
Abstract
Background
A comprehensive understanding of the genetic basis of rare diseases and their regulatory mechanisms is essential for human molecular genetics. However, the genetic mutant spectrum of pathogenic genes within the Chinese population remains underrepresented. Here, we reported previously unreported functional ABHD12 variants in two Chinese families and explored the correlation between genetic polymorphisms and phenotypes linked to PHARC syndrome.
Methods
Participants with biallelic pathogenic ABHD12 variants were recruited from the Chinese Deafness Genetics Cohort. These participants underwent whole-genome sequencing. Subsequently, a comprehensive literature review was conducted.
Results
Two Han Chinese families were identified, one with a compound heterozygous variant and the other with a novel homozygous variant in ABHD12. Among 65 PHARC patients, including 62 from the literature and 3 from this study, approximately 90% (57 out of 63) exhibited hearing loss, 82% (50 out of 61) had cataracts, 82% (46 out of 56) presented with retinitis pigmentosa, 79% (42 out of 53) experienced polyneuropathy, and 63% (36 out of 57) displayed ataxia. Seventeen different patterns were observed in the five main phenotypes of PHARC syndrome. A total of 33 pathogenic variants were identified in the ABHD12. Compared with other genotypes, individuals with biallelic truncating variants showed a higher incidence of polyneuropathy (p = 0.006), but no statistically significant differences were observed in the incidence of hearing loss, ataxia, retinitis pigmentosa and cataracts.
Conclusions
The diagnosis of PHARC syndrome is challenging because of its genetic heterogeneity. Therefore, exploring novel variants and establishing genotype-phenotype correlations can significantly enhance gene diagnosis and genetic counseling for this complex disease.
Similar content being viewed by others

Background
PHARC syndrome (OMIM 612674) is an autosomal recessive neurodegenerative disorder characterized by polyneuropathy, hearing loss, ataxia, retinitis pigmentosa, and cataracts. The worldwide incidence of this syndrome is less than 1 in 1,000,000 (https://www.orpha.net). In 2009, Fiskerstrand et al. first described this syndrome as a Refsum-like disorder and mapped the causative gene to a 15.96 Mb region on chromosome 20 (20p11.21-q12) [1]. Then, in 2010, they identified ABHD12 as the causative gene of PHARC syndrome [2]. The ABHD12 gene encodes a 45 kDa protein with 398 residues, termed lysophosphatidylserine lipase, a key enzyme in the regulation of nervous and immune processes. It is predominantly expressed in the central nervous system and innate immune cells and facilitates the hydrolysis of 2-arachidonoyl glycerol (2-AG) [3] ABHD12 belongs to the α/β-hydrolase folding domain (ABHD) superfamily, which encompasses diverse protein families such as proteases, lipases, esterases, dehalogenases, peroxidases, and epoxide hydrolases, making it one of the most diverse protein families [4]. Age-related symptoms appeared in ABHD12 gene knockout mice, which were similar to human PHARC syndrome, indicating that the interruption of lysophosphatidylserine lipase metabolism and induced neuroinflammation may be the molecular basis for PHARC syndrome [3].
Clinically, PHARC syndrome typically appears in early childhood or adolescence, impacting multiple body systems such as the eyes, ears, muscles, and nerves, with variable onset and severity [5]. In the eyes, the main symptoms include cataracts, retinitis pigmentosa (RP), optic atrophy, and nystagmus. RP typically begins in the second or third decade of life, leading to nyctalopia, constricted visual fields, and blindness when the fovea is affected [2, 6, 7]. Sensorineural hearing loss is postlingual and progressively worsens, with the degree of hearing loss ranging from moderate to profound [2, 6]. Polyneuropathy usually presents in childhood with symptoms such as distal muscle weakness, sensory disturbances, arched feet, and Achilles tendon contracture. In the late stages, patients often require a wheelchair for mobility [2, 8, 9]. PHARC syndrome encompasses neurological, auditory, and ophthalmic symptoms, although not all of them appear at the initial onset of symptoms. Patients with PHARC syndrome exhibit a heterogeneous clinical phenotype, making diagnosis challenging. Comprehensive phenotype recognition and genetic testing are essential for addressing this diagnostic challenge. The development of next-generation sequencing techniques has accelerated the discovery of gene variants responsible for rare diseases. In this study, we utilized whole-genome sequencing (WGS) to identify ABHD12 pathogenic variants in a Chinese hearing loss cohort. Additionally, we conducted a comprehensive analysis of the genotype-phenotype spectrum and correlation between PHARC syndrome and pathogenic variants in ABHD12 based on previously reported literature.
Methods
Pedigrees and participators
We enrolled three patients from two families as part of a Chinese hearing loss cohort. A comprehensive medical history was obtained, including information on the age of onset, relevant clinical manifestations, clinical tests, severity of hearing loss, history of cochlear implant surgery, rehabilitative outcomes, noise exposure, ototoxic drug exposure, and family history. Ethical approval for this study was granted by the Institutional Review Board of the Ethics Committee of West China Hospital of Sichuan University (2021 − 190), and informed consent was obtained from all participants or their parents.
WGS and data analysis
Peripheral blood DNA was extracted from both patients and family members, and the proband of each family underwent WGS. The sequencing procedure was conducted using DNBSEQ-T7 sequencing instruments (MGI, Shenzhen, China) with paired-end reads of 150 bp in length. Burrows-Wheeler Aligner (BWA, v0.5.10) was utilized for mapping the reads to the Human genome (Hg19/GRCh37). We developed an in-house variations calling workflow based on the GATK best practice. Variations were annotated, including variant quality, details, gene information, sequence database, allele state, and allele frequency [10]. For variant analysis, we applied the following filters: (1) Genotype Quality ≥ 20, Read Depth ≥ 6, and Alternate Allele Frequency > 0.1; (2) Minor Allele Frequencies (MAF) ≤ 0.5% in the Genome Aggregation Database (gnomAD) (https://gnomad.broadinstitute.org/) and the retention of coding variations, splice donor or acceptor site variations, or selected pathogenic or likely pathogenic variants in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar), Human Gene Mutation Database (HGMD) (https://www.hgmd.cf.ac.uk), and Deafness Variation Database (DVD) (https://deafnessvariationdatabase.org). The pathogenic potential of our identified variants was classified and interpreted according to the expert specifications of the ACMG/AMP variant interpretation guidelines for genetic hearing loss [11]. We also used CNVnator and Manta to detect copy number variants (CNVs) in WGS data. Filtering conditions included allele frequencies < 1% in gnomAD and the 1000 Genomes Project, ≤ 50% reciprocal overlap with the Database of Genomic Variants, and ≤ 25% overlap of the CNV with repetitive and low-complexity regions. The interpretation of variant pathogenicity for copy number variants followed the expert specifications of the ACMG/AMP variant interpretation guidelines [12].
Sanger sequencing and segregation analysis
The variations identified in the proband through WGS were validated in both the affected patients and their unaffected family members using Sanger sequencing. Primer sequences for polymerase chain reaction (PCR) amplification were designed using Primer3 software (http://primer3.ut.ee/), and the primer details can be found in Supplementary Material Table 1. Segregation analysis was performed to assess the concordance of phenotypic patterns within families and confirm the transmission of the candidate gene associated with each phenotype.
Statistics
The data were analyzed using SPSS 25.0. Continuous variables with a normal distribution were presented as mean ± standard deviation (SD). Fisher’s exact test was utilized to assess the presence of a significant association between two rates of categorical variables. A significance level of p < 0.05 was considered statistically significant.
Results
Clinical findings
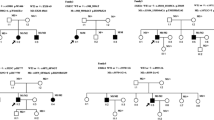
In Family 1, the proband (V-2), a 26-year-old woman, experienced the onset of hearing loss at the age of 12, which progressed to a profound level by the time she turned 21. As a result, she underwent cochlear implant surgery. At the age of 16, she started to exhibit symptoms of night blindness and constricted vision. Additionally, at the age of 22, she underwent surgery for cataracts. Subsequently, at 24 years old, she presented with ataxia. The proband’s younger brother (V-3), 25 years old, also experienced hearing loss at the age of 8, which progressed to a profound level at 19, but did not exhibit any other symptoms. It is worth noting that the proband’s parents are in a consanguineous marriage. In Family 2, the proband (II-1), a 19-year-old woman, developed hearing loss at the age of 13 and night blindness at the age of 18. By the age of 14, her hearing loss had progressed to a severe level (Fig. 1). None of the other family members displayed any symptoms. A summary of all clinical details can be found in Table 1.

The pedigree, Sanger sequencing results, and audiogram of Family 1 and Family 2 are presented
Genetic findings
In Family 1, a homozygous variant in ABHD12 gene (NM_001042472.3: c.690G > A, NP_001035937.1: p.Trp230Ter) was detected in the proband (V-2) and her younger brother (V-3). This homozygous variant was inherited from their unaffected parents with a heterozygous variant (Fig. 1). Notably, this variant was not detected in our normal hearing controls, which consist of 7,254 unrelated adults over the age of 18 years without self-reported hearing impairment, nor in the gnomAD database. According to the ACMG guidelines, the c.690G > A variant (PVS1 + PM2 + PP1_moderate) is classified as a pathogenic variant. In Family 2, a compound heterozygous variant in the ABHD12 gene was identified. The proband (II-1) carried the variants NM_001042472.3: c.874 C > T (NP_001035937.1: p.Arg292Ter) and NM_001042472.3: c.205_206del (NP_001035937.1: p.Trp69ValfsTer44). The proband’s mother was a carrier of the c.205_206del variant, while the father and sister (II-2) were carriers of the c.874 C > T variant (Fig. 1). Both variants in Family 2 were not observed in our normal hearing controls and the gnomAD database. Following the ACMG guidelines, the c.874 C > T variant (PVS1 + PM2 + PP1) and the c.205_206del variant (PVS1 + PM2 + PP1) are classified as pathogenic variants. No CNVs or other coding region variations (SNV/Indels) were detected in the ABHD12 gene in either of the probands.
Cochlear implant outcomes
In this study, both patients V-2 and V-3 from Family 1 underwent left-sided cochlear implantation (CI) at the ages of 21 and 19, respectively. Following the surgical intervention and speech rehabilitation, both patients achieved favorable outcomes in terms of auditory perception and speech performance.
Genotype and phenotype correlation in PHARC patients
Based on a comprehensive literature review, we identified 62 patients with PHARC from 43 families, including 9 consanguineous families. Affected individuals may experience a variety of symptoms, including hearing loss (mean onset age: 19.15 ± 1.73 years old, onset age range: 5–44 years old, n = 34), retinitis pigmentosa (mean onset age: 28.38 ± 1.55 years old, onset age range: 14–46 years old, n = 32), cataracts (mean onset age: 27.52 ± 3.52 years old, onset age range: 0–77 years old, n = 31), ataxia (mean onset age: 21.16 ± 3.64 years old, onset age range: 2–49 years old, n = 19), and polyneuropathy (mean onset age: 26.04 ± 3.03 years old, onset age range: 4–53 years old, n = 25). Detailed information is summarized in Table 2. Among the 65 PHARC syndrome patients, which includes the 62 patients from the literature and 3 patients from our study, approximately 90% (57 of 63) had hearing loss, 82% (50 of 61) had cataracts, 82% (46 of 56) had retinitis pigmentosa, 79% (42 of 53) had polyneuropathy, and 63% (36 of 57) had ataxia. When comparing biallelic truncating ABHD12 variants with other genotype groups, a statistically significant difference was observed in the rate of the polyneuropathy phenotype (p = 0.006). However, no statistically significant differences were found in the rates of hearing loss, cataracts, retinitis pigmentosa, and ataxia phenotypes (p > 0.05). Further details are provided in Table 3. The five main phenotypes of PHARC may combine to form 17 phenotypic combinations. Among the patients, 26% (17 of 65) exhibited all five characteristic phenotypes, 17% (11 of 65) exhibited a combination of hearing loss, cataracts, and retinitis pigmentosa, 11% (7 of 65) exhibited a combination of hearing loss, cataracts, polyneuropathy, and ataxia, 8% (5 of 65) exhibited a combination of hearing loss, cataracts, retinitis pigmentosa, polyneuropathy, and ataxia, 6% (4 of 65) exhibited a combination of hearing loss, cataracts, polyneuropathy, and ataxia, 6% (4 of 65) exhibited a combination of hearing loss, cataracts, and polyneuropathy, 5% (3 of 65) exhibited a combination of hearing loss, polyneuropathy, and ataxia, 5% (3 of 65) exhibited a combination of hearing loss and retinitis pigmentosa, 3% (2 of 65) exhibited a combination of polyneuropathy and ataxia, 3% (2 of 65) exhibited only retinitis pigmentosa, and the remaining seven phenotype combinations were each observed once. Details are summarized in Fig. 2.

The phenotypic combinations in a total of 65 PHARC syndrome patients. Note1: The height of the columns represents the number of patients for each phenotype combination. Note2: The rate in parentheses is the penetrance of each phenotype, refer to Table 3
Among the 65 PHARC syndrome patients, we identified a total of 33 ABHD12 pathogenic variations, including 11 (33%) nonsense variants, 8 (24%) missense variants, 7 (21%) frameshift variants, 5 (15%) splicing variants, and 2 (6%) CNVs. Truncating variants accounted for 76% (25 of 33), while the remaining 24% (8 of 33) were missense variants (Supplementary Fig. 1). ABHD12 pathogenic variants were found throughout the gene, with the exception of exon 11 and 13 (Figs. 3 and 4).

The diagram represents the distribution of variants across the gene structure of ABHD12 associated with PHARC syndrome

The diagram illustrates the variants (frameshift, nonsense, missense variants) in ABHD12 protein associated with PHARC syndrome
Discussion
To date, we have identified a total of 65 patients from 45 families with PHARC, including this study. Our study revealed that among the five main phenotypes of PHARC, approximately 90% (57 out of 63) exhibited hearing loss, 82% (50 out of 61) had cataracts, 82% (46 out of 56) experienced retinitis pigmentosa, 79% (42 out of 53) displayed polyneuropathy, and 63% (36 out of 57) demonstrated ataxia. These five phenotypes resulted in a total of 17 phenotypic combinations. However, only 26% (17 out of 65) of patients presented with all five characteristic phenotypes. This suggests that certain phenotypes may be overlooked without long-term follow-up or specific examinations, such as retinitis pigmentosa, polyneuropathy, and ataxia. Previous literature has indicated that the onset age of PHARC syndrome ranges from 0 to 77 years old, usually occurring during childhood or around the age of 20 years. In Family 1, the proband’s initial symptom was hearing loss, followed by retinitis pigmentosa, cataracts, and ataxia at the age of 26. However, by the age of 25, the proband’s younger brother had only complained of hearing loss. In Family 2, hearing loss was the first symptom, followed by night blindness. Similarly, other reported Chinese patients experienced the same situation. Li et al. reported two PHARC cases in a Chinese family, where both patients had hearing loss as their first symptom, followed by retinitis pigmentosa, with neither displaying neurological signs until the age of 33 [13]. Nevertheless, in some reported cases, the first phenotype may be a neurological or ophthalmic disorder [5, 14,15,16]. Hence, the genetic heterogeneity and onset time of PHARC syndrome vary among patients, even within the same family. It is worth noting that we also identified additional phenotypes coexisting with PHARC patients. Marina et al. reported two Spanish siblings who exhibited heel-knee dysmetria [17], and Li et al. reported two brothers with olfactory decline [13]. Other phenotypes reported included epilepsy [2, 18], learning difficulties [5], mental retardation [2], and congenital convergent strabismus of the eyes [19]. In the current situation, it is uncertain if these phenotypes are related to ABHD12 gene variations. However, after making a clear molecular diagnosis, we should also pay attention to these phenotypes to further expand the spectrum of this syndrome. Considering the diverse phenotypes and onset patterns of this syndrome, patients may initially seek consultation with ophthalmology, otorhinolaryngology, or neurology, making the diagnosis of PHARC syndrome challenging. In this study, the probands from Family 1 and Family 2 initially presented to the otorhinolaryngology department with preliminary diagnoses of Usher syndrome and non-syndromic hearing loss, respectively, prior to undergoing WGS. Upon establishing a definitive molecular diagnosis, the diagnoses for both patients were revised to PHARC syndrome. Thus, the differential diagnosis of PHARC syndrome primarily involves distinguishing it from diseases related to polyneuropathy, hearing loss, ataxia, retinitis pigmentosa, and cataracts, such as Usher syndrome (type I, type II, type III), Cockayne syndrome (OMIM 133540, OMIM 216400), Alström syndrome (OMIM 203800), mitochondrial diseases (e.g., NARP syndrome: neuropathy, ataxia, retinitis pigmentosa, OMIM 551500), and peroxisomal metabolism defect diseases (e.g., Refsum disease: retinitis pigmentosa, peripheral neuropathy, cerebellar ataxia, and elevated protein levels in the cerebrospinal fluid without an increase in the number of cells; OMIM 266500) [2, 13]. Therefore, the key to achieving a molecular diagnosis of this syndrome lies in conducting sequencing technology based on the characteristics of each phenotype, including onset age, progression, and severity.
The ABHD12 gene consists of 13 exons that encode a transmembrane protein with an extracellular catalytic domain in the C-terminal region [15]. In this study, we reported three PHARC syndrome patients from two Chinese families with ABHD12 variants, thereby expanding the genotype spectrum of this disorder. Out of the 65 patients identified, we found 33 ABHD12 mutations, including 11 (33%) nonsense variants, 8 (24%) missense variants, 7 (21%) frameshift variants, 5 (15%) splicing variants, and 2 (6%) CNVs. These variants are distributed throughout the gene, except for exons 11 and 13, with some clustered near or within the Hydrolase-4-Serine aminopeptidase domain. To further investigate the genotype-phenotype correlation of this syndrome, we grouped biallelic truncating genotypes as a proxy for genotypes that would cause a total loss of function, and biallelic missense and mixed genotypes as a proxy for genotypes that may retain some protein function. As a result, our findings revealed a notable association between biallelic truncating ABHD12 variants and a higher incidence rate of the polyneuropathy phenotype. This result aligns with previous studies that have demonstrated the deleterious effects of truncating variants on protein function, leading to severe clinical manifestations [20]. The observed higher incidence rate of polyneuropathy in individuals with biallelic truncating genotypes supports the hypothesis that these variants have a more pronounced impact on the development of this particular phenotype. In contrast, we did not observe a significant difference in the incidence rates of the other four phenotypes between the biallelic truncating genotypes and other genotype groups. This suggests that missense and mixed genotypes, which were presumed to retain some protein function, did not exhibit a distinct phenotype compared to other genotypes. Additional investigations are warranted to analyze the precise molecular mechanisms disrupted by different genotypes which could provide valuable insights into the pathogenesis of the various phenotypes observed.
Currently, there is no effective drug for treating PHARC syndrome, and symptomatic supportive treatment remains the main approach. In terms of hearing loss, CI is generally considered one of the best treatments for individuals with profound or severe hearing loss, provided that their auditory nerves and spiral ganglion are intact. Since the exact location of the ABHD12 gene in the inner ear and the biochemical basis of how ABHD12 gene variations lead to hearing loss are still unknown, further studies are needed to understand the function of ABHD12 in the inner ear [5]. Typically, CI yields good results in very young children with congenital hearing loss or in adults with post-lingual hearing loss [21]. Previous literature has reported the use of CI in ten post-lingual moderate to severe hearing loss patients caused by ABHD12 gene mutations [6, 8, 9, 13, 16,17,18]. Two of these patients reported positive CI outcomes, while the remaining eight did not mention the CI outcome. In our study, patients V-2 and V-3 from Family 1 underwent CI and achieved good auditory and speech outcomes. Therefore, PHARC patients with severe to profound hearing loss may benefit from CI.
In this study, we reported two Chinese families and conducted a genotype-phenotype correlation analysis of PHARC. This will be beneficial for ABHD12 variant interpretation and counseling of patient families. However, this study’s reliance on a retrospective analysis of existing literature limits our capacity to conduct long-term follow-ups to monitor the progression of phenotypic changes in patients. Consequently, our conclusions are based solely on the data currently available. The current mechanistic studies of PHARC mainly focus on the nervous system, revealing that abnormal lysophospholipase metabolism leads to the accumulation of proinflammatory lipids, thereby promoting microglial and neurobehavioral abnormalities [22, 23]. Considering the involvement of visual and hearing disturbances in PHARC, it is also essential to investigate the role of lysophospholipase in the development of these two organs. Moreover, ABHD12 has posed challenges for functional studies due to the lack of available crystal structures. As ABHD12 is an integral membrane protein and has been difficult to purify, analyzing its protein structure would be helpful for functional studies and the design of molecular drugs.
Conclusions
In conclusion, this study has identified one compound heterozygous variant and one novel homozygous variant in two families presenting syndromic hearing loss, thereby broadening the spectrum of ABHD12 variants. The accumulated knowledge from prior investigations strengthens our comprehension of the correlation between ABHD12 genotypes and phenotypes. Our study further substantiates the genotype-phenotype correlation in individuals affected by this syndrome. The findings of this research have potential implications for genetic counseling, carrier screening, and prenatal genetic diagnosis in patients diagnosed with PHARC syndrome. Consequently, our study offers new insights for the diagnosis and genetic counseling of PHARC syndrome, thereby contributing to the advancement of clinical practice in this field.
Data availability
The variants analysed in the current study are available in the Genetic Deafness Commons (GDC) repository, accessible via http://www.gdcdb.net/gene/ABHD12/variant.
References
Fiskerstrand T, Knappskog P, Majewski J, Wanders RJ, Boman H, Bindoff LA. A novel Refsum-like disorder that maps to chromosome 20. Neurology. 2009;72(1):20–7.
Fiskerstrand T, H’mida-Ben Brahim D, Johansson S, M’zahem A, Haukanes BI, Drouot N, Zimmermann J, Cole AJ, Vedeler C, Bredrup C, Assoum M, Tazir M, Klockgether T, Hamri A, Steen VM, Boman H, Bindoff LA, Koenig M, Knappskog PM. Mutations in ABHD12 cause the neurodegenerative disease PHARC: an inborn error of endocannabinoid metabolism. Am J Hum Genet. 2010;87(3):410–7.
Blankman JL, Long JZ, Trauger SA, Siuzdak G, Cravatt BF. ABHD12 controls brain lysophosphatidylserine pathways that are deregulated in a murine model of the neurodegenerative disease PHARC. Proc Natl Acad Sci U S A. 2013;110(4):1500–5.
Lord CC, Thomas G, Brown JM. Mammalian alpha beta hydrolase domain (ABHD) proteins: lipid metabolizing enzymes at the interface of cell signaling and energy metabolism. Biochim Biophys Acta. 2013;1831(4):792–802.
Nguyen XT, Almushattat H, Strubbe I, Georgiou M, Li CHZ, van Schooneveld MJ, Joniau I, De Baere E, Florijn RJ, Bergen AA, Hoyng CB, Michaelides M, Leroy BP, Boon CJF. The phenotypic spectrum of patients with PHARC Syndrome due to variants in ABHD12: an Ophthalmic Perspective. Genes (Basel). 2021;12(9):1404.
Chen DH, Naydenov A, Blankman JL, Mefford HC, Davis M, Sul Y, Barloon AS, Bonkowski E, Wolff J, Matsushita M, Smith C, Cravatt BF, Mackie K, Raskind WH, Stella N, Bird TD. Two novel mutations in ABHD12: expansion of the mutation spectrum in PHARC and assessment of their functional effects. Hum Mutat. 2013;34(12):1672–8.
Verbakel SK, van Huet RAC, Boon CJF, den Hollander AI, Collin RWJ, Klaver CCW, Hoyng CB, Roepman R, Klevering BJ. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res. 2018;66:157–86.
Eisenberger T, Slim R, Mansour A, Nauck M, Nürnberg G, Nürnberg P, Decker C, Dafinger C, Ebermann I, Bergmann C, Bolz HJ. Targeted next-generation sequencing identifies a homozygous nonsense mutation in ABHD12, the gene underlying PHARC, in a family clinically diagnosed with Usher syndrome type 3. Orphanet J Rare Dis. 2012;7:59.
Thimm A, Rahal A, Schoen U, Abicht A, Klebe S, Kleinschnitz C, Hagenacker T, Stettner M. Genotype-phenotype correlation in a novel ABHD12 mutation underlying PHARC syndrome. J Peripher Nerv Syst. 2020;25(2):112–6.
Austin-Tse CA, Jobanputra V, Perry DL, Bick D, Taft RJ, Venner E, Gibbs RA, Young T, Barnett S, Belmont JW, Boczek N, Chowdhury S, Ellsworth KA, Guha S, Kulkarni S, Marcou C, Meng L, Murdock DR, Rehman AU, Spiteri E, Thomas-Wilson A, Kearney HM, Rehm HL. Medical Genome Initiative*. Best practices for the interpretation and reporting of clinical whole genome sequencing. NPJ Genom Med. 2022;7(1):27.
Patel MJ, DiStefano MT, Oza AM, Hughes MY, Wilcox EH, Hemphill SE, Cushman BJ, Grant AR, Siegert RK, Shen J, Chapin A, Boczek NJ, Schimmenti LA, Nara K, Kenna M, Azaiez H, Booth KT, Avraham KB, Kremer H, Griffith AJ, Rehm HL, Amr SS, Tayoun ANA. ClinGen Hearing Loss Clinical Domain Working Group. Disease-specific ACMG/AMP guidelines improve sequence variant interpretation for hearing loss. Genet Med. 2021;23(11):2208–12.
Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, Raca G, Ritter DI, South ST, Thorland EC, Pineda-Alvarez D, Aradhya S, Martin CL. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. 2020;22(2):245–57.
Li T, Feng Y, Liu Y, He C, Liu J, Chen H, Deng Y, Li M, Li W, Song J, Niu Z, Sang S, Wen J, Men M, Chen X, Li J, Liu X, Ling J. A novel ABHD12 nonsense variant in Usher syndrome type 3 family with genotype-phenotype spectrum review. Gene. 2019;704:113–20.
Nishiguchi KM, Avila-Fernandez A, van Huet RA, Corton M, Pérez-Carro R, Martín-Garrido E, López-Molina MI, Blanco-Kelly F, Hoefsloot LH, van Zelst-Stams WA, García-Ruiz PJ, Del Val J, Di Gioia SA, Klevering BJ, van de Warrenburg BP, Vazquez C, Cremers FP, García-Sandoval B, Hoyng CB, Collin RW, Rivolta C, Ayuso C. Exome sequencing extends the phenotypic spectrum for ABHD12 mutations: from syndromic to nonsyndromic retinal degeneration. Ophthalmology. 2014;121(8):1620–7.
Lerat J, Cintas P, Beauvais-Dzugan H, Magdelaine C, et al. A complex homozygous mutation in ABHD12 responsible for PHARC syndrome discovered with NGS and review of the literature. J Peripher Nerv Syst. 2017;22(2):77–84.
Lerat J, Cintas P, Beauvais-Dzugan H, Magdelaine C, Sturtz F, Lia AS. Functional validation of ABHD12 mutations in the neurodegenerative disease PHARC. Neurobiol Dis. 2017;98:36–51.
Frasquet M, Lupo V, Chumillas MJ, Vázquez-Costa JF, Espinós C, Sevilla T. Phenotypical features of two patients diagnosed with PHARC syndrome and carriers of a new homozygous mutation in the ABHD12 gene. J Neurol Sci. 2018;387:134–8.
Yoshimura H, Hashimoto T, Murata T, Fukushima K, Sugaya A, Nishio SY, Usami S. Novel ABHD12 mutations in PHARC patients: the differential diagnosis of deaf-blindness. Ann Otol Rhinol Laryngol. 2015;124(Suppl 1):S77–83.
Dias Bastos PA, Mendonça M, Lampreia T, Magriço M, Oliveira J, Barbosa R. PHARC Syndrome, a Rare Genetic disorder-case report. Mov Disord Clin Pract. 2021;8(6):977–9.
Dias Bastos PA, Mendonça M, Lampreia T, Magriço M, Oliveira J, Barbosa R. Human genomics. Effect of predicted protein-truncating genetic variants on the human transcriptome. Science. 2015;348(6235):666–9.
Lazard DS, Lee HJ, Gaebler M, Kell CA, Truy E, Giraud AL. Phonological processing in post-lingual deafness and cochlear implant outcome. NeuroImage. 2010;49(4):3443–51.
Thomas G, Brown AL, Brown JM. In vivo metabolite profiling as a means to identify uncharacterized lipase function: recent success stories within the alpha beta hydrolase domain (ABHD) enzyme family. Biochim Biophys Acta. 2014;1841(8):1097–101.
Minamihata T, Takano K, Nakamura Y, Seto R, Moriyama M. Increase in Cellular Lysophosphatidylserine Content exacerbates inflammatory responses in LPS-Activated Microglia. Neurochem Res. 2022;47(9):2602–16.
Acknowledgements
The authors express their gratitude to the patients, families, and scientists who made valuable contributions to this study.
Funding
National Key R&D Program of China (2021YFC10053101); 1.3.5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (ZYJC20002).
Author information
Authors and Affiliations
Contributions
The work was conceived by HY, YL, JC, and FB. Data acquisition and analysis were assisted by XW, JG, MZ, ML, and YH. WX was responsible for the follow-up of case data. The manuscript was drafted by XL and subsequently revised and approved by all authors.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Ethical approval for this study was granted by the Institutional Review Board of the Ethics Committee of West China Hospital of Sichuan University (2021 − 190). Informed consent was obtained from all participants or their parents.
Consent for publication
Not Applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Long, X., Xiong, W., Wang, X. et al. Genotype-phenotype spectrum and correlation of PHARC Syndrome due to pathogenic ABHD12 variants. BMC Med Genomics 17, 203 (2024). https://doi.org/10.1186/s12920-024-01984-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-024-01984-7