
Abstract
Background
Liver fibrosis is a major global healths problem; nevertheless, its molecular mechanism are not completely clear. This study aimed to build a lncRNA-miRNA-mRNA network, identify potentially related lncRNAs, and explore the pathogenesis of liver fibrosis.
Materials and methods
We used the Gene Expression Omnibus databases and bioinformatics analysis to identify differentially expressed genes (DEGs) between liver fibrosis and normal tissues. The ceRNA network was constructed according to the interactions between DElncRNA, miRNA, and DEmRNA. Then, these DEGs were identified using functional enrichment analysis, and a protein–protein interaction (PPI) network was established. The critical lncRNAs were verified using the quantitative real-time polymerase chain reaction (qRT-PCR).
Results
The ceRNA network was composed of three lncRNAs, five miRNAs, and 93 mRNAs. Gene Ontology functional enrichment analysis revealed significant enhancement in cell components, molecular function, and biological process. Kyoto Encyclopedia of Genes and Genomes pathway analysis revealed pathways associated with transcriptional misregulation in cancer, including the Rap1 signaling pathway, proteoglycans in cancer, mineral absorption, HTLV-l infection, and central carbon metabolism in cancer. According to the PPI network and the GSE84044 database, seven hub genes associated with liver fibrosis were identified. In addition, qRT-PCR revealed that lncRNA AC100861 (lncRNA TNFRSF10A-DT) was explicitly decreased in liver fibrosis tissues and activated hepatic stellate cells.
Conclusions
In summary, this study preliminarily found that lncRNA TNFRSF10A-DT may be a biomarker for the diagnosis and outcome of liver fibrosis. We uncovered a novel lncRNA-mediated ceRNA regulatory mechanism in the pathogenesis of liver fibrosis.
Similar content being viewed by others


Introduction
Liver fibrosis is a common event that triggers wound-healing in the context of chronic injury. Fibrosis may progress to cirrhosis and primary liver cancer [1]. Studies reported that liver fibrosis is associated with the deposition of extracellular matrix (ECM) proteins [2]. The activation of hepatic stellate cells (HSCs) is thought to be the primary factor causing liver fibrosis [3,4,5,6]. Liver fibrosis can be detected early and reversed by treatments [7]. In the past few decades, investigators have focused substantial attention on the molecular mechanisms of liver fibrosis [8, 9]. Nevertheless, precise pathogenesis requires further exploration [10]. Definitive treatment of liver fibrosis requires identifying novel therapeutic strategies that include novel biomarkers and targets.
Long non-coding RNAs (lncRNAs) are regulated by several mechanisms, including chromatin regulation, epigenetic modification promoter activity regulation, and post-transcriptional mechanisms [11,12,13,14]. Studies indicated that lncRNA participates in the formation or inhibition of liver fibrosis [15,16,17]. LncRNAs regulate gene expression in acis or trans manner [18, 19]. Exploring the mechanisms of interactions between lncRNA and miRNA can generate novel strategies for liver fibrosis treatment [20].
The number of lncRNA families is vast, and it remains unknown whether there are other lncRNAs involved in liver fibrosis. Therefore, in this study, the expression of lncRNAs and mRNAs in liver fibrosis were identified using the Gene Expression Omnibus (GEO) database, and a co-expression network was constructed to elucidate the regulation and targets of lncRNAs. Then, we used Gene Ontology (GO) terminology and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis to identify the functions of the lncRNAs associated with liver fibrosis. Finally, we used quantitative real-time polymerase chain reaction (qRT-PCR) to confirm the results.
Material and methods
Data collection
This study intended to preliminarily explore the regulatory mechanisms of ceRNA during liver fibrosis progression by analyzing the gene expression differences and co-expression analysis between the healthy group and a liver fibrosis group. GEO, a public international and functional genomics data repository, is used for high-throughput microarray and next-generation sequences [21]. Therefore, GSE12392 and GSE84044 were mined for bioinformatics analysis by searching the keywords "liver fibrosis" and "Homo sapiens" in the GEO database. In the dataset, lncRNA and mRNA expression in six healthy individuals and six patients were measured using the Agilent MicroArray V4 platform.
DEGs analysis
The DEGs were located using edgeR [22] in the R Bioconductor package. The expression data in GSE12392 were analyzed using the LIMMA package of the R language, and the probe ID was converted into GeneSymbol according to the CORRESPONDING GPL file of the probe. The mRNA or lncRNA were selected from the probe for differential expression and gene co-expression network analysis. The criteria for selecting DEGs were P < 0.05 and log2 Fold-Change less than -1 or greater than 1.
WGCNA analysis
Gene co-expression network analysis was used to analyze gene expression in the two groups at the mRNA and lncRNA levels in combination with basic clinical information of the samples (i.e., age, gender, and whether liver fibrosis occurred) aiming to explore gene co-expression modules related to disease occurrence.
LncRNAs target gene prediction
The results of co-expression analysis and differential expression analysis were combined to obtain common lncRNAs, that is, to identify DEGs and co-expressed genes associated with clinicopathological features. StarBase and bioinformatics were used to predict the target genes of lncRNAs.
MiRNA target gene prediction
StarBase and bioinformatics methods were used to predict the target genes of human miRNAs, and the default parameters of the official database were adopted.
Establishment of the ceRNA regulatory network
An lncRNA-miRNA-mRNA network was constructed according to ceRNA theory [23]. Based on the prediction results of these target genes, a ceRNA regulatory network of lncRNA-miRNA-mRNA was constructed by combining mRNA co-expression network analysis (i.e., genes co-expressed between mRNA level and liver fibrosis) and mRNA differential expression analysis (i.e., genes with differential expression). We used Cytoscape 3.7.2 [24] to visualize the lncRNA-miRNA-mRNA network, and the CytoHubba plugin in Cytoscape 3.7.2 [25] was used to calculate all node degrees.
GO and pathway enrichment analysis
GO analysis [26] included biological process, cellular component, and molecular function, generated using the bioinformatics tool DAVID [27] (P < 0.05). This analysis was used to examine the unique biological significance of the high-throughput transcriptome. KEGG enrichment analysis was used to predict the biological pathways [28]. The results were visualized using the R language.
Analysis of the PPI network
The PPI information for the common DEG network was evaluated using the search tool STRING [29]. Any potential correlations between these DEGs were analyzed using Cytoscape [30]. The PPI network modules were recognized using the Cytoscape plugin MCODE, and only those were presented based on a node degree ≥ 3.
Hub gene selection
We analyzed the PPI network using the Cyto-Hubba plugin of Cytoscape 3.7.2 and selected candidate hub genes with top node degrees. Subsequently, hub genes associated with liver fibrosis were identified using GSE84044.
Human samples
From the Xiangya Hospital of Central South University, ten healthy subjects and 16 patients with liver fibrosis were recruited. Informed consent was obtained, and ethical approval was obtained from the Ethics Committee of Xiangya Hospital. Human liver tissues were immediately frozen in liquid nitrogen and stored at − 80 °C.
Cell culture experiments
LX-2 cells were obtained from our laboratory and cultured in DMEM containing 1% fetal bovine serum and 1% penicillin/streptomycin. LX-2 cells were passaged using trypsin. LX-2 cells were stimulated by TGF-β1 (0, 5, 10, 20, 40 ng/ml; Sigma, Cat No. SAB4502958) for 48 h. All cells were stored in an incubator at 37 °C in 5% CO2.
QRT-PCR
Total RNA was isolated from liver fibrosis human samples and LX-2 cell lines using TRIzol reagent (Invitrogen, CA, USA). The cDNAs were synthesized using a commercial kit (Bio-Rad, Hercules, CA). Gene expression was measured using the ABI 7900HT Fast Real-Time PCR System. Relative mRNA expression levels were calculated using the 2−ΔΔCT method. The following primer sequences were used: lncRNA PCBP1-AS1, forward, 5'-ACTACTCAGTCAATTGCTCCA-3', reverse, 5'-ATTTCCTTACTGACCTGCAT-3'; lncRNA AC100861, forward, 5'-GCAC ATGACACGGGATGAGA-3', reverse, 5'-GGCTTTCGGGAGGCTGATT A-3'; lncRNA TEX41, forward, 5'-TGGCCAAGAGACAACACCAA-3', reverse, 5'-GGCAGAGTGAGTCCAAAGG-3'; GAPDH, forward, 5'-TGGAAATCCCATCACCATCT-3', reverse, 5'-TGGACTCCACGACGTACTCA-3'.
Statistical analysis
Data obtained from three independent experiments were expressed as mean ± standard deviation. The t-test was performed to compare differences between two groups, and one-way analysis of variance was used for multi-group comparisons. Differences with p < 0.05 were considered statistically significant in multiple testing.
Results
Identification of differentially expressed genes (DEGs)
The differential expressions of mRNA and lncRNA in the GSE123932 data set were analyzed using the R language's LIMMA package. The standard of differential gene screening was P < 0.05, log2 fold change > 1 or < -1. There were 210 cases with significantly differentially expressed lncRNAs (Figs. 1A and 2A); there were 333 cases of significantly differentially expressed mRNAs (Figs. 1B and 2B).

Volcano map demonstrating the differential expression analysis of lncRNAs (A) and mRNAs (B). Red and blue dots were significantly high and low expression in the disease group, respectively

Heat map illustrating differential expression analysis of lncRNAs (A) and mRNAs (B)
Weighted gene co-expression network analysis(WGNA)
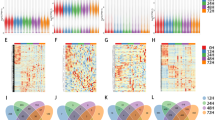
mRNA. None of the 12 samples showed abnormal gene expression (Fig. 3A). Cluster analysis of gene expression profiles was performed by combining clinical information of samples (Fig. 3B). The experimental results converged when the Soft threshold (SFT) value was 8 (Fig. 3C, D). Based on the WGCNA documentation, the SFT value was set to 8 to construct the co-expression network, and 11 modules were obtained (Fig. 3E, F). Genes in brown, yellow, and turquoise modules are related to disease occurrence (P < 0.05, Fig. 3G).

Sample cluster analysis (A and B); Calculation and selection of soft threshold (C and D); Cluster analysis of modules (E and F); Correlation analysis between modules and traits (G).
A total of 246 DEGs were obtained from disease-related modules by exporting the genes from these three modules and combining them with differential expression analysis (Data collection Section) (Fig. 4).

Venn diagram of joint analysis
LncRNA
None of the 12 samples showed abnormal gene expression (Fig. 5A). Cluster analysis of gene expression profiles was performed by combining clinical information of samples (Fig. 5B). The experimental results converged when the SFT value was 8 (Figs. 5C, D). According to the WGCNA documentation, the SFT value was set to 7 to construct the co-expression network, and eight modules were obtained (Figs. 5E, F). Genes under the green module were significantly correlated with disease occurrence (P < 0.05, Fig. 5G).

Sample cluster analysis (A and B); Calculation and selection of soft threshold (C and D); Cluster analysis of modules (E and F); Correlation analysis between modules and traits (G)
The co-expressed genes in the green module were exported and combined with the differential expression analysis results of lncRNA (Data collection Section), which obtained 57 DEGs (Fig. 6).

Venn diagram of joint analysis
Competing endogenous RNA (ceRNA) network construction
LncRNA target gene prediction
StarBase was used to predict 57 lncRNAs target genes to obtain the predicted results of nine lncRNA (Table 1).
Prediction of miRNA target genes
StarBase was used to predict lncRNA target genes-miRNAs (Table 2).
Establishment of the ceRNA regulatory network
Combined with the prediction results and difference analysis results, lncRNA-miRNA and miRNA-mRNA were identified, and Cytoscape software was used for ceRNA-network visualization (Fig. 7).

CeRNA network (hexagon: lncRNAs; inverted triangle: miRNAs; circle: mRNA)
GO and KEGG pathway analyses
GO and KEGG pathway analyses were performed on mRNA in the ceRNA network using DAVID, and the results were visualized using the R language (Fig. 8).

GO and KEGG enrichment analysis: (A) Cell component; (B) Molecular function; (C) biological process; and (D) KEGG pathway
PPI analysis
PPI analysis was performed for genes in the lncRNA-miRNA-mRNA regulatory network using STRING. The analysis results were visualized using Cytoscape (Fig. 9).

PPI (blue dots: low expression; red dots: high expression)
Hub gene validation
Using PPI network analysis of genes in the lncRNA-miRNA-mRNA regulatory network, the following hub genes were obtained (Table 3). The gene expression data in dataset GSE84044 were used to verify the expression of hub genes (genes with degree > 20 in the PPI network).
The expressions of hub genes in the eight cases were verified using GSE840044. The expression differences of genes (CCNB1, TOP2A, KIF20A, KIF4A, KIF14, MYCN, CDCA7) were found (except MET) (p < 0.05) (Fig. 10).

The expression of hub genes: A. CCNB1; B. TOP2A; C. KIF20A; D. KIF4A; E. KIF14; F. MYCN; G. CDCA7; H. MET
The downregulation of lncRNA AC100861
To verify the critical lncRNAs involved in liver fibrosis, qRT-PCR was performed in fibrotic and healthy livers. LncRNA AC100861 mRNA expression was much lower in samples from liver fibrosis patients than healthy liver tissues (Fig. 11).

Downregulation of lncRNA AC100861 in liver fibrosis. All data are expressed as mean ± standard deviation (SD).The experiment was repeated three times. * p < 0.05, ** p < 0.01
The downregulation of lncRNA AC100861 in activated HSCs
Because activated HSCs (LX-2) are thought to be the significant cellular participants in accelerating the deposition of ECM proteins, we determined whether lncRNA AC100861 participated in HSC activation. QRT-PCR analysis showed that compared with quiescent HSCs, lncRNA AC100861 expression was lower in activated HSCs treated with various concentrations of TGF-β1 (Fig. 12).

Downregulation of lncRNA AC100861 in activated HSCs. All data are expressed as mean ± standard deviation (SD).The experiment was repeated three times. * p < 0.05, ** p < 0.01
Discussion
Liver fibrosis is considered a severe health problem worldwide. Liver fibrosis is an intense and reversible wound-healing process resulting from tissue necrosis [31, 32]. The resting HSCs transdifferentiate into myofibroblasts responsible for the deposition of ECM proteins (or collagen fibers), causing tissue scarring [7, 33]. Therefore, it is of great significance to study biomarkers for the diagnosis of liver fibrosis.
LncRNA, a small RNA molecule with 200 nucleotides, is closely involved in critical processes of cell development, proliferation, differentiation, and pluripotency [34]. With the improvement of deep transcriptome sequencing technology, the study of RNA molecules has significantly increased [35]. Although lncRNAs without protein-coding ability and bio-function, previous studies have provided ample evidence that these RNA molecules play a crucial function in controlling the expression of a gene by a series of mechanisms (such as targeted transcription) [36], affecting splicing function [37], targeting cis-acting promoter RNAs [38]. In recent decades, lncRNAs are involved in developing liver fibrosis [39,40,41,42].
LncRNAs act by regulating the binding of micro RNAs to targeted mRNA. The mechanism of co-expression and interaction between mRNA miRNAs and lncRNAs in disease regulation requires clarification. In this study, using the gene expression data from the GEO database and R language LIMMA package, we conducted bioinformatics mining on GSE12392 and GSE84044 datasets and analyzed the significantly expressed lncRNA and mRNA in tissues of patients with liver fibrosis. Then, 57 lncRNAs and 246 mRNAs were changed in liver fibrosis compared to the control group. Finally, we constructed the lncRNA-miRNA-mRNA network that included three lncRNAs, five miRNAs, and 93 mRNAs.
To further understand the effect of lncRNA activity on function, the possible biological mechanisms of lncRNA in liver fibrosis tissues were clarified through GO and KEGG. The analysis of GO functional enrichment were mainly manifested in cytosol, cytoplasm and other cellular components, protein binding, protein dimer activity, protein kinase binding and other molecular functions, as well as positive transcriptional regulation of DNA templatization and significant enhancement of extracellular matrix tissues. These results suggested that liver fibrosis-related enrichment of GO is mainly related to cell growth and tissue hyperplasia. KEGG pathway analysis revealed that this pathway is generally enriched in tumor transcriptional dysregulation, Rap1 signaling, tumor proteoglycan, mineral absorption, HTLVL infection and central carbon metabolism.
We found eight candidate hub genes using PPI analysis of genes in the lncRNA-miRNA-mRNA regulatory network using STRING. The expression of the eight hub genes was verified using GSE84044, and seven hub genes (CCNB1, TOP2A, KIF20A, KIF4A, KIF14, MYCN, CDCA7) (p < 0.05) showed significant differences between the groups (except MET). A study showed that CCNB1 and KIF20A are related to the activation of HSCs [43].
LncRNAs with more edges are hubs that participate in more ceRNA interactions, suggesting that lncRNAs participate in network organization. According to node degree and relationship, we selected the critical lncRNAs (TEX41, PCBP1-AS1, and AC100861). These lncRNAs can be used as biomarkers for the diagnosis of liver fibrosis. To speculate about the potential functions of lncRNAs, the function of annotated miRNAs or mRNAs can be studied based on the ceRNA theory. LncRNA-miRNA-mRNA can offer a holistic view of the ceRNA interactions, which permit the study of the regulatory properties of lncRNAs; however, the subnetwork of the key lncRNA-miRNA-mRNA detailed how key lncRNAs work in conjunction with competing mRNAs [44]. A growing body of evidence suggests that lncRNAs and miRNAs ‘chat’ with one another in ceRNA processes [45]. In the present study case, lncRNAs may act as sponges to isolate miRNAs from their targeted mRNAs, causing changes in the expression of their target genes [46]. The CeRNA network showed that DE lncRNAs in liver fibrosis bound to miR-103a-3p, miR-525-5p, miR-103a-3p, miR-107, and miR-873-5p, can effectively regulate a large of target genes. Of the three lncRNAs, some may competitively bind to these miRNAs through the ceRNA affect and regulate the expression of hub genes (ACCNB1, TOP2A, kif20a, dkif4a, kif14, MYCN, and CDH2).
QRT-PCR results indicated no significant differences in expression of lncRNA TEX41 or PCBP1-AS1 between liver fibrosis tissue control liver tissue; however, the mRNA level of lncRNA AC100861 (lncRNA TNFRSF10A-DT) was significantly decreased. At present, there are no reports on the correlation between the lncRNA TNFRSF10A-DT and liver fibrosis. The mRNA expression of lncRNA TNFRSF10A-DT in activated HSCs was significantly lower than in resting HSCs. Our study demonstrated that the lncRNA TNFRSF10A-DT expression is correlated with liver fibrosis and may be helpful for diagnosis and outcome prediction.
We used StarBase to predict the interactions between lncRNA and miRNA and found that the predicted target miRNA of lncRNA TNFRSF10A-DT was miR-873–5. Next, we used StarBase to obtain miRNA-targeted mRNAs and found that the predicted target genes of miR-873–5 were RCC2, UBR4 and AKR7A2. Based on the ceRNA network, lncRNA TNFRSF10A-DT/has-mir-873–5/RCC2, UBR4, and the AKR7A2 axis may construct a ceRNA network to participate in the progression of liver fibrosis.
In summary, we constructed a liver fibrosis-related lncRNA-miRNA-mRNA network to study the biological functions of lncRNA in liver fibrosis development according to ceRNA theory. LncRNA TNFRSF10A-DT was the critical lncRNA in the ceRNA network. QRT-PCR indicated that the mRNA level of lncRNA TNFRSF10A-DT in liver fibrosis patients and the expression level of activated HSCs were much lower than in healthy people and resting HSCs. These findings suggest that lncRNA TNFRSF10A-DT may serve as a biomarker and therapeutic target for liver fibrosis. We speculate that lncRNA TNFRSF10A-DT may compete with miR-873–5 to regulate the target mRNA genes (RCC2, UBR2, and AKR7A2) in the ceRNA network. The ceRNA network is helpful to improve understanding of the pathogenesis of liver fibrosis. However, the specific ceRNA mechanism of lncRNA TNFRSF10A-DT requires further research.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available in the Gene Expression Omnibus (GEO) datasets (accession no. GSE12392 and GSE84044; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE12392; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE84044).
Abbreviations
- DEGs:
-
Diferentially expressed genes
- GEO:
-
Gene Expression Omnibus
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- PPI:
-
Protein–protein interaction
- CC:
-
Cellular component
- MF:
-
Molecular function
- BP:
-
Biological process
- STRING:
-
Search Tool for Retrieval of interacting Genes
- WGCNA:
-
Weighted Gene Co-expression Network Analysis
- qRT-PCR:
-
Quantitative Real-Time PCR
References
Weiskirchen R, Tacke F. Liver fibrosis: from pathogenesis to novel therapies. Dig Dis. 2016;34(4):410–22.
Friedman SL. Hepatic fibrosis-overview. Toxicology. 2008;254(3):120–9.
Rezvani M, Español-Suñer R, Malato Y, Dumont L, Grimm AA, Kienle E, Bindman JG, Wiedtke E, Hsu BY, Naqvi SJ, et al. In vivo hepatic reprogramming of myofibroblasts with AAV vectors as a therapeutic strategy for liver fibrosis. Cell Stem Cell. 2016;18(6):809–16.
Ray K. HAstening the development of liver fibrosis. Nat Rev Gastroenterol Hepatol. 2019;16(8):455.
Zhang F, Kong D, Lu Y, Zheng S. Peroxisome proliferator-activated receptor-gamma as a therapeutic target for hepatic fibrosis: from bench to bedside. Cell Mol Life Sci. 2013;70(2):259–76.
Wang XQ, Gao YZ, Li Y, Huang YQ, Zhu YW, Lv W, Wang RZ, Gou LS, Cheng C, Feng ZJ, et al. Roseotoxin B alleviates cholestatic liver fibrosis through inhibiting PDGF-B/PDGFR-beta pathway in hepatic stellate cells. Cell Death Dis. 2020;11(6):458.
Bataller R, Brenner DA. Liver fibrosis. J Clin Investig. 2005;115(2):209–18.
Panebianco C, Oben JA, Vinciguerra M, Pazienza V. Senescence in hepatic stellate cells as a mechanism of liver fibrosis reversal: A putative synergy between retinoic acid and PPAR-gamma signalings. Clin Exp Med. 2017;17(3):269–80.
Tomita K, Teratani T, Suzuki T, Shimizu M, Sato H, Narimatsu K, Okada Y, Kurihara C, Irie R, Yokoyama H, et al. Free cholesterol accumulation in hepatic stellate cells: Mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology. 2014;59(1):154–69.
He Y, Jin L, Wang J, Yan Z, Chen T, Zhao Y. Mechanisms of fibrosis in acute liver failure. Liver Int. 2015;35(7):1877–85.
Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–66.
Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152:1298–307.
Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17(1):47–62.
Tao Z, Yuan Y, Liao Q. Alleviation of lipopolysaccharides induced acute lung injury by MiR-454. Cell Physiol Biochem. 2016;38(1):65–74.
Xiao Y, Liu R, Li X, Gurley EC, Hylemon PB, Lu Y, Zhou H, Cai W. Long noncoding RNA H19 contributes to cholangiocyte proliferation and cholestatic liver fibrosis in biliary atresia. Hepatology. 2019;70(5):1658–73.
Peng H, Wan LY, Liang JJ, Zhang YQ, Ai WB, Wu JF. The roles of lncRNA in hepatic fibrosis. Cell Biosci. 2018;8:63.
Chen MJ, Wang XG, Sun ZX, Liu XC. Diagnostic value of LncRNA-MEG3 as a serum biomarker in patients with hepatitis B complicated with liver fibrosis. Eur Rev Med Pharmacol Sci. 2019;23(10):4360–7.
Elcheva IA, Spiegelman VS. The role of cis-and transacting RNA regulatory elements in leukemia. Cancers (Basel). 2020;12(12):3854.
Rinn JL, Chang HY. Long noncoding RNAs: molecular modalites to organismal functions. Annu Rev Biochem. 2020;89:283–308.
Bian EB, Xiong ZG, Li J. New advances of lncRNAs in liver fibrosis, with specific focus on lncRNA-miRNA interactions. J Cell Physiol. 2019;234(3):2194–203.
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013;41:D991–5.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40.
Jiang H, Ma R, Zou S, Wang Y, Li Z, Li W. Reconstruction and analysis of the lncRNA-miRNA-mRNA network based on competitive endogenous RNA reveal functional lncRNAs in rheumatoid arthritis. Mol Biosyst. 2017;13(6):1182–92.
Shannon P, Markiel A, Ozieretal O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: identifying hub objects and subnetworks from complex interactome. BMC Syst Biol. 2014;8(Suppl 4):S11.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium Nat Genet. 2000;25(1):25–9.
da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57.
Altermann E, Klaenhammer TR. Pathway Voyager: Pathway mapping using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. BMC Genomics. 2005;6:60.
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447-452.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Sun M, Kisseleva T. Reversibility of liver fibrosis. Clin Res Hepatol Gastroenterol. 2015;39(Suppl 1):S60–3.
Atta HM. Reversibility and heritability of liver fibrosis: implications for research and therapy. World J Gastroentero. 2015;21(17):5138–48.
Zhao YL, Ma X, Wang JB, He X, Hu Y, Zhang P, Wang RL, Li RS, Gong M, Luo SQ, et al. Curcumin protects against CCl4-induced liver fibrosis in rats by inhibiting HIF-1alpha through an ERK-dependent pathway. Molecules. 2014;19(11):18767–80.
Wei W, Liu Y, Lu Y, Yang B, Tang L. LncRNA XIST Promotes Pancreatic Cancer Proliferation Through miR133a/EGFR. J Cell Biochem. 2017;118(10):3349–58.
Matsui M, Corey DR. Non-coding RNAs as drug targets. Nat Rev Drug Discovery. 2017;16(3):167–79.
Matsui M, Chu Y, Zhang H, Gagnon KT, Shaikh S, Kuchimanchi S, Manoharan M, Corey DR, Janowski BA. Promoter RNA links transcriptional regulation of infammatory pathway genes. Nucleic Acids Res. 2013;41(22):10086–109.
Liu J, Hu J, Corey DR. Expanding the action of duplex RNAs into the nucleus: redirecting alternative splicing. Nucleic Acids Res. 2012;40(3):1240–50.
Rougeulle C, Cardoso C, Fontés M, Colleaux L, Lalande M. An imprinted antisense RNA overlaps UBE3A and a second maternally expressed transcript. Nat Genet. 1998;19(1):15–6.
Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, Freier SM, Bennett CF, Sharma A, Bubulya PA, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. 2010;39(6):925–38.
Leti F, Legendre C, Still CD, Chu X, Petrick A, Gerhard GS, DiStefano JK. Altered expression of MALAT1 lncRNA in nonalcoholic steatohepatitis fibrosis regulates CXCL5 in hepatic stellate cells. Transl Res. 2017;190(25–39): e21.
Yu F, Zheng J, Mao Y, Dong P, Li G, Lu Z, Guo CY, Liu ZJ, Fan XM. Long non-coding RNA APTR promotes the activation of hepatic stellate cells and the progression of liver fibrosis. Biochem Biophys Res Commun. 2015;4634(4):679–85.
Fu N, Zhao SX, Kong LB, Du JH, Ren WG, Han F, Zhang QS, Li WC, Cui P, Wang RQ. LncRNA-ATB/microRNA-200a/beta-catenin regulatory axis involved in the progression of HCV-related hepatic fibrosis. Gene. 2017;618:1–7.
Yuan BY, Chen YH, Wu ZF, Zhang L, Zhuang Y, Zhao XM, Niu H, Cheng JCH, Zeng ZC. Proteomic Profiling of Human Hepatic Stellate Cell Line LX2 Responses to Irradiation and TGF-β1. J Proteome Res. 2019;18(1):508–21.
Cao Y, Wang P, Ning S, Xiao W, Xiao B, Li X. Identification of prognostic biomarkers in glioblastoma using a long non-coding RNA-mediated, competitive endogenous RNA network. Oncotarget. 2016;7(27):41737–47.
Tay Y, Rinn J. Pandolf PP The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505(7483):344–52.
Smillie CL, Sirey T, Ponting CP. Complexities of post-transcriptional regulation and the modeling of ceRNA crosstalk. Crit Rev Biochem Mol Biol. 2018;53(3):231–45.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the Natural Science Foundation of Hunan Province (grant no.2021JJ70148) and 2023 Science and Technology Project of Hunan Provincial Health Commission (grant no.B202319019225).
Author information
Authors and Affiliations
Contributions
M.F.X. contribute to the concept and design. F.Z. conducted all bioinformatic analyses. S.Y.P performed the validation experiments. M.F.X. wrote the manuscript. All authors read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All experiments were performed in accordance with relevant guidelines of the Ethics Committee of Xiangya Hospital and regulations of the the Declaration of Helsinki. Approval of the study and informed consent of the participants was obtained. Ethical approval was obtained from the Ethics Committee of Xiangya Hospital.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Table S1. The expression data of mRNA and lncRNA in GSE123932.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, F., Pei, S. & Xiao, M. Identification of functional genes in liver fibrosis based on bioinformatics analysis of a lncRNA-mediated ceRNA network. BMC Med Genomics 17, 56 (2024). https://doi.org/10.1186/s12920-024-01813-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-024-01813-x