
Abstract
Background
People with autosomal recessive disorders often were born without awareness of the carrier status of their parents. The American College of Medical Genetics and Genomics (ACMG) recommends screening 113 genes known to cause autosomal recessive and X-linked conditions in couples seeking to learn about their risk of having children with these disorders to have an appropriate reproductive plan.
Methods
We analyzed the exome sequencing data of 1,642 unrelated Thai individuals to identify the pathogenic variant (PV) frequencies in genes recommended by ACMG.
Results
In the 113 ACMG-recommended genes, 165 PV and likely PVs in 60 genes of 559 exomes (34%, 559/1642) were identified. The carrier rate was increased to 39% when glucose-6-phosphate dehydrogenase (G6PD) was added. The carrier rate was still as high as 14.7% when thalassemia and hemoglobinopathies were excluded. In addition to thalassemia, hemoglobinopathies, and G6PD deficiency, carrier frequencies of > 1% were found for Gaucher disease, primary hyperoxaluria, Pendred syndrome, and Wilson disease. Nearly 2% of the couples were at risk of having offsprings with the tested autosomal recessive conditions.
Conclusions
Based on the study samples, the expanded carrier screening, which specifically targeted common autosomal recessive conditions in Thai individuals, will benefit clinical outcomes, regarding preconception/prenatal genetic carrier screening.
Similar content being viewed by others


Background
Although each Mendelian disease is generally uncommon, with > 5,000 known heritable disorders, they are accounted for ~ 20% of infant mortality, ~ 18% of pediatric hospitalizations, and substantial numbers in adults in the United States [1]. At least 2,000 autosomal recessive diseases [2] and 500 X-linked diseases [3] have been identified. The preconception carrier screening for these disorders and proper reproductive option counseling have been shown to be cost-effective measures that reduce the burden of Mendelian diseases [4,5,6,7]. The studies of the carrier rate of these disorders are highly varied with the number of tested genes and the genes included in the studies among different populations with no consensus agreement [8,9,10,11]. Until recently, the American College of Medical Genetics and Genomics (ACMG) released a set of 113 genes, both autosomal recessive and X-linked conditions, proposed as a standard list of genes for carrier screening [12]. Because X-linked glucose-6-phosphate dehydrogenase (G6PD) deficiency is the most common genetic disorder in Thailand and Southeast Asia [13], we added G6PD into the list and determined the carrier frequency and variant distribution of 114 recessive genes in the Thai population using the exomes of 1,642 unrelated Thais.
Methods
Study samples
In our study, we collected a sample of 1642 exomes from unrelated healthy Thai individuals, consisting of 811 men and 831 women. Prior to participation, informed consent was obtained from each individual. The study was conducted in accordance with ethical guidelines and was approved by the Institutional Review Board of the Faculty of Medicine, Chulalongkorn University (IRB No. 264/62). Our research with human subjects conforms with the Declaration of Helsinki (1964). The data analyzed in our study were part of the Thai Reference Exome database, although not all data from the database were used for our analysis [14]. It is important to note that for the patient cohort, we specifically excluded variants in genes known to be responsible for the patients' diagnosed conditions. This ensured that our analysis focused on carriers of pathogenic variants unrelated to the diagnosed diseases in the patient cohort.
Sequencing and bioinformatics analysis
Genomic DNA was extracted from peripheral blood leukocytes. Exomes were captured using either a TruSeq® Exome Kit (Illumina, San Diego, CA) on the NextSeq 500 system or SureSelect Human All Exons (Agilent Inc., Santa Clara, CA) on the Hiseq 4000 and Novaseq 6000 Systems. The reagents are according to the manufacturer's standard protocol. Sequences were aligned to the human reference genome (GRCh37) using the Burrows–Wheeler Aligner package version 0.7.15 [15]. Variant calling was performed using the Genome Analysis Tool Kit (GATK Best Practice V3.7; Broad Institute), which is called by HaplotypeCaller [16]. ANNOVAR was used to annotate genetic variants. The variant interpretation was limited to the 113 ACMG-recommended genes and G6PD. We included variants listed as PV and LPV according to VarSome's ACMG classification [17] with a final manual curation by the authors.
Statistical analysis
The frequency and 95% confidence interval were calculated using the proportion of carriers in total individuals tested (n/1642) and the Wilson method, respectively.
Results
Using 1,642 exome sequencing data of unaffected parents whose children had rare diseases, we first excluded pathogenic and likely pathogenic variants (PV and LPV, respectively) found in these exomes, which caused the diseases in their children. We excluded 13 PV and LPV in 18 exomes (1%, 18/1642) (S1 Table).
Of the 113 ACMG-recommended genes, 165 PV and LPV in 60 genes of 559 exomes were found (559/1642: 34%). For G6PD, 7 PV and LPV of 127 exomes (female) were identified. In the analysis of a total of 114 genes, 172 PV and LPV in 61 genes of 640 exomes (640/1642: 39%) were identified (Table 1). Although we excluded HBA1, HBA2, and HBB genes causing thalassemia and hemoglobinopathies, the carrier rate of at least one screened disorder was still as high as 14.7% (241/1642) in our cohort. Of the samples, 7.4% (121/1642) carried more than one PV and LPV (Fig. 1 and S2 Table).

Carrier frequencies of one or more genetic disorders in 114 genes of 1,642 unrelated Thais. Orange color represents carrier frequencies in 113 ACMG recommended genes. Blue color represents those in 114 genes, which are the 113 genes and G6PD. Gray color represents those in 110 genes, which are the 113 genes excluding three genes underlying thalassemia and hemoglobinopathies (HBB, HBA1, and HBA2)
Gene carrier rate
In our cohort, all top three gene carrier rates were in hemolytic disorders with positive selection from malaria. The gene with the highest carrier rate is HBB, with 50.2% (321/640) and 19.6% (291/1642) of all carriers and participants, respectively. The second most common carrier gene is G6PD, with 19.8% (127/640) and 7.7% (127/1642) of all carriers and participants, respectively. HBA2 is the third most common gene, with 10.2% (65/640) and 4% (65/1642) of all carriers and participants, respectively.
There were 17 autosomal recessive genes with carrier frequencies of at least 1/250, an expected prevalence at birth of at least 1 in 250,000. As shown in Fig. 2, three, five, and nine of these genes are related to hemolytic disorders (G6PD, HBB, and HBA2), inborn errors of metabolism (AGXT, ATP7B, CYP27A1, GBA, and PAH), and in the miscellaneous group (CEP290, CFTR, CLCN1, GJB2, MCPH1, NEB, PKHD1, SLC26A4, and USH2A), respectively.

The 17 genes underlying autosomal recessive disorders with carrier frequencies of ≥ 1/250 (0.4%) in the 1,642 unrelated Thai individuals
Variant carrier rate
The most common PV is HBB; c.79G > A (p.Glu27Lys) responsible for hemoglobin E (HbE), representing 45.5% (291/640) and 17.7% (291/1642) of all carriers and participants, respectively. HBA2; c.427 T > C (p. Ter143Glnext*31, also known as Hb Constant Spring) is the second most common variant, representing 9.2% (59/640) and 3.6% (59/1642) of all carriers and participants, respectively. The third most common variant is G6PD; c.961G > A (p.Val321Met) causing G6PD deficiency, with 8.6% (55/640) and 3.4% (55/1642) of all carriers and participants, respectively. The variant carrier rates are shown in S3 Table.
Thalassemia and hemoglobinopathies
Although, the most common type of α-thalassemia mutation is the deletion of one or more of the α-globin genes, HBA1 and HBA2 [18], we did not attempt to analyze this type of genetic variant due to the inherent limitation of exome sequencing. The carrier rates of nondeletional α-thalassemia were detected in 5.1% (71/1642; 6 in HBA1 and 65 in HBA2) of our cohort. The most common variant is c.427 T > C 3.6% (59/1642) in Hb Constant Spring, followed by Hb Paksé c.429A > T 0.3% (5/1642). The frequency rate of β-thalassemia carriers is 19.6% (321/1642). The most prevalent variant, c.79G > A (p.Glu27Lys [HbE]), accounts for 17.7% (291/1642) of cases in the HBB genes. The next two most prevalent variants are c.52A > T (0.79%, 13/1642) and c.126_129del (0.67%, 11/1642).
G6PD deficiency
The carrier rate of G6PD variants was 7.7% (127/1642). The prevalence of heterozygous/carrier G6PD variant and homozygous females in our cohort is 15.28% (127/831) and 0.8% (7/831), respectively (S4 Table). The percentage of symptomatic or biochemical G6PD deficiency in these samples was not known. The prevalence of hemizygous G6PD variant males in our cohort is 8% (65/811). The female carrier rate corresponds with the predicted prevalence calculated with male hemizygous prevalence using the Hardy–Weinberg formula. c.961G > A (p.Val321Met) (G6PD Viantien) and c.577G > A (p.Gly193Ser) (G6PD Mahidol) are the two most common G6PD variants found in our cohort.
Other autosomal recessive disorder carrier rate and common variants
The carrier frequency of PV and LPV in GBA causing Gaucher disease is 1.6% (27/1642) with c.605G > A (p.Arg202Gln) (0.54%, 9/1642) and c.1448 T > C (p.Leu483Pro) (0.42%, 7/1642) being the two most common variants.
The carrier frequency of PV and PLV in genes associated with hereditary syndromic hearing loss SLC26A2 is 1.1% (18/1642). c.919-2A > G was the most common variant (0.3%, 5/1642). The second most common gene causing syndromic hearing loss carriers was USH2A (Usher syndrome) (0.97%, 16/1642). The carrier frequency of PV and PLV in GJB2, one of the most common genes causing nonsyndromic deafness is 0.6% (9/1642). All cases identified carried c.235delC (p.Leu79CysfsTer3) in GJB2.
Besides Gaucher disease, the most common inherited metabolic disease gene carriers in our cohort are hyperoxaluria (AGXT; 1.1%, 18/1642), Wilson disease (ATP7B; 1%, 17/1642), and phenylketonuria (PAH; 0.79%, 13/1642).
Discussion
Every approach possesses both advantages and limitations. In our study, we chose to engage parents whose children were afflicted with rare diseases rather than employing a population-based screening methodology. This strategy, akin to "killing two birds with one stone," aimed to optimize the benefits derived from trio exome testing. Specifically, children with rare diseases stood a higher chance of obtaining molecular diagnoses through trio exomes as opposed to singleton testing. Furthermore, the exomes of the parents provided valuable insights into a spectrum of other biological and medical issues. To mitigate bias, we deliberately excluded genes identified as causative for rare diseases in the children. Notably, our approach is characterized by a lower ethical burden. The apparently healthy parents, whose children were affected by rare diseases in our cohort, received genetic counseling and were well-informed about the implications of these tests. They made a conscious choice to permit the use of their genomic information for purposes beyond the primary focus of the testing.Conversely, a population-based approach is not without its constraints. Recent evidence [19, 20], suggests the presence of participation bias. Additionally, ascertainment bias may manifest, with individuals referred or participating in the project having a known family history of rare diseases showing heightened interest. The ethical complexities surrounding population screening pose a challenge. Carrier screening currently remains optional and not a standard mandatory test. While couples undergoing population screening could potentially benefit from counseling, extending genetic counseling to research subjects who are apparently normal may be cumbersome and miss the opportunity to support those genuinely in need. Moreover, the practical medical advantages of counseling may be constrained by the exorbitant costs associated with reproductive choices such as in vitro fertilization (IVF) and preimplantation genetic diagnosis (PGD).
Prior investigations into the carrier rates of these disorders in various countries have exhibited substantial variability in terms of the number of genes tested and the specific genes encompassed within the studies across diverse populations. Notably, within the Thai population, genetic disorders that manifest with a comparatively higher prevalence than in other populations encompass thalassemia, hemoglobinopathies, and glucose-6-phosphate dehydrogenase (G6PD) deficiency [21]. Carriers of these hematological disorders in Thais demonstrate a relative resistance to malaria, affording them a selective advantage. Since HBA and HBB are already incorporated into the ACMG 113-gene list, we have augmented our gene panel to encompass G6PD deficiency, resulting in a total of 114 genes. This expansion aligns with our commitment to comprehensively address the genetic landscape relevant to carrier screening, particularly in the context of the prevalent hematological disorders observed in the Thai population.
Next-Generation Sequencing (NGS) has been increasingly used for expanded genetic carrier screening for multiple genes in a highly efficient manner in clinical laboratories [22]. When screening for carriers in individuals with an average risk, it is deemed acceptable or advisable to report solely (likely) pathogenic variants [13]. The carrier rate ranges from as high as 43.6% in Ashkenazi Jewish individuals to 8.5% in East Asians [23], although the number of genes tested and which genes are included are varied among these studies. In our study using 1642 exomes of a Thai population, 39% of Thai individuals carried a PV and an LPV in at least one of 114 genes (113 genes have been recommended by ACMG [12]) plus G6PD, causing G6PD deficiency, which is most common in the Thai population. We observed that 23.8% of individuals were carriers of thalassemia and hemoglobinopathies (HBB, HBA1, and HBA2), followed by 7.7% who were carries of G6PD deficiency. The carrier rate observed in our cohort is considered high, comparable to that of the Ashkenazi Jewish group. The majority of these variants are found in the Hb genes and G6PD, which are believed to be the result of heterozygous advantage in regions with a high prevalence of malaria [24]. This differs from the Ashkenazi Jewish population, where the high carrier rate is primarily attributed to in-group mating.
Consanguineous marriage is generally considered taboo in the majority of the Thai population, with exceptions observed in certain minor communities such as Muslims and hill tribes. Our assertions are substantiated by evidence gleaned from our analysis of inheritance patterns among patients referred for genetic testing at our institution. Notably, our findings reveal that only 25% of positive tests exhibit inheritance in an autosomal recessive pattern, in stark contrast to Middle Eastern countries where the autosomal recessive pattern can reach up to 80%, particularly in regions where consanguinity is commonplace [25]. It is imperative to underscore that thalassemia and hemoglobinopathies are highly prevalent in our region, and the pseudoautosomal dominant pattern observed in our study is reflective of this prevalence rather than indicative of prevailing marriage patterns.
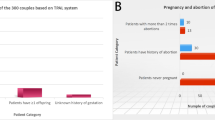
Even after excluding thalassemia and hemoglobinopathies, the carrier rate in our cohort remained significantly high at 14.7%. We observed 14 couples (1.7%) who were carriers of PV and LPV in the same gene (S5 Table). Of these couples, seven (0.9%, 7/821) were non thalassemia carrier couples at risk of having children affected with recessive disorders. Thalassemia carrier screening is already included and available in the screening program of Thai pregnant women; therefore, it is estimated that nearly 1% of the couples could benefit from expanded carrier screening.
The prevalence of common recessive disorders in our cohort can be estimated and, in general, is comparable to previous prevalence rates reported for common autosomal recessive disorders, such as Thalassemia and G6PD deficiency [18, 26,27,28,29,30,31]. This suggests that our approach of excluding samples carrying genetic variants responsible for the diseases in their offspring during ascertainment and analysis helps eliminate bias and ensures that the frequency estimates are comparable to those of non-disease or population cohorts. We have previously demonstrated the effectiveness of this strategy in our analysis of the prevalence of secondary findings, particularly since the majority of reportable diseases are inherited in an autosomal dominant pattern.
The most common carriers of inherited metabolic disease identified in our cohort are individuals with Gaucher disease caused by mutations in the GBA gene. However, interpreting the results can be challenging due to the presence of a pseudogene associated with GBA. The GBA pseudogene exhibits the highest homology with GBA between exons 8 and 11, which is likely a result of recombination events [32]. Many clinical exome sequencing laboratories do not analyze or report the variants in GBA. However, within the exome, there exists a mappable region of GBA that encompasses known recurrent pathogenic variants, including the c.1448 T > C mutation. This region can be analyzed with a high degree of sensitivity and specificity. This methodology has been detailed in our recent publication on early-onset Parkinson’s disease in Thai cohorts [33]. In addition, the variants commonly identified in our cohort are outside these highly homologous areas and are therefore less likely to be affected in our analysis. Conversely, the prevalence of the carrier of GBA variant is likely to be underestimated in our cohort or a similar cohort using exome sequencing. Nevertheless, it is crucial to acknowledge the limitation associated with utilizing exome sequencing for GBA carrier screening. While this approach offers high sensitivity and specificity for certain regions, its effectiveness in reducing the posttest probability of being a GBA carrier for other areas is comparatively low. This limitation underscores the importance of considering alternative testing methods for a comprehensive and accurate assessment of GBA carrier status.
In our study, carriers of primary hyperoxaluria and Wilson disease PV and PLV were found to be the next most common. Founder variants in East Asian populations (c.2 T > C (p.Met1Thr) in AGXT [34,35,36,37] and c.2333G > T (p.Arg778Leu) in ATP7B [38,39,40,41,42,43] were also identified in our cohort. Our study further supports the existing newborn screening program in Thailand for Phenylketonuria (PKU). The estimated carrier rate of 1 in 127 individuals is comparable to rates reported in other countries [44,45,46]. With an annual live birth rate of 600,000, our findings suggest that approximately 8–10 children will be born with PKU each year in Thailand. These findings emphasize the importance of continued implementation and monitoring of the newborn screening program for PKU in the country.
Hearing loss is one of the major disability problems in childhood, with hereditary causes, both syndromic and nonsyndromic, being the primary etiologies in affected children. In our study, we identified carriers of Pendred syndrome, Usher syndrome, and GJB2 (nonsyndromic hereditary neurosensory hearing loss) to be common in the Thai cohort, aligning with previous estimations [47,48,49,50,51,52,53,54,55,56,57,58]. These findings highlight the relevance of these hereditary conditions as significant contributors to hearing loss in the Thai population.
Compared to the common recommendation of screening for cystic fibrosis carrier status in Caucasians [59,60,61], our study revealed that only 0.97% of our sample carried pathogenic variants (PV) and likely pathogenic variants (LPV) in the CFTR gene. This suggests that the estimated prevalence of cystic fibrosis among Thais is lower than that among Caucasians (1 in 2,000 in Northern European descent vs. 1 in 400,000). Interestingly, we observed three females (0.18%) who carried the 5T variant (c.1210–12 [5]) in CFTR. While this variant is associated with mild cystic fibrosis, it can result in male infertility due to congenital bilateral absence of the vas deferens [62].
Limitations
Our study had some limitations. Whole Exome Sequencing (WES) cannot reliably detect structural variants that are common in α-thalassemia. Spinal muscular atrophy, a common muscular disorder, most commonly caused by a copy number change in the SMN1 gene and modified by the copy numbers of SMN2, is also missed by WES. The genome analysis with structural variant detection will help estimate these common disorders. Additionally, the frequency of variants based on NGS analysis may be underestimated without validation using Sanger sequencing, particularly for complex recombinant alleles of GBA pseudogenes. The other common PVs that will be missed by WES are trinucleotide repeat expansion. PV in FMR1 causing fragile X syndrome, the most common cause of intellectual disability especially in males, cannot be detected with exome sequencing data. It is worth mentioning that our sample size provides over 80% power to detect alleles with a frequency of 1%. However, for the detection of uncommon variants, a larger sample size would be necessary [63].
Conclusion
Among the 114 genes analyzed, which included the 113 ACMG-recommended genes along with G6PD, our study revealed that 39% (640/1642) of Thai individuals in our cohort were carriers of pathogenic variants (PVs). Through a combination of computational filtering and manual curation, we were able to unveil the landscape of these PVs that are commonly observed in the Thai population. This information will serve as a foundation for designing gene lists for individual-level carrier screening and future public health plans aimed at addressing inherited genetic disorders.
Availability of data and materials
The datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.
Abbreviations
- ACMG:
-
The American College of Medical Genetics and Genomics
- LPV:
-
Likely pathogenic variants
- PV:
-
Pathogenic variants
- NGS:
-
Next-Generation Sequencing
- WES:
-
Whole exome sequencing
References
Kingsmore S. Comprehensive carrier screening and molecular diagnostic testing for recessive childhood diseases. PLoS Curr. 2012:e4f9877ab8ffa9.
Xiao Q, Lauschke VM. The prevalence, genetic complexity and population-specific founder effects of human autosomal recessive disorders. NPJ Genom Med. 2021;6(1):41.
Migeon BR. X-linked diseases: susceptible females. Genet Med. 2020;22(7):1156–74.
Shotelersuk V, Limwongse C, Mahasirimongkol S. Genetics and genomics in Thailand: challenges and opportunities. Mol Genet Genomic Med. 2014;2(3):210–6.
Ministry of Public Health Thailand. Thalassemia in Thailand: 2018 [Available from: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0246736#pone.0246736.ref006.
Cowan RS. Moving up the slippery slope: mandated genetic screening on Cyprus. Am J Med Genet C Semin Med Genet. 2009;151C(1):95–103.
Bozkurt G. Results from the North Cyprus thalassemia prevention program. Hemoglobin. 2007;31(2):257–64.
Singh K, Bijarnia-Mahay S, Ramprasad VL, Puri RD, Nair S, Sharda S, et al. NGS-based expanded carrier screening for genetic disorders in North Indian population reveals unexpected results - a pilot study. BMC Med Genet. 2020;21(1):216.
Chan OYM, Leung TY, Cao Y, Shi MM, Kwan AHW, Chung JPW, et al. Expanded carrier screening using next-generation sequencing of 123 Hong Kong Chinese families: a pilot study. Hong Kong Med J. 2021;27(1):177–83.
Hernandez-Nieto C, Alkon-Meadows T, Lee J, Cacchione T, Iyune-Cojab E, Garza-Galvan M, et al. Expanded carrier screening for preconception reproductive risk assessment: prevalence of carrier status in a Mexican population. Prenat Diagn. 2020;40(5):635–43.
Chau JFT, Yu MHC, Chui MMC, Yeung CCW, Kwok AWC, Zhuang X, et al. Comprehensive analysis of recessive carrier status using exome and genome sequencing data in 1543 Southern Chinese. NPJ Genom Med. 2022;7(1):23.
Gregg AR, Aarabi M, Klugman S, Leach NT, Bashford MT, Goldwaser T, et al. Screening for autosomal recessive and X-linked conditions during pregnancy and preconception: a practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021;23(10):1793–806.
Sathupak S, Leecharoenkiat K, Kampuansai J. Prevalence and molecular characterization of glucose-6-phosphate dehydrogenase deficiency in the Lue ethnic group of northern Thailand. Sci Rep. 2021;11(1):2956.
Shotelersuk V, Wichadakul D, Ngamphiw C, et al. The Thai reference exome (T-REx) variant database. Clin Genet. 2021;100(6):703–12. https://doi.org/10.1111/cge.14060.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303.
Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, et al. VarSome: the human genomic variant search engine. Bioinformatics. 2019;35(11):1978–80.
Farashi S, Harteveld CL. Molecular basis of alpha-thalassemia. Blood Cells Mol Dis. 2018;70:43–53.
Pirastu N, Cordioli M, Nandakumar P, Mignogna G, Abdellaoui A, Hollis B, Kanai M, Rajagopal VM, Parolo PD, Baya N, Carey CE. Genetic analyses identify widespread sex-differential participation bias. Nat Genet. 2021;53(5):663–71.
Schoeler T, Speed D, Porcu E, Pirastu N, Pingault JB, Kutalik Z. Participation bias in the UK biobank distorts genetic associations and downstream analyses. Nat Hum Behav. 2023;27:1–2.
Banyatsuppasin W, Jindadamrongwech S, Limrungsikul A, Butthep P. Prevalence of thalassemia and glucose-6-phosphate dehydrogenase deficiency in newborns and adults at the Ramathibodi hospital, Bangkok. Thailand Hemoglobin. 2017;41(4–6):260–6.
Rowe CA, Wright CF. Expanded universal carrier screening and its implementation within a publicly funded healthcare service. J Community Genet. 2020;11(1):21–38.
Lazarin GA, Haque IS, Nazareth S, Iori K, Patterson AS, Jacobson JL, et al. An empirical estimate of carrier frequencies for 400+ causal Mendelian variants: results from an ethnically diverse clinical sample of 23,453 individuals. Genet Med. 2013;15(3):178–86.
Ha J, Martinson R, Iwamoto SK, Nishi A. Hemoglobin E, malaria and natural selection. Evol Med Public Health. 2019;2019(1):232–41.
Aleissa M, Aloraini T, Alsubaie LF, Hassoun M, Abdulrahman G, Swaid A, Eyaid WA, Mutairi FA, Ababneh F, Alfadhel M, Alfares A. Common disease-associated gene variants in a Saudi Arabian population. Ann Saudi Med. 2022;42(1):29–35.
Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86(6):480–7.
Lai K, Huang G, Su L, He Y. The prevalence of thalassemia in mainland China: evidence from epidemiological surveys. Sci Rep. 2017;7(1):920.
Colah R, Italia K, Gorakshakar A. Burden of thalassemia in India: the road map for control. Pediatr Hematol Oncol J. 2017;2(4):79–84.
Luzzatto L, Nannelli C, Notaro R. Glucose-6-phosphate dehydrogenase deficiency. Hematol Oncol Clin North Am. 2016;30(2):373–93.
Boonyuen U, Songdej D, Tanyaratsrisakul S, Phuanukoonnon S, Chamchoy K, Praoparotai A, et al. Glucose-6-phosphate dehydrogenase mutations in malaria endemic area of Thailand by multiplexed high-resolution melting curve analysis. Malar J. 2021;20(1):194.
Phompradit P, Kuesap J, Chaijaroenkul W, Rueangweerayut R, Hongkaew Y, Yamnuan R, et al. Prevalence and distribution of glucose-6-phosphate dehydrogenase (G6PD) variants in Thai and Burmese populations in malaria endemic areas of Thailand. Malar J. 2011;10:368.
Leija-Salazar M, Sedlazeck FJ, Toffoli M, Mullin S, Mokretar K, Athanasopoulou M, et al. Evaluation of the detection of GBA missense mutations and other variants using the Oxford Nanopore MinION. Mol Genet Genomic Med. 2019;7(3):e564.
Thanprasertsuk S, Phowthongkum P, Hopetrungraung T, Poorirerngpoom C, Sathirapatya T, Wichit P, Phokaewvarangkul O, Vongpaisarnsin K, Bongsebandhu-Phubhakdi S, Bhidayasiri R. Levodopa-induced dyskinesia in early-onset Parkinson’s disease (EOPD) associates with glucocerebrosidase mutation: a next-generation sequencing study in EOPD patients in Thailand. PLoS ONE. 2023;18(10): e0293516.
Hulton S-A. Lumasiran: expanding the treatment options for patients with primary hyperoxaluria type 1. Expert Opin Orphan Drugs. 2021;9(7–10):189–98.
Li GM, Xu H, Shen Q, Gong YN, Fang XY, Sun L, et al. Mutational analysis of AGXT in two Chinese families with primary hyperoxaluria type 1. BMC Nephrol. 2014;15:92.
Yuen YP, Lai CK, Tong GM, Wong PN, Wong FK, Mak SK, et al. Novel mutations of the AGXT gene causing primary hyperoxaluria type 1. J Nephrol. 2004;17(3):436–40.
Tammachote R, Kingsuwannapong N, Tongkobpetch S, Srichomthong C, Yeetong P, Kingwatanakul P, et al. Primary hyperoxaluria type 1 and brachydactyly mental retardation syndrome caused by a novel mutation in AGXT and a terminal deletion of chromosome 2. Am J Med Genet A. 2012;158A(9):2124–30.
Kumar M, Gaharwar U, Paul S, Poojary M, Pandhare K, Scaria V, et al. WilsonGen a comprehensive clinically annotated genomic variant resource for Wilson’s disease. Sci Rep. 2020;10(1):9037.
Jang JH, Lee T, Bang S, Kim YE, Cho EH. Carrier frequency of Wilson’s disease in the Korean population: a DNA-based approach. J Hum Genet. 2017;62(9):815–8.
Qiao L, Ge J, Li C, Liu Y, Hu C, Hu S, et al. Pathogenic gene variation spectrum and carrier screening for Wilson’s disease in Qingdao area. Mol Genet Genomic Med. 2021;9(8):e1741.
Hua R, Hua F, Jiao Y, Pan Y, Yang X, Peng S, et al. Mutational analysis of ATP7B in Chinese Wilson disease patients. Am J Transl Res. 2016;8(6):2851–61.
Panichareon B, Taweechue K, Thongnoppakhun W, Aksornworanart M, Pithukpakorn M, Yenchitsomanus PT, et al. Six novel ATP7B mutations in Thai patients with Wilson disease. Eur J Med Genet. 2011;54(2):103–7.
Treepongkaruna S, Pienvichit P, Phuapradit P, Kodcharin P, Wattanasirichaigoon D. Mutations of ATP7B gene in two Thai siblings with Wilson disease. Asian Biomedicine. 2010;4(1):163–9.
Hillert A, Anikster Y, Belanger-Quintana A, Burlina A, Burton BK, Carducci C, et al. The genetic landscape and epidemiology of phenylketonuria. Am J Hum Genet. 2020;107(2):234–50.
Wang T, Okano Y, Eisensmith RC, Lo WH, Huang SZ, Zeng YT, et al. Missense mutations prevalent in orientals with phenylketonuria: molecular characterization and clinical implications. Genomics. 1991;10(2):449–56.
Chaiyasap P, Ittiwut C, Srichomthong C, Sangsin A, Suphapeetiporn K, Shotelersuk V. Massive parallel sequencing as a new diagnostic approach for phenylketonuria and tetrahydrobiopterin-deficiency in Thailand. BMC Med Genet. 2017;18(1):102.
Petit C, Levilliers J, Hardelin JP. Molecular genetics of hearing loss. Annu Rev Genet. 2001;35:589–646.
Hu H, Zhou P, Wu J, Lei W, Wang Y, Yang Y, et al. Genetic testing involving 100 common mutations for antenatal diagnosis of hereditary hearing loss in Chongqing, China. Medicine (Baltimore). 2021;100(17):e25647.
Klumsathian S, Lorlipiwong W, Iemwimangsa N, Sensorn I, Panthan B, Chareonsirisuthigul T, et al. AB034. Carrier frequency of inherited genetic disorders in Thai population: implication for designing expanded carrier screening panel. Ann Transl Med. 2017;5(S2):AB034-AB.
Snabboon T, Plengpanich W, Saengpanich S, Sirisalipoch S, Keelawat S, Sunthornyothin S, et al. Two common and three novel PDS mutations in Thai patients with Pendred syndrome. J Endocrinol Invest. 2007;30(11):907–13.
Wattanasirichaigoon D, Limwongse C, Jariengprasert C, Yenchitsomanus PT, Tocharoenthanaphol C, Thongnoppakhun W, et al. High prevalence of V37I genetic variant in the connexin-26 (GJB2) gene among non-syndromic hearing-impaired and control Thai individuals. Clin Genet. 2004;66(5):452–60.
Choy KW, Cao Y, Lam ST, Lo FM, Morton CC, Leung TY. Target-enriched massively parallel sequencing for genetic diagnosis of hereditary hearing loss in patients with normal array CGH result. Hong Kong Med J. 2018;24 Suppl 3(3):11–4.
Taniguchi M, Matsuo H, Shimizu S, Nakayama A, Suzuki K, Hamajima N, et al. Carrier frequency of the GJB2 mutations that cause hereditary hearing loss in the Japanese population. J Hum Genet. 2015;60(10):613–7.
Han SH, Park HJ, Kang EJ, Ryu JS, Lee A, Yang YH, et al. Carrier frequency of GJB2 (connexin-26) mutations causing inherited deafness in the Korean population. J Hum Genet. 2008;53(11–12):1022–8.
Kim SY, Park G, Han KH, Kim A, Koo JW, Chang SO, et al. Prevalence of p.V37I variant of GJB2 in mild or moderate hearing loss in a pediatric population and the interpretation of its pathogenicity. PLoS One. 2013;8(4):e61592.
Pollak A, Skorka A, Mueller-Malesinska M, Kostrzewa G, Kisiel B, Waligora J, et al. M34T and V37I mutations in GJB2 associated hearing impairment: evidence for pathogenicity and reduced penetrance. Am J Med Genet A. 2007;143A(21):2534–43.
Lentz J, Keats B. Usher syndrome type II. GeneReviews®[Internet]. 2016.
Xi Y, Chen G, Lei C, Wu J, Zhang S, Xiao M, et al. Expanded carrier screening in Chinese patients seeking the help of assisted reproductive technology. Mol Genet Genomic Med. 2020;8(9):e1340.
Antoniou S, Elston C. Cystic fibrosis. Medicine. 2016;44(5):321–5.
Scotet V, L'Hostis C, Ferec C. The changing epidemiology of cystic fibrosis: incidence, survival and impact of the CFTR gene discovery. Genes (Basel). 2020;11(6):589.
Grody WW, Desnick RJ. Cystic fibrosis population carrier screening: here at last–are we ready? Genet Med. 2001;3(2):87–90.
Sun W, Anderson B, Redman J, Milunsky A, Buller A, McGinniss MJ, et al. CFTR 5T variant has a low penetrance in females that is partially attributable to its haplotype. Genet Med. 2006;8(6):339–45.
Fujikura K. Global carrier rates of rare inherited disorders using population exome sequences. PLoS ONE. 2016;11(5):e0155552. https://doi.org/10.1371/journal.pone.0155552.
Acknowledgements
Not applicable.
Funding
This study was supported by the Health Systems Research Institute (65–040).
Author information
Authors and Affiliations
Contributions
WC Data curation, analysis, and writing the original draft. PP Conceptualization and editing the manuscript. VS Conceptualization, funding acquisition, and editing the manuscript. All authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Institutional Review Board of Faculty of Medicine, Chulalongkorn University, Thailand (IRB No. 264/62). Written informed consent was obtained from parents or legal guardians of the participants.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: S1 Table.
Genetic variants of the 18 parents which were responsible for the presenting symptoms in their children. S2 Table. Carrier frequencies of one or more genetic disorders in 114 genes of 1,642 unrelated Thais. S3 Table. Variant carrier rate (VCR) of each variant. S4 Table. Characteristics of G6PD phenotypes in 1,642 unrelated Thais. S5 Table. Pathogenic or likely pathogenic variants in the same genes which were harbored by the couples.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chetruengchai, W., Phowthongkum, P. & Shotelersuk, V. Carrier frequency estimation of pathogenic variants of autosomal recessive and X-linked recessive mendelian disorders using exome sequencing data in 1,642 Thais. BMC Med Genomics 17, 9 (2024). https://doi.org/10.1186/s12920-023-01771-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-023-01771-w