
Abstract
Background
Waardenburg syndrome type 2 (WS2) has been reported to be a rare hereditary disorder, which is distinguished by vivid blue eyes, varying degrees of hearing impairment, and abnormal pigment deposition in the skin and hair. Variants in the sex-determining region Y-box containing gene 10 (SOXl0) gene may cause congenital deafness and have been demonstrated to be important during the development of WS2.
Methods
Complete clinical data of the proband and her family members (her parents and 2 sisters) was collected and physical examinations were performed in the hospital. The laboratory examination including hemoglobin, Coomb’s test, urine protein, ENA, autoimmune hepatitis-related autoantibodies and ultrasonography were all conducted. We obtained the peripheral blood samples from all the participants and performed whole exome sequencing and sanger sequencing validation.
Results
The present study identified a family of 5 members, and only the proband exhibited typical WS2. Beyond the characteristics of WS2, the proband also manifested absence of puberty. The proband and her younger sister manifested systemic lupus erythematosus (SLE). Whole exome sequencing revealed a de novo variant in the SOX10 gene. The variant c.175 C > T was located in exon 2 of the SOX10 gene, which is anticipated to result in early termination of protein translation.
Conclusion
The present study is the first to report a case of both WS2 and SLE, and the present findings may provide a new insight into WS2.
Similar content being viewed by others


Introduction
A rare hereditary condition called Waardenburg syndrome (WS) mainly causes following typical symptoms: accumulated pigment in the skin, eyes and hair, various degrees of sensorineural hearing impairment [1]. There are four subtypes of the typical syndrome (WS1-4) based on their additional symptoms except for the typical symptoms above. The type I of WS is characterized by dystopia canthorum. (WS1, OMIM193500). There are no additional symptoms in WS type 2 (WS2, OMIM 193,510, 600,193, 606,662, 608,890 and 611,584). The type III of WS represents dystopia canthorum and aberrant musculoskeletal function of the upper extremities (WS3, OMIM148820). The type IV of WS is featured with Hirschsprung Disease (WS4, OMIM 277,580, 613,265 and 613,266). Variable phenotype of WS can be observed in both interfamily and intrafamily, suggesting that there is an interplay between genetic modifiers and environmental factors [2].
WS is genetically heterogeneous. Several genes are involved in the pathogenetic process of WS, such as melanocyte inducing tyrosinase related protein 1, transcription factor (MITF), endothelin (EDN) 3, paired box 3 (PAX3) transcription factor, EDN receptor type B (EDNRB), Snail homolog 2 (SNAI2) and sex-determining region Y-box containing gene 10 (SOX10), accompanied with varying frequencies [3]. Approximately 15% of WS2 cases are caused by SOX10 variants [4, 5]. In 2002, two patients diagnosed with WS and SOX10 variants were reported to exhibit semicircular canals agenesis for the first time [6].
The SOX10 gene is composed of five exons, and it belongs to the SOX family of transcription factors. The typical gene was also reported to determine the cell fate and to participate in the development of cell lineage, which plays a significant role in the early developmental stages of the peripheral nervous system and neural crest. The SOX10 protein contains a DNA-binding domain named high mobility group (HMG), which is highly conserved and consists of 80 amino acids [5]. SOX10 was also reported to regulate embryonic development [7].
The present report describes a 28-year-old female with bilateral hearing damage, pigmentation abnormalities and hypogonadism. Her condition was considered to be a clinical phenotype 2 of WS with a rare SOX10 variant complicated with systemic lupus erythematosus (SLE). The purpose of the study was to report this case for its rarity and to review the relevant literature.
Case description
Complete clinical data of the patient and her family members (her parents and two sisters) were collected, and physical examinations were performed at the Tongji Hospital (1095 Jiefang Avenue, Wuhan, Hubei). We performed experiments according to the Declaration of Helsinki. Consent was obtained from each participant enrolled in our study.
A 28-year-old Chinese woman presented to our hospital for further diagnosis and therapy on June 30, 2020. She had a 6-month history of polyarticular pain, which mainly covered bilateral shoulders, wrist, knee, hip, elbow and proximal interphalangeal joints. Meanwhile, she noted with hair loss, oral ulcer, xerostomia and Raynaud’s phenomenon, but denied morning stiffness, fever, cough, expectoration or chest pain during the course of the disease. Upon physical examination, she was found to have hearing loss of the left ear, irises in blue color and premature grayed hair. Estimated pure-tone audiogram was conducted in our hospital, which indicated bilateral hearing loss (right ear > 100 dB hearing level, left ear > 30 dB hearing level) and left sensorineural hearing loss of the proband (Fig. 1). Further investigation of her medical history revealed that she had been diagnosed with primary amenorrhea and exhibited no signs of pubertal sex development. The whole medical history of her family was gathered, and physical examinations were performed on all the family members. Environmental factors were not considered as a possibility.

Multi-frequency stimuli based pure-tone audiogram demonstrated bilateral hearing loss (right ear > 100 dB hearing level, left ear > 30 dB hearing level) and left sensorineural hearing loss of the proband
Through the clinical manifestations above, the woman presented joint, skin and mucous membrane involvement. According to the findings, the patient can be inferred as connective tissue disease (CTD). But her hearing loss, blue irises, premature grayed hair and delayed sex development could not be elucidated by CTD. Then we conducted laboratory examinations for further exploration.
Laboratory examinations, including hemoglobin levels, Coombs test, protein in urine, extractable nuclear antigen(ENA), autoimmune hepatitis-related autoantibodies and ultrasonography, were all conducted in Tongji Hospital (1095 Jiefang Avenue, Wuhan, Hubei). Peripheral blood samples were obtained from all the participants. The expression of adrenocorticotropic hormone (ACTH) was below 5.0 pg/ml. The level of 17α-hydroxyprogesterone was 0.03 nmol/l and of androstenedione was 0.21 nmol/l, which were both below the lower limit of the reference interval (Table SI). Gynecology ultrasonography showed infantile uterus and endometrial cavity fluid. Hemoglobin was moderately reduced and Coombs test was positive. Urinary protein was positive and globulin was slightly elevated. ENA showed antinuclear antibodies (ANA) 1:3,200, which is a nuclear particle type. In addition, anti double-stranded deoxyribonucleic acid (anti-dsDNA), anti ribonucleoprotein (anti-RNP), anti-Smith, anti-Ro/SSA antibodies were positive. Autoimmune hepatitis-related autoantibodies, including anti-mitochondrial M2, anti-GP210 and anti-Ro-52 antibodies, were positive. Erythrocyte sedimentation rate (ESR) (77 mm/h) and IgG (32.08 g/l) were increased. Based on her clinical symptoms and laboratory examination, the patient was diagnosed with SLE. After prednisone (40 mg, qd) and mycophenolate mofetil (0.5 g, bid) standard therapy, the SLE of the patient was kept in a stable condition. Then the prednisone gradually decreased to 7.5 mg (qd) to avoid the side effects. The intervention adherence and tolerability were assessed by testing hemogram, hepatic function, renal function, urine routine once a month and all were kept well. During the course of disease, there existed no adverse and unanticipated events. Timeline of the medical diagnosis and treatment process had been shown in Fig. 2, 3. Unexpectedly, her younger sister also had a history of SLE, whereas no other abnormal clinical manifestations were found in her father, her mother and her older sister.

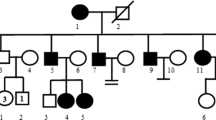
Identification of a de novo variant in the SOX10 gene [transcript ID: NM_006941.3; variant c. 175 C > T (p. Q59X)]. (A) Sanger sequencing of the family. (a) The proband was identified to have the novel variant, while her (b) father, (c) mother and (d and e) two sisters exhibited the wild-type genotype. (B) Family tree of the Waardenburg syndrome type 2 pedigree. The arrow indicates the proband. +/- represents the heterozygous SOX10-p. Q59X variant. -/- represents wild-type. (C) Affected region of the novel variant. HMG, high mobility group DNA-binding domain; D, dimerization domain; E, conserved domain of SOX10; TA, transactivation domain; SOX10, sex-determining region Y-box containing gene 10

Timeline of the medical diagnosis and treatment process
Genetic analysis
Apart from the SLE manifestation, the additional symptoms such as hearing loss of the left ear, irises in blue color and premature grayed hair can not be explained. Then the genetic analyses were performed on our patient as a part of diagnostic procedure.
Firstly, we constructed the deoxyribonucleic acid (DNA) library. The extraction and purification of the genomic DNA in this family were performed with a DNA extraction kit (Tiangen Biotech Co., Ltd.) and Tiangen Automatic Nucleic Acid Extractor (Tiangen Biotech Co., Ltd.). µThe DNA purity was detected with Nanodrop One (NanoDrop Technologies; Thermo Fisher Scientific, Inc.). The absorbance (A)260/280 and A260/230 values were 1.6-2.0. After purification, 150 ng genomic DNA was employed for enzymolysis by fragmentation enzyme. End repair and ‘A-tail’ adding were then conducted. The fragment length was 250–300 bp. Briefly, the following steps included splice ligation, pre-PCR and library purification, library quantification and pooling, concentration, hybrid capture, enrichment and recovery of the target area, post-PCR amplification and purification, library quantification and quality assessment. XGen™ Exome Research Panel v1 (Integrated DNA Technologies, Inc.), HiFi Ready Mix Enzyme (Kapa Biosystems; Roche Diagnostics) and other library construction reagents (Vazyme Biotech Co., Ltd.) were used. The final concentration of the library was evaluated by Qubit 4.0 (Thermo Fisher Scientific, Inc.) and Qubit™ dsDNA HS Assay Kit (concentration > 10 ng/µl). Library fragment length (300–550 bp) was detected using QSeq400 Fragment Analyzer (BIOptic Inc.).
Secondly, the synthetic DNA library was used for whole exome sequencing. µµµWhole exome sequencing was performed using the NovaSeq 6000 platform, NovaSeq 6000 S4 Reagent Kit (Illumina, Inc.) and S4 flowcell with PE150 (read length 150 bp) mode. Point variants and small fragment deletion/insertion variants within 20 bp were detected. The raw BCL data were converted to FASTQ format, and then annotated and analyzed.
Thirdly, we conducted the bioinformatics analysis with the raw data. All data were filtered by fastp software (HaploX Inc.), compared with the hg19/GRCh37 human reference genome by using the bwa software, and corrected by public dbsnp159 database. Only on-target reads were used to call variants using gatk 3.8. Sequencing depth was detected by samtools depth. Statistics of bed file region coverage and 20X sequencing depth was conducted by gatk depth of coverage. Mosdepth 0.2.5 software was used for GC content statistics. The reported mutations were detected using the Clinical Significance for Variants Relative to Phenotypes (ClinVar) (https://www.ncbi.nlm.nih.gov/clinvar/) and human gene mutation database (HGMD) (http://www.hgmd.cf.ac.uk/ac/search.php) database. The data that covered < 20-fold and variants whose minimum allele frequency (MAF) was > 0.01% in the Exome Aggregation Consortium database were removed. Next, the noncoding and synonymous variants were removed. Furthermore, the potential pathogenic missense variants were identified by the Polyphen-2 (score > 0.85) (http://genetics.bwh.harvard.edu/pph2/) and sorts intolerant from tolerant (SIFT) (score < 0.05) (http://sift.jcvi.org). Point variants were annotated using ANNOVAR software, and further annotation was analyzed with local databases such as HGMD, human phenotype ontology (HPO) and internal carryover rate. The pathogenic variant was referred to the American College of Medical Genetics and Genomics (ACMG) grading criteria [4].
The whole exome sequencing above was performed for screening possible variants of genes associated with metabolic, endocrine and immune diseases. A total of 1,397 known or novel variations were detected by semiconductor sequencing (Table SII). A heterozygous nonsense variant of SOX10 (Fig. 2), c. 175 C > T (p. Q59X), was predicted to be pathogenic according to the ACMG grading criteria. The SOX10 variant has an effect on hearing and pigmentation [5], which was considered to be the cause of WS2 in the patient. The diagnosis of WS2 was made on the basis of the clinical manifestations above and the international criteria raised by the Waardenburg Consortium [7]. No other variants related to WS were identified in the patient.
Furthermore, the sanger sequencing validation of the SOX10 gene was then performed with all the relatives of the patient. After enzymatic lysis, the PCR products were labeled with fluorescence signals via the terminal dideoxygenation method for capillary electrophoresis separation [6]. The forward primer of the SOX10 gene is 5’-GTTGGACTCTTTGCGAGGAC-3’ and the reverse primer is 5’-TGACGTGCGGCTTGCTTTTG-3’. PCR amplifications were performed with an ABI 9700 (Thermo Fisher Scientific, Inc.) as follows: denaturation of 96˚C for 3 min, followed by 30 cycles of denaturation at 96˚C for 15 s, annealing at 56˚C for 15 s and extension at 60˚C for 1 min, and a final extension of 72˚C for 5 min. The purified PCR products with a size of a 391 bp were sequenced by Sanger sequencing. The result showed that SOX10 variant in the patient does not exist in her family members and it was considered to be a de novo variant. In the meantime, no other SOX10 variants were identified in other family members.
Finally, the proband was diagnosed with WS2 containing a rare SOX10 heterozygous variant and she suffered from SLE simultaneously.
Discussion
The present study reports a Chinese woman diagnosed with WS2, who had a c. 175 C > T (p. Q59X) heterozygous variant in exon 2 of the SOX10 gene. The patient manifested auditory malfunction of both ears, blue-colored irises, premature graying of the hair which are characteristics of WS2. In addition, the patient was absent of pubertal sex development, and exhibited primary amenorrhea, low gonadal hormone, infantile uterus and breast dysplasia. The additional manifestation may be explained by the fact that distinct categories of variants in the SOX10 gene may display various clinical features by affecting different signaling pathways in WS. It has been reported that SOX10 gene variants may result in pubertal delay [8, 9]. One case was identified as co-existence of WS2 and Kallmann syndrome associated with SOX10 gene variants [10]. Meanwhile, in the present study, no other gene variants were associated with the absence of pubertal sex development.
The ACTH level of the patient was under the lower limit of detection. ACTH has been reported to be important in the regulation of melanocytes [11]. ACTH plays a crucial role in melanocyte differentiation and melanogenesis regulation through the cAMP signaling pathway. It has been suggested that melanogenesis upregulation is associated with cAMP [12]. In the present study, SOX10 gene variant and low level of ACTH were detected. Thus, it was speculated that SOX10 gene variants may cause melanocyte dysfunction through downregulating the expression of ACTH. However, further research on the pathogenesis of WS is required in the future.
Notably, the patient also presented SLE characteristics, including polyarticular pain, hair loss, oral ulcer, xerostomia and Raynaud’s phenomenon. To date, no cases diagnosed with WS and SLE have been reported. Besides, the family may have a genetic predisposition to SLE. But no studies have been reported the association between the predisposition to SLE and SOX10 gene. The coexistence of WS and SLE may be a coincidence, and it was also speculated that other gene variants may exist in the proband and one of her sisters. As previously reported, there are various genes linked to monogenic lupus [13, 14]. Through searching these genes in the proband, gene variants of interferon stimulated gene 15 (ISG15), ribonuclease H2, subunit B (RNASEH2B) and SOS Ras/Rac guanine nucleotide exchange factor 1 (SOS1) were found in the proband, and the dysfunction of these genes may be related to SLE pathogenesis in the proband. Thus, it was speculated that, in a patient with WS, this may combine with any genetic or non-hereditary disease.
The c. 175 C > T (p. Q59X) variant was detected as a heterozygous variant located in the second exon of SOX10. After analyzing her family members, only the proband contained the variant, indicating that it was a de novo pathogenic variant. The variant of this type was evaluated as pathogenic, and this new variant was absent in HGMD and Clinvar databases. No other WS associated gene (such as MITF, PAX3 or SNAI2) variants were identified. The current study is the first to report the c. 175 C > T (p. Q59X) variant in the SOX10 gene. The truncating heterozygous variant of the SOX10 gene in the present study is a de novo pathogenic variant which caused WS2 associated manifestation in a Chinese woman. Previous studies have shown that ∼ 15% of WS2 pathogenesis is related to variants in the SOX10 gene, and several SOX10 gene heterozygous variants have been reported in WS2 in Chinese family [15,16,17,18]. The present findings expand the knowledge of SOX10 gene variants in WS2.
The SOX10 gene is situated on chromosome 22q 13.1 and contains five exons. Among the five exons, only exons 3, 4 and 5 encode protein. The SOX10 protein consists of 466 amino acids. It has a size of 51 kDa and belongs to the SOX gene family [19]. SOX10 contains a central DNA-binding domain named high mobility group (HMG) domain, which is a highly conserved and active domain, and a C-terminal transactivation domain [20]. The HMG domain can specifically identify and bind to the promoters of the target genes, which allows SOX10 to act as a key transcription factor during the process of migration and differentiation of neural crest cell, including melanocytes, olfactory ensheathing cells, intestinal ganglion cells, and numerous sensory and autonomic ganglion cells [21,22,23]. The transactivation domain of SOX10 activates the transcription of the MITF, tyrosinase and EDNRB genes [24]. The target genes of SOX10 (including MITF, tyrosinase and EDNRB) participate in melanin synthesis [21, 22]. SOXl0 plays a crucial role in the pathogenesis of WS, and SOXl0 variants may lead to WS2 and WS4, which can result in hearing impairment and pigmentary abnormalities of the hair, iris and skin [15]. In addition, variants in SOX10 may cause other neural crest-related diseases, including central demyelinating leukodystrophy, peripheral demyelinating neuropathy and Hirschsprung disease [25, 26].
The present study identified a de novo variant in the SOX10 gene, which provides a new perspective into the mechanism of WS and contributes to updating the HGMD and Clinvar databases. Moreover, the current study is the first to report a case of WS2 complicated with SLE, and the present findings may lay a vital foundation for non-invasive prenatal genetic diagnosis and genetic counseling. Furthermore, the co-occurrence of WS2 and SLE in the current case broadens the recognition that a patient with WS may combine any genetic or non-hereditary disease, which may contribute to the field of disease diagnosis in the future. The association between WS2 and SLE should be further explored. In addition, targeted gene therapy should be emphasized for SOX10 and other genes in patients with WS2. Moreover, it is necessary to pay particular attention to the sexual development of patients with WS2 with SOX10 variants during adolescence.
Data availability
All data generated or analyzed during the present study are included in this published article.
References
Kiani R, Gangadharan SK, Miller H. Case Report: Association of Waardenburg Syndrome with Intellectual disability, autistic spectrum disorder and unprovoked aggressive outbursts: a new behavioural phenotype? Br J Dev Disabil. 2013;53:53–62.
Sun L, Li X, Shi J, et al. Molecular etiology and genotype-phenotype correlation of Chinese Han deaf patients with type I and type II Waardenburg Syndrome. Sci Rep. 2016;6:35498.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Medicine: Official J Am Coll Med Genet. 2015;17:405–24.
Pingault V, Ente D, Dastot-Le Moal F, Goossens M, Marlin S, Bondurand N. Review and update of mutations causing Waardenburg syndrome. Hum Mutat. 2010;31:391–406.
Haldin CE, LaBonne C. SoxE factors as multifunctional neural crest regulatory factors. Int J Biochem Cell Biol. 2010;42:441–4.
Nai YH, Powell SM, Breadmore MC. Capillary electrophoretic system of ribonucleic acid molecules. J Chromatogr A. 2012;1267:2–9.
Liu XZ, Newton VE, Read AP. Waardenburg syndrome type II: phenotypic findings and diagnostic criteria. Am J Med Genet. 1995;55:95–100.
Wang SQ, Chen Y, Luo KH, et al. Diagnosis and genetic analysis of a case of Waardenburg syndrome type 2 with hypogonadotropic hypogonadism caused by SOX10 gene deletion. Yi Chuan = Hereditas. 2022;44:1158–66.
Sreenivasan R, Gonen N, Sinclair A. SOX genes and their role in disorders of Sex Development. Sex Development: Genet Mol Biology Evol Endocrinol Embryol Pathol sex Determ Differ. 2022;16:80–91.
Suzuki E, Izumi Y, Chiba Y, et al. Loss-of-function SOX10 mutation in a patient with Kallmann Syndrome, hearing loss, and Iris Hypopigmentation. Hormone Res Paediatrics. 2015;84:212–6.
Busca R, Ballotti R. Cyclic AMP a key messenger in the regulation of skin pigmentation. Pigment Cell Res. 2000;13:60–9.
Buscà R, Ballotti R. Cyclic AMP a key messenger in the regulation of skin pigmentation. Pigment Cell Res. 2000;13:60–9.
Alperin JM, Ortiz-Fernández L, Sawalha AH. Monogenic lupus: a developing paradigm of Disease. Front Immunol. 2018;9:2496.
Kwon YC, Chun S, Kim K, Mak A. Update on the Genetics of systemic Lupus Erythematosus: genome-wide Association studies and Beyond. Cells 8: 2019.
Bondurand N, Dastot-Le Moal F, Stanchina L, et al. Deletions at the SOX10 gene locus cause Waardenburg syndrome types 2 and 4. Am J Hum Genet. 2007;81:1169–85.
Wang Y, Chai Y, Zhang P, Zang W. A novel variant of the SOX10 gene associated with Waardenburg syndrome type IV. BMC Med Genom. 2023;16:147.
Guo M, Li Q, Jiang C, Li S, Ruan B. A De Novo Mutation in SOX10 in a Chinese boy with Waardenburg Syndrome Type 2. J Int Adv Otology. 2023;19:255–9.
Bogdanova-Mihaylova P, Alexander MD, Murphy RPJ, Murphy SM. Waardenburg syndrome: a rare cause of inherited neuropathy due to SOX10 mutation. J Peripheral Nerv System: JPNS. 2017;22:219–23.
Harris ML, Baxter LL, Loftus SK, Pavan WJ. Sox proteins in melanocyte development and melanoma. Pigment Cell Melanoma Res. 2010;23:496–513.
Chan KK, Wong CK, Lui VC, Tam PK, Sham MH. Analysis of SOX10 mutations identified in Waardenburg-Hirschsprung patients: Differential effects on target gene regulation. J Cell Biochem. 2003;90:573–85.
Murisier F, Guichard S, Beermann F. The tyrosinase enhancer is activated by Sox10 and Mitf in mouse melanocytes. Pigment Cell Res. 2007;20:173–84.
Hou L, Arnheiter H, Pavan WJ. Interspecies difference in the regulation of melanocyte development by SOX10 and MITF. Proc Natl Acad Sci U S A. 2006;103:9081–5.
Barraud P, St John JA, Stolt CC, Wegner M, Baker CV. Olfactory ensheathing glia are required for embryonic olfactory axon targeting and the migration of gonadotropin-releasing hormone neurons. Biol Open. 2013;2:750–9.
Kuhlbrodt K, Schmidt C, Sock E, et al. Functional analysis of Sox10 mutations found in human Waardenburg-Hirschsprung patients. J Biol Chem. 1998;273:23033–8.
Inoue K, Khajavi M, Ohyama T, et al. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat Genet. 2004;36:361–9.
Pingault V, Guiochon-Mantel A, Bondurand N, et al. Peripheral neuropathy with hypomyelination, chronic intestinal pseudo-obstruction and deafness: a developmental neural crest syndrome related to a SOX10 mutation. Ann Neurol. 2000;48:671–6.
Acknowledgements
We thank all the participants for consenting to participate in this study.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
GS, ZL and SL participated in the conception and supervision of the project. GS, LD and YL were involved in the design of the study. YL and YC conducted the experiments. YL and YC saw and verified all the raw data. YL and YS analyzed the results. YL drafted the manuscript. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Informed consent from all subjects and their legal guardians for study participation was obtained. The study was approved by the ethics committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology.
Consent for publication
Informed consent from all subjects and their legal guardians for publication of identifying information/images in an online open-access publication was obtained.
Conflict of interest
We declare that we have no financial and personal relationships with other people or organizations. There is no professional or other personal interest of any product, service or company.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, Y., Chen, Y., Sun, Y. et al. Waardenburg syndrome type 2 with a de novo variant of the SOX10 gene: a case report. BMC Med Genomics 17, 104 (2024). https://doi.org/10.1186/s12920-024-01877-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-024-01877-9