back of MI mice reduced MI induced cardiac fibrosis [91]. Inhibition of E1 enzyme limited both the SUMOylation of PML, hampering the TGFβ1 pathway as well as angiotensin II induced fibroblast to myofibroblast transformation [91]. Inhibitors of SUMO E2 enzyme like Spectomycin B1, directly bind to Ubc9 and inhibit its SUMO binding [38]. Treating the MCF7 breast cancer line with Spectomycin B1 restrains estrogen dependent proliferation of these cells [38]. Hemopoietic lineage switch 5, a RING finger B-box protein with a coiled coil domain, get SUMOylated by SUMO1 [114, 125]. Interestingly, HLS5 can also bind to both Ubc9 and PIAS1 and degrades the SUMO E2 and E3 via its coiled coil domain. SENP inhibitors generally bind to the catalytic site of SENPs impeding their action [125]. These inhibitors can be used to reduce viral infectivity and are also candidates for anticancer therapy [125]. Poly-SUMO chain inhibitors normally have two components, a metal nanoparticle and a modified SIM mimic conjugated to each other via a thiol tail [66]. These compounds get attached to SAE and then get transferred to Ubc9 and eventually to the SUMO substrate, restricting the entry of SUMO molecules in the cascade [125]. Since SUMO 2/3 also contain SUMO binding sites, the metal nanoparticles attach to SUMO 2/3, hampering the poly-SUMOylation process [125].
Another example is sumoylation of SERCA2a, which has long been in the focus of investigators working on heart failure, since it regulates the calcium fluxes in cardiomyocytes necessary for proper contractile function. In 2015, the compound “N106” was identified which could increase SUMOylation of SERCA2a in a heart failure mouse model. N106 increases SUMOylation by activating SUMO E1 enzymes, thereby increasing SERCA2a activity and subsequently contractile function. But targeting a specific SUMO E3 enzyme could turn out to be more definitive as SUMO E1s fail to convey specific effects.
Cardiomyocyte metabolism is tightly regulated, as a slight dysfunction in the energy balance could be catastrophic for a cell type with such a high energy consumption. AMP activated protein kinase (AMPK) is an enzyme which plays a pivotal role in the metabolic homeostasis of a cell with the AMPK pathway shown to be associated with cardiovascular diseases as well [3]. AMPK is activated via interaction with the upstream molecule liver kinase B1 (LKB1), cardiomyocyte-specific deletion of which leads impaired AMPK activation and cardiac dysfunction [46]. Interestingly, during an energy crisis, LKB1 gets sumoylated by SUMO1 at lysine 178, thus enabling its interaction with AMPK [94]. Site-directed mutagenesis studies have demonstrated that the SUMOylation-deficient LKB1 resulted in defective AMPK signaling and malformed mitochondrial function, inducing death in energy-deprived cells [94]. Targeting LKB1 sumoylation could thus be an effective measure against heart diseases. Protection against ischemia is also a common physiological phenomenon in ground squirrels during the state of torpor. Interestingly, the reduction in blood flow to the brain in this context is accompanied with a drastic increase in Ubc9 expression along with a spike in global SUMOylation in the brain and other organs, playing a cytoprotective role. In vitro studies also show a similar response of cells upon oxygen/nutrient deprivation. In conjunction to this, Lee et al. used a flavonoid Quercetin which inhibits SENPs and rescued neuronal cells against ischemia in vitro. It would be fascinating to know if Quercetin could serve the same protective role in the heart against ischemic injury.
Another study suggested the presence of giant fibrillar centres (GFC), large stores of ribosomal gene transcriptional machinery components, in rat sensory ganglia neurons, and reported that the number of GFCs increased with the increase in the cell size [13]. The point of interest here is that they also observed an upsurge of Ubc9 and SUMO1 with GFCs [13]. This data along with other studies suggest a direct role of SUMO in cellular growth. Further investigations regarding targeting SUMO as a therapeutic measure against cardiomyocyte hypertrophy are therefore necessary.
Alterations in mitochondrial dynamics have been reported in almost all cardiovascular diseases and DRP1 thus seems to be an attractive candidate for therapy. After a successful pilot study, a clinical trial on inhibition of the mitochondrial fission protein, drp1, for the prevention of atherosclerosis is under way [ClinicalTrials.gov Identifier: NCT03980548]. Recently, SUMO entered the world of clinical trials when an inhibitor of SUMO conjugation cycle TAK-981 was tested in patients with neoplasms or lymphomas in a phase 1 clinical trial [ClinicalTrials.gov Identifier: NCT03648372]. TAK-981 is derived from ML-792, and both inhibit the SUMO cycle by binding to the C terminal of the SUMO molecule, rendering SUMO E1 impotent to transfer the SUMO protein to E2.
An important point to emphasize is the necessity to find the difference between the exact roles of each of the SUMO molecules along with the upstream regulators of the respective SUMO pathway to impartially target any pathophysiological condition. Another important aspect which needs further clarification are the mechanisms involved in substrate specificity as well as SUMO isoform preference. Also, as we have seen earlier, on how the SUMOylation–ubiquitination axis and the SUMOylation–phosphorylation axis decides the fate of their substrate, unearthing more such interplays would definitely provide a better understanding.
Conclusions
So, is SUMO ‘a friend or a foe’? It would be too early to comment as SUMOylation studies still have a lot to uncover. As of now, it looks like SUMO has context-dependent roles, so SUMO can either be a friend or a foe. The SUMO machinery could definitely be pertinent in understanding the mechanics of cardiovascular diseases as well as finding plausible targets for therapy.
Availability of data and materials
Not applicable.
References
Abe J, Ko KA, Kotla S, Wang Y, Paez-Mayorga J, Shin IJ, Imanishi M, Vu HT, Tao PT, Leiva-Juarez MM, Thomas TN, Medina JL, Won JH, Fujii Y, Giancursio CJ, McBeath E, Shin JH, Guzman L, Abe RJ, Taunton J, Mochizuki N, Faubion W, Cooke JP, Fujiwara K, Evans SE, Le NT. MAGI1 as a link between endothelial activation and ER stress drives atherosclerosis. JCI Insight. 2019;4(7):e125570.
Akaike M, Che W, Marmarosh NL, Ohta S, Osawa M, Ding B, Berk BC, Yan C, Abe J. The hinge-helix 1 region of peroxisome proliferator-activated receptor gamma1 (PPARgamma1) mediates interaction with extracellular signal-regulated kinase 5 and PPARgamma1 transcriptional activation: involvement in flow-induced PPARgamma activation in endothelial cells. Mol Cell Biol. 2004;24(19):8691–704.
Bairwa SC, Parajuli N, Dyck JR. The role of AMPK in cardiomyocyte health and survival. Biochim Biophys Acta. 2016;1862(12):2199–210.
Beketaev I, Kim EY, Zhang Y, Yu W, Qian L, Wang J. Potentiation of Tbx5-mediated transactivation by SUMO conjugation and protein inhibitor of activated STAT 1 (PIAS1). Int J Biochem Cell B. 2014;50:82–92.
Belaguli NS, Zhang M, Garcia AH, Berger DH. PIAS1 is a GATA4 SUMO ligase that regulates GATA4-dependent intestinal promoters independent of SUMO ligase activity and GATA4 Sumoylation. PLoS ONE. 2012;7(4):e35717.
Benson MD, Li QJ, Kieckhafer K, Dudek D, Whorton MR, Sunahara RK, Iniguez-Lluhi JA, Martens JR. SUMO modification regulates inactivation of the voltage-gated potassium channel Kv1.5. Proc Natl Acad Sci USA. 2007;104(6):1805–10.
Bernt A, Rangrez AY, Eden M, Jungmann A, Katz S, Rohr C, Muller OJ, Katus HA, Sossalla ST, Williams T, Ritter O, Frank D, Frey N. Sumoylation-independent activation of Calcineurin-NFAT-signaling via SUMO2 mediates cardiomyocyte hypertrophy. Sci Rep. 2016;6:35758.
Bertke MM, Dubiak KM, Cronin L, Zeng EL, Huber PW. A deficiency in SUMOylation activity disrupts multiple pathways leading to neural tube and heart defects in Xenopus embryos. BMC Genomics. 2019;20:386.
Bian XY, Xu JM, Zhao HH, Zheng Q, Xiao XL, Ma XF, Li YX, Du XP, Liu XZ. Zinc-induced SUMOylation of dynamin-related protein 1 protects the heart against ischemia-reperfusion injury. Oxid Med Cell Longev. 2019. https://doi.org/10.1155/2019/1232146.
Boddy MN, Howe K, Etkin LD, Solomon E, Freemont PS. PIC 1, a novel ubiquitin-like protein which interacts with the PML component of a multiprotein complex that is disrupted in acute promyelocytic leukaemia. Oncogene. 1996;13(5):971–82.
Boggio R, Passafaro A, Chiocca S. Targeting SUMO E1 to ubiquitin ligases—a viral strategy to counteract sumoylation. J Biol Chem. 2007;282(21):15376–82.
Bohren KM, Nadkarni V, Song JH, Gabbay KH, Owerbach D. A M55V polymorphism in a novel SUMO gene (SUMO-4) differentially activates heat shock transcription factors and is associated with susceptibility to type I diabetes mellitus. J Biol Chem. 2004;279(26):27233–8.
Casafont I, Bengoechea R, Navascues J, Pena E, Berciano MT, Lafarga M. The giant fibrillar center: a nucleolar structure enriched in upstream binding factor (UBF) that appears in transcriptionally more active sensory ganglia neurons. J Struct Biol. 2007;159(3):451–61.
Chang E, Heo KS, Woo CH, Lee H, Le NT, Thomas TN, Fujiwara K, Abe J. MK2 SUMOylation regulates actin filament remodeling and subsequent migration in endothelial cells by inhibiting MK2 kinase and HSP27 phosphorylation. Blood. 2011;117(8):2527–37.
Cox OF, Huber PW. Developing practical therapeutic strategies that target protein SUMOylation. Curr Drug Targets. 2019;20(9):960–9.
David G, Neptune MA, DePinho RA. SUMO-1 modification of histone deacetylase 1 (HDAC1) modulates its biological activities. J Biol Chem. 2002;277(26):23658–63.
de la Pompa JL, Timmerman LA, Takimoto H, Yoshida H, Elia AJ, Samper E, Potter J, Wakeham A, Marengere L, Langille BL, Crabtree GR, Mak TW. Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature. 1998;392(6672):182–6.
Dehnavi S, Sadeghi M, Penson PE, Banach M, Jamialahmadi T, Sahebkar A. The role of protein SUMOylation in the pathogenesis of atherosclerosis. J Clin Med. 2019;8(11):1856.
Desterro JMP, Rodriguez MS, Hay RT. SUMO-1 modification of I kappa B alpha inhibits NF-kappa B activation. Mol Cell. 1998;2(2):233–9.
Desterro JMP, Rodriguez MS, Kemp GD, Hay RT. Identification of the enzyme required for activation of the small ubiquitin-like protein SUMO-1. J Biol Chem. 1999;274(15):10618–24.
Du YP, Liu P, Xu TD, Pan DF, Zhu H, Zhai NN, Zhang YB, Li DY. Luteolin modulates SERCA2a leading to attenuation of myocardial ischemia/reperfusion injury via sumoylation at lysine 585 in mice. Cell Physiol Biochem. 2018;45(3):883–98.
Duncan JS, Litchfield DW. Too much of a good thing: the role of protein kinase CK2 in tumorigenesis and prospects for therapeutic inhibition of CK2. BBA-Proteins Proteom. 2008;1784(1):33–47.
Eisenhardt N, Chaugule VK, Koidl S, Droescher M, Dogan E, Rettich J, Sutinen P, Imanishi SY, Hofmann K, Palvimo JJ, Pichler A. A new vertebrate SUMO enzyme family reveals insights into SUMO-chain assembly. Nat Struct Mol Biol. 2015;22(12):959–67.
Feliciangeli S, Bendahhou S, Sandoz G, Gounon P, Reichold M, Warth R, Lazdunski M, Barhanin J, Lesage F. Does sumoylation control K2P1/TWIK1 background K+ channels? Cell. 2007;130(3):563–9.
Gareau JR, Lima CD. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol. 2010;11(12):861–71.
Gong LM, Kamitani T, Fujise K, Caskey LS, Yeh ETH. Communication—Preferential interaction of sentrin with a ubiquitin-conjugating enzyme, Ubc9. J Biol Chem. 1997;272(45):28198–201.
Goodson ML, Hong Y, Rogers R, Matunis MJ, Park-Sarge OK, Sarge KD. SUMO-1 modification regulates the DNA binding activity of heat shock transcription factor 2, a promyelocytic leukemia nuclear body associated transcription factor. J Biol Chem. 2001;276(21):18513–8.
Gregoire S, Yang XJ. Association with class IIa histone deacetylases upregulates the sumoylation of MEF2 transcription factors. Mol Cell Biol. 2005;25(6):2273–87.
Gu JM, Fan YQ, Liu XB, Zhou LH, Cheng JK, Cai R, Xue S. SENP1 protects against myocardial ischaemia/reperfusion injury via a HIF1 alpha-dependent pathway. Cardiovasc Res. 2014;104(1):83–92.
Guo D, Li M, Zhang Y, Yang P, Eckenrode S, Hopkins D, Zheng W, Purohit S, Podolsky RH, Muir A, Wang J, Dong Z, Brusko T, Atkinson M, Pozzilli P, Zeidler A, Raffel LJ, Jacob CO, Park Y, Serrano-Rios M, Larrad MT, Zhang Z, Garchon HJ, Bach JF, Rotter JI, She JX, Wang CY. A functional variant of SUMO4, a new I kappa B alpha modifier, is associated with type 1 diabetes. Nat Genet. 2004;36(8):837–41.
Gupta AK, Ross EA, Myers JN, Civen M, Maeda T, Gadhe BM, Kashyap ML. Reverse cholesterol transport is increased in athletes. Clin Res. 1990;38(2):A252–A252.
Hasselberg NE, Haland TF, Saberniak J, Brekke PH, Berge KE, Leren TP, Edvardsen T, Haugaa KH. Lamin A/C cardiomyopathy: young onset, high penetrance, and frequent need for heart transplantation. Eur Heart J. 2018;39(10):853–60.
Hendriks IA, Vertegaal ACO. SUMO in the DNA damage response. Oncotarget. 2015;6(18):15734–5.
Heo KS, Chang E, Le NT, Cushman H, Yeh ETH, Fujiwara K, Abe J. De-SUMOylation enzyme of sentrin/SUMO-specific protease 2 regulates disturbed flow-induced SUMOylation of ERK5 and p53 that leads to endothelial dysfunction and atherosclerosis. Circ Res. 2013;112(6):911-U132.
Heo KS, Cushman HJ, Akaike M, Woo CH, Wang X, Qiu X, Fujiwara K, Abe J. ERK5 activation in macrophages promotes efferocytosis and inhibits atherosclerosis. Circulation. 2014;130(2):180-U184.
Heo KS, Le NT, Cushman HJ, Giancursio CJ, Chang E, Woo CH, Sullivan MA, Taunton J, Yeh ET, Fujiwara K, Abe J. Disturbed flow-activated p90RSK kinase accelerates atherosclerosis by inhibiting SENP2 function. J Clin Invest. 2015;125(3):1299–310.
Heo KS, Lee H, Nigro P, Thomas T, Le NT, Chang E, McClain C, Reinhart-King CA, King MR, Berk BC, Fujiwara K, Woo CH, Abe J. PKCzeta mediates disturbed flow-induced endothelial apoptosis via p53 SUMOylation. J Cell Biol. 2011;193(5):867–84.
Hirohama M, Kumar A, Fukuda I, Matsuoka S, Igarashi Y, Saitoh H, Takagi M, Shin-ya K, Honda K, Kondoh Y, Saito T, Nakao Y, Osada H, Zhang KYJ, Yoshida M, Ito A. Spectomycin B1 as a novel SUMOylation inhibitor that directly binds to SUMO E2. ACS Chem Biol. 2013;8(12):2635–42.
Hochstrasser M. SP-RING for SUMO: new functions bloom for a ubiquitin-like protein. Cell. 2001;107(1):5–8.
Hsieh YL, Tsai YL, Shibu MA, Su CC, Chung LC, Pai PY, Kuo CH, Yeh YL, Viswanadha VP, Huang CY. ZAK induces cardiomyocyte hypertrophy and brain natriuretic peptide expression via p38/JNK signaling and GATA4/c-Jun transcriptional factor activation. Mol Cell Biochem. 2015;405(1–2):1–9.
Hu WJ, Xu TD, Wu P, Pan DF, Chen JH, Chen J, Zhang BC, Zhu H, Li DY. Luteolin improves cardiac dysfunction in heart failure rats by regulating sarcoplasmic reticulum Ca2+-ATPase 2a. Sci Rep. 2017;7:41017.
Huang CY, Kuo CH, Pai PY, Ho TJ, Lin YM, Chen RJ, Tsai FJ, Padma VV, Kuo WW, Huang CY. Inhibition of HSF2 SUMOylation via MEL18 upregulates IGF-IIR and leads to hypertension-induced cardiac hypertrophy. Int J Cardiol. 2018;257:283–90.
Huang JH, Cheng L, Li J, Chen M, Zhou DY, Lu MM, Proweller A, Epstein JA, Parmacek MS. Myocardin regulates expression of contractile genes in smooth muscle cells and is required for closure of the ductus arteriosus in mice. J Clin Invest. 2008;118(2):515–25.
Huang JH, Lu MM, Cheng L, Yuan LJ, Zhu XQ, Stout AL, Chen M, Li J, Parmacek MS. Myocardin is required for cardiomyocyte survival and maintenance of heart function. Proc Natl Acad Sci USA. 2009;106(44):18734–9.
Huang TT, Wuerzbrger-Davis SM, Wu ZH, Miyamoto S. Sequential modification of NEMO/IKK gamma by SUMO-1 and ubiquitin mediates NF-kappa B activation by genotoxic stress. Cell. 2003;115(5):565–76.
Ikeda Y, Sato K, Pimentel DR, Sam F, Shaw RJ, Dyck JR, Walsh K. Cardiac-specific deletion of LKB1 leads to hypertrophy and dysfunction. J Biol Chem. 2009;284(51):35839–49.
Jagavelu K, Tietge UJF, Gaestel M, Drexler H, Schieffer B, Bavendiek U. Systemic deficiency of the MAP kinase-activated protein kinase 2 reduces atherosclerosis in hypercholesterolemic mice. Circ Res. 2007;101(11):1104–12.
Johnson ES, Blobel G. Ubc9p is the conjugating enzyme for the ubiquitin-like protein Smt3p. J Biol Chem. 1997;272(43):26799–802.
Johnson ES, Schwienhorst I, Dohmen RJ, Blobel G. The ubiquitin-like protein Smt3p is activated for conjugation to other proteins by an Aos1p/Uba2p heterodimer. EMBO J. 1997;16(18):5509–19.
Kagey MH, Melhuish TA, Wotton D. The polycomb protein Pc2 is a SUMO E3. Cell. 2003;113(1):127–37.
Kamitani T, Kito K, Nguyen HP, Fukuda-Kamitani T, Yeh ETH. Characterization of a second member of the sentrin family of ubiquitin-like proteins. J Biol Chem. 1998;273(18):11349–53.
Karvonen U, Jaaskelainen T, Rytinki M, Kaikkonen S, Palvimo JJ. ZNF451 is a novel PML body- and SUMO-associated transcriptional coregulator. J Mol Biol. 2008;382(3):585–600.
Kerscher O. SUMO junction—what’s your function? New insights through SUMO-interacting motifs. EMBO Rep. 2007;8(6):550–5.
Kho C, Lee A, Jeong D, Oh JG, Chaanine AH, Kizana E, Park WJ, Hajjar RJ. SUMO1-dependent modulation of SERCA2a in heart failure. Nature. 2011;477(7366):601-U263.
Kim D, Kang D. The two-pore domain K+ channel K2P1.1 (TWIK-1) is non-functional in the heart. FASEB J. 2008;22:971–6.
Kim EY, Chen L, Ma YL, Yu W, Chang J, Moskowitz IP, Wang J. Enhanced desumoylation in murine hearts by overexpressed SENP2 leads to congenital heart defects and cardiac dysfunction. J Mol Cell Cardiol. 2012;52(3):638–49.
Kim EY, Chen L, Ma YL, Yu W, Chang J, Moskowitz IP, Wang J. Expression of sumoylation deficient Nkx2.5 mutant in Nkx2.5 haploinsufficient mice leads to congenital heart defects. PLoS ONE. 2011;6(6):e20803.
Kim EY, Zhang Y, Beketaev I, Segura AM, Yu W, Xi YT, Chang J, Wang J. SENP5, a SUMO isopeptidase, induces apoptosis and cardiomyopathy. J Mol Cell Cardiol. 2015;78:154–64.
Kim EY, Zhang Y, Ye B, Segura AM, Beketaev I, Xi YT, Yu W, Chang J, Li FQ, Wang J. Involvement of activated SUMO-2 conjugation in cardiomyopathy. BBA-Mol Basis Dis. 2015;1852(7):1388–99.
Kroetz MB. SUMO: a ubiquitin-like protein modifier. Yale J Biol Med. 2005;78(4):197–201.
Kruse M, Schulze-Bahr E, Corfield V, Beckmann A, Stallmeyer B, Kurtbay G, Ohmert I, Schulze-Bahr E, Brink P, Pongs O. Impaired endocytosis of the ion channel TRPM4 is associated with human progressive familial heart block type I. J Clin Invest. 2009;119(9):2737–44.
Lapenta V, Chiurazzi P, van der Spek P, Pizzuti A, Hanaoka F, Brahe C. SMT3A, a human homologue of the S-cerevisiae SMT3 gene, maps to chromosome 21qter and defines a novel gene family. Genomics. 1997;40(2):362–6.
Lee GW, Melchior F, Matunis MJ, Mahajan R, Tian QS, Anderson P. Modification of Ran GTPase-activating protein by the small ubiquitin-related modifier SUMO-1 requires Ubc9, an E2-type ubiquitin-conjugating enzyme homologue. J Biol Chem. 1998;273(11):6503–7.
Lee JY, Won HY, Park JH, Kim HY, Choi HJ, Shin DH, Mang JH, Woo JK, Oh SH, Son T, Choi JW, Kim S, Kim HY, Yi K, Jang KS, Oh YH, Kong G. MEL-18 loss mediates estrogen receptor-alpha downregulation and hormone independence. J Clin Invest. 2015;125(5):1801–14.
Li W, Ye Y. Polyubiquitin chains: functions, structures, and mechanisms. Cell Mol Life Sci. 2008;65(15):2397–406.
Li YJ, Perkins AL, Su Y, Ma YL, Colson L, Horne DA, Chen Y. Gold nanoparticles as a platform for creating a multivalent poly-SUMO chain inhibitor that also augments ionizing radiation. Proc Natl Acad Sci USA. 2012;109(11):4092–7.
Liang YC, Lee CC, Yao YL, Lai CC, Schmitz ML, Yang WM. SUMO5, a novel poly-SUMO isoform regulates PML nuclear bodies. Sci Rep. 2016;6:26509.
Liao XH, Wang N, Zhao DW, Zheng DL, Zheng L, Xing WJ, Zhou H, Cao DS, Zhang TC. NF-kappa B (p65) negatively regulates myocardin-induced cardiomyocyte hypertrophy through multiple mechanisms. Cell Signal. 2014;26(12):2738–48.
Lin Q, Schwarz J, Bucana C, Olson EN. Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science. 1997;276(5317):1404–7.
Liu HW, Zhang J, Heine GF, Arora M, Ozer HG, Onti-Srinivasan R, Huang K, Parvin JD. Chromatin modification by SUMO-1 stimulates the promoters of translation machinery genes. Nucleic Acids Res. 2012;40(20):10172–86.
Long JY, Wang GN, He DM, Liu F. Repression of Smad4 transcriptional activity by SUMO modification. Biochem J. 2004;379:23–9.
Mahajan R, Delphin C, Guan TL, Gerace L, Melchior F. A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell. 1997;88(1):97–107.
Matsuzaki K, Minami T, Tojo M, Honda Y, Uchimura Y, Saitoh H, Yasuda H, Nagahiro S, Saya H, Nakao M. Serum response factor is modulated by the SUMO-1 conjugation system. Biochem Biophys Res Commun. 2003;306(1):32–8.
Matunis MJ, Coutavas E, Blobel G. A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein RanGAP1 between the cytosol and the nuclear pore complex. J Cell Biol. 1996;135(6):1457–70.
Minty A, Dumont X, Kaghad M, Caput D. Covalent modification of p73 alpha by SUMO-1—two-hybrid screening with p73 identifies novel SUMO-1-interacting proteins and a SUMO-1 interaction motif. J Biol Chem. 2000;275(46):36316–23.
Mohideen F, Capili AD, Bilimoria PM, Yamada T, Bonni A, Lima CD. A molecular basis for phosphorylation-dependent SUMO conjugation by the E2 UBC9. Nat Struct Mol Biol. 2009;16(9):945-U968.
Molkentin JD. Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc Res. 2004;63(3):467–75.
Moskowitz IP, Wang J, Peterson MA, Pu WT, Mackinnon AC, Oxburgh L, Chu GC, Sarkar M, Berul C, Smoot L, Robertson EJ, Schwartz R, Seidman JG, Seidman CE. Cardiac-specific transcription factor genes Smad4 and Gata4 cooperatively regulate cardiac valve development (vol 108, pg 4006, 2011). Proc Natl Acad Sci USA. 2011;108(14):5921.
Nayak A, Glockner-Pagel J, Vaeth M, Schumann JE, Buttmann M, Bopp T, Schmitt E, Serfling E, Berberich-Siebelt F. Sumoylation of the transcription factor NFATc1 leads to its subnuclear relocalization and interleukin-2 repression by histone deacetylase. J Biol Chem. 2009;284(16):10935–46.
Nayak L, Lin ZY, Jain MK. “Go with the flow”: how Kruppel-like factor 2 regulates the vasoprotective effects of shear stress. Antioxid Redox Sign. 2011;15(5):1449–61.
Nishimoto T. A new role of Ran GTPase. Biochem Biophys Res Commun. 1999;262(3):571–4.
Niu Z, Iyer D, Conway SJ, Martin JF, Ivey K, Srivastava D, Nordheim A, Schwartz RJ. Serum response factor orchestrates nascent sarcomerogenesis and silences the biomineralization gene program in the heart. Proc Natl Acad Sci USA. 2008;105(46):17824–9.
Ohnesorge N, Viemann D, Schmidt N, Czymai T, Spiering D, Schmolke M, Ludwig S, Roth J, Goebeler M, Schmidt M. Erk5 activation elicits a vasoprotective endothelial phenotype via induction of Kruppel-like Factor 4 (KLF4). J Biol Chem. 2010;285(34):26199–210.
Okuma T, Honda R, Ichikawa G, Tsumagari N, Yasuda H. In vitro SUMO-1 modification requires two enzymatic steps, E1 and E2. Biochem Biophys Res Commun. 1999;254(3):693–8.
Okura T, Gong L, Kamitani T, Wada T, Okura I, Wei CF, Chang HM, Yeh ET. Protection against Fas/APO-1- and tumor necrosis factor-mediated cell death by a novel protein, sentrin. J Immunol. 1996;157(10):4277–81.
Pai PY, Shibu MA, Chang RL, Yang JJ, Su CC, Lai CH, Liao HE, Viswanadha VP, Kuo WW, Huang CY. ER targets ZAK and attenuates cellular hypertrophy via SUMO-1 modification in H9c2 cells. J Cell Biochem. 2018;119(9):7855–64.
Palvimo JJ. PIAS proteins as regulators of small ubiquitin-related modifier (SUMO) modifications and transcription. Biochem Soc Trans. 2007;35:1405–8.
Pan MR, Chang TM, Chang HC, Su JL, Wang HW, Hung WC. Sumoylation of Prox1 controls its ability to induce VEGFR3 expression and lymphatic phenotypes in endothelial cells. J Cell Sci. 2009;122(18):3358–64.
Parmar KM, Larman HB, Dai GH, Zhang YH, Wang ET, Moorthy SN, Kratz JR, Lin ZY, Jain MK, Gimbrone MA, Garcia-Cardena G. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J Clin Invest. 2006;116(1):49–58.
Psakhye I, Castellucci F, Branzei D. SUMO-chain-regulated proteasomal degradation timing exemplified in DNA replication initiation. Mol Cell. 2019;76(4):632.
Qiu F, Dong CJ, Liu YX, Shao XQ, Huang D, Han YN, Wang B, Liu YL, Huo R, Paulo P, Zhang ZR, Zhao D, Chu WF. Pharmacological inhibition of SUMO-1 with ginkgolic acid alleviates cardiac fibrosis induced by myocardial infarction in mice. Toxicol Appl Pharm. 2018;345:1–9.
Rajan S, Plant LD, Rabin ML, Butler MH, Goldstein SAN. Sumoylation silences the plasma membrane leak K+ channel K2P1 (vol 121, pg 37, 2005). Cell. 2010;141(2):368–368.
Risebro CA, Searles RG, Melville AAD, Ehler E, Jina N, Shah S, Pallas J, Hubank M, Dillard M, Harvey NL, Schwartz RJ, Chien KR, Oliver G, Riley PR. Prox1 maintains muscle structure and growth in the developing heart (vol 136, pg 495, 2008). Development. 2009;136(4):699.
Ritho J, Arold ST, Yeh ET. A critical SUMO1 modification of LKB1 regulates AMPK activity during energy stress. Cell Rep. 2015;12(5):734–42.
Rodriguez MS, Dargemont C, Hay RT. SUMO-1 conjugation in vivo requires both a consensus modification motif and nuclear targeting. J Biol Chem. 2001;276(16):12654–9.
Saitoh H, Hinchey J. Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J Biol Chem. 2000;275(9):6252–8.
Saitoh H, Pu R, Cavenagh M, Dasso M. RanBP2 associates with Ubc9p and a modified form of RanGAP1. Proc Natl Acad Sci USA. 1997;94(8):3736–41.
Schulman BA, Harper JW. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat Rev Mol Cell Biol. 2009;10(5):319–31.
Schwarz SE, Matuschewski K, Liakopoulos D, Scheffner M, Jentsch S. The ubiquitin-like proteins SMT3 and SUMO-1 are conjugated by the UBC9 E2 enzyme. Proc Natl Acad Sci USA. 1998;95(2):560–4.
Seufert W, Futcher B, Jentsch S. Role of a ubiquitin-conjugating enzyme in degradation of S-Phase and M-Phase cyclins. Nature. 1995;373(6509):78–81.
Shen Z, Pardington-Purtymun PE, Comeaux JC, Moyzis RK, Chen DJ. UBL1, a human ubiquitin-like protein associating with human RAD51/RAD52 proteins. Genomics. 1996;36(2):271–9.
Shimizu Y, Lambert JP, Nicholson CK, Kim JJ, Wolfson DW, Cho HC, Husain A, Naqvi N, Chin LS, Li L, Calvert JW. DJ-1 protects the heart against ischemia-reperfusion injury by regulating mitochondrial fission. J Mol Cell Cardiol. 2016;97:56–66.
Song J, Durrin LK, Wilkinson TA, Krontiris TG, Chen Y. Identification of a SUMO-binding motif that recognizes SUMO modified proteins. Mol Biol Cell. 2004;15:119a–119a.
Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, Cullis CA, Doucette A, Garnsey JJ, Gaulin JL, Gershman RE, Lublinsky AR, McDonald A, Mizutani H, Narayanan U, Olhava EJ, Peluso S, Rezaei M, Sintchak MD, Talreja T, Thomas MP, Traore T, Vyskocil S, Weatherhead GS, Yu J, Zhang J, Dick LR, Claiborne CF, Rolfe M, Bolen JB, Langston SP. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458(7239):732-U767.
Sriramachandran AM, Dohmen RJ. SUMO-targeted ubiquitin ligases. BBA-Mol Cell Res. 2014;1843(1):75–85.
Stein S, Oosterveer MH, Mataki C, Xu P, Lemos V, Havinga R, Dittner C, Ryu D, Menzies KJ, Wang X, Perino A, Houten SM, Melchior F, Schoonjans K. SUMOylation-dependent LRH-1/PROX1 interaction promotes atherosclerosis by decreasing hepatic reverse cholesterol transport. Cell Metab. 2014;20(4):603–13.
Tilemann L, Lee A, Ishikawa K, Aguero J, Rapti K, Santos-Gallego C, Kohlbrenner E, Fish KM, Kho C, Hajjar RJ. SUMO-1 gene transfer improves cardiac function in a large-animal model of heart failure. Sci Transl Med. 2013;5(211):211ra159.
Verger A, Perdomo J, Crossley M. Modification with SUMO—a role in transcriptional regulation. EMBO Rep. 2003;4(2):137–42.
Wang J, Chen L, Wen S, Zhu HP, Yu W, Moskowitz IP, Shaw GM, Finnell RH, Schwartz RJ. Defective sumoylation pathway directs congenital heart disease. Birth Defects Res A. 2011;91(6):468–76.
Wang J, Feng XH, Schwartz RJ. SUMO-1 modification activated GATA4-dependent cardiogenic gene activity. J Biol Chem. 2004;279(47):49091–8.
Wang J, Li AK, Wang ZG, Feng XH, Olson EN, Schwartz RJ. Myocardin sumoylation transactivates cardiogenic genes in pluripotent 10T1/2 fibroblasts. Mol Cell Biol. 2007;27(2):622–32.
Wang J, Zhang H, Iyer D, Feng XH, Schwartz RJ. Regulation of cardiac specific nkx2.5 gene activity by small ubiquitin-like modifier. J Biol Chem. 2008;283(34):23235–43.
Wang XR, Liu XH, Wang S, Luan K. Myofibrillogenesis regulator 1 induces hypertrophy by promoting sarcomere organization in neonatal rat cardiomyocytes. Hypertens Res. 2012;35(6):597–603.
Winteringham LN, Endersby R, Beaumont J, Lalonde JP, Crossley M, Klinken SP. Hls5, a novel ubiquitin E3 ligase, modulates levels of sumoylated GATA-1. Blood. 2009;114(22):108–108.
Woo CH, Massett MP, Shishido T, Itoh S, Ding B, McClain C, Che W, Vulapalli SR, Yan C, Abe J. ERK5 activation inhibits inflammatory responses via peroxisome proliferator-activated receptor delta (PPARdelta) stimulation. J Biol Chem. 2006;281(43):32164–74.
Wu SY, Chiang CM. p53 sumoylation Mechanistic insights from reconstitution studies. Epigenetics-US. 2009;4(7):445–51.
Xie B, Liu XY, Yang J, Cheng JK, Gu JM, Xue S. PIAS1 protects against myocardial ischemia-reperfusion injury by stimulating PPAR SUMOylation. BMC Cell Biol. 2018;19:24.
Xiong DZ, Li T, Dai H, Arena AF, Plant LD, Goldstein SAN. SUMOylation determines the voltage required to activate cardiac I-Ks channels. Proc Natl Acad Sci USA. 2017;114(32):E6686–94.
Yamashita D, Yamaguchi T, Shimizu M, Nakata N, Hirose F, Osumi T. The transactivating function of peroxisome proliferator-activated receptor gamma is negatively regulated by SUMO conjugation in the amino-terminal domain. Genes Cells. 2004;9(11):1017–29.
Yang SH, Galanis A, Witty J, Sharrocks AD. An extended consensus motif enhances the specificity of substrate modification by SUMO. EMBO J. 2006;25(21):5083–93.
Zhang YQ, Sarge KD. Sumoylation regulates lamin A function and is lost in lamin A mutants associated with familial cardiomyopathies. J Cell Biol. 2008;182(1):35–9.
Zhang ZB, Ruan CC, Chen DR, Zhang K, Yan C, Gao PJ. Activating transcription factor 3 SUMOylation is involved in angiotensin II-induced endothelial cell inflammation and dysfunction. J Mol Cell Cardiol. 2016;92:149–57.
Zhao YF, Ponnusamy M, Liu CY, Tian J, Dong YH, Gao JN, Wang CQ, Zhang Y, Zhang L, Wang K, Li PF. MiR-485-5p modulates mitochondrial fission through targeting mitochondria anchored protein ligase in cardiac hypertrophy. BBA-Mol Basis Dis. 2017;1863(11):2871–81.
Zhong S, Muller S, Ronchetti S, Freemont PS, Dejean A, Pandolfi PP. Role of SUMO-1-modified PML in nuclear body formation. Blood. 2000;95(9):2748–53.
Zhou YJ, Ji CM, Cao MD, Guo M, Huang W, Ni WW, Meng L, Yang HW, Wei JF. Inhibitors targeting the SUMOylation pathway: a patent review 2012–2015 (Review). Int J Mol Med. 2018;41(1):3–12.
Acknowledgements
None.
Funding
This work was funded by the grant from DZHK (German Centre for Cardiovascular Research) to NF.
Author information
Authors and Affiliations
Contributions
PS, AR and NF drafted, revised and approved the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shetty, P.M.V., Rangrez, A.Y. & Frey, N. SUMO proteins in the cardiovascular system: friend or foe?. J Biomed Sci 27, 98 (2020). https://doi.org/10.1186/s12929-020-00689-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-020-00689-0