
Abstract
Neoadjuvant chemotherapy (NAC) is a well-established treatment modality for locally advanced breast cancer (BC). However, it can also result in severe toxicities while controlling tumors. Therefore, reliable predictive biomarkers are urgently needed to objectively and accurately predict NAC response. In this study, we integrated single-cell and bulk RNA-seq data to identify nine genes associated with the prognostic response to NAC: NDRG1, CXCL14, HOXB2, NAT1, EVL, FBP1, MAGED2, AR and CIRBP. Furthermore, we constructed a prognostic risk model specifically linked to NAC. The clinical independence and generalizability of this model were effectively demonstrated. Additionally, we explore the underlying cancer hallmarks and microenvironment features of this NAC response-related risk score, and further assess the potential impact of risk score on drug response. In summary, our study constructed and validated a nine-gene signature associated with NAC prognosis, which was accomplished through the integration of single-cell and bulk RNA data. The results of our study are of crucial significance in the prediction of the efficacy of NAC in BC, and may have implications for the clinical management of this disease.
Similar content being viewed by others


Background
Breast cancer (BC) is the most common cancer among women and a leading cause of cancer-related deaths worldwide, accounting for approximately 11.6% of all cancer deaths [1, 2]. Neoadjuvant chemotherapy (NAC) has emerged as the standard treatment for stage II – III BC in women, as the postoperative pathological complete response (pCR) status have been used for individualized systemic adjuvant treatment [3]. NAC can provide systemic chemotherapy for naive BC patients without metastasis before planned surgical treatment or local surgery plus radiotherapy, and additionally, and it is also the standard approach for locally advanced BC patients, enabling downstaging of inoperable tumors and facilitating breast-conserving surgery [4]. Moreover, NAC can obtain information related to drug sensitivity in vivo, guiding follow-up treatment and improve the prognosis of patients. Nonetheless, some patients may face significant challenges to their survival in cases where NAC proves ineffective. This is due to the fact that their prognosis may worsen, and they may experience severe toxic side effects [5]. Those patients who will not benefit from NAC will experience side effects of chemotherapy without any benefit, so we need biomarkers of NAC response to identify subgroups of patients who may benefit from NAC. Therefore, the identification of biomarkers predicting NAC is of great importance in treatment guidance based on NAC as well as other alternatives combination therapy [6].
Studies by Li J et al. revealed that the higher the level of aldehyde dehydrogenase 1 (ALDH1) in BC patients are associated with poorer response to NAC [7]. Wang et al. demonstrated that measuring the expression of matrix metalloproteinase-9 (MMP-9) in tumor tissues helps to identify TNBC patients who respond well to NAC [8]. In addition, targeting key molecules in signaling pathways, such as AKT/pERK and Fas/FasL, has shown potential in BC sensitivity to chemotherapy [8, 9]. However, most previous studies have mainly focused on individual biomarker, while integrating high-throughput multi-omics data can provide a more comprehensive understanding of the mechanisms underlying BC [10], reveal cellular heterogeneity and diversity, and identify biomarkers reflecting the complexity of these processes.
Genomic tests, such as Mammaprint and Oncotype DX, are used in the management of BC [11, 12]. Mammaprint examines the activity of 70 genes to categorize breast cancer as low or high risk of recurrence, providing a recurrence score indicating the likelihood of cancer returning. However, its validation primarily pertains to early-stage, ER+, LN-, and untreated patients. Mammaprint’s limitations include its limited applicability to other breast cancer subtypes, relatively high cost, and absence of direct treatment benefit information. Oncotype DX analyzes the expression of 21 genes and provides a recurrence score to predict distant recurrence and potential chemotherapy benefits in ER+, LN-, and HER2-negative breast cancers. However, it may not be suitable for HER2-positive or triple-negative breast cancer patients. Oncotype DX’s limitations include its limited application to certain breast cancer subtypes, challenges in interpreting intermediate recurrence scores, and concerns about cost and insurance coverage.
Currently, reliable biomarkers for predicting NAC in BC remain limited [13, 14]. Single-cell RNA sequencing (scRNA-seq) can contribute to identifying distinct cell populations involves in carcinogenesis and profiling marker genes at single-cell level [15, 16]. Understanding the heterogeneity of tumor microenvironment (TME) in drug resistance mechanisms and identify more effective targets for individualized management [17]. In this study, we aimed to identify potential NAC related prognostic signatures for predicting response to NAC in BC patients through integrated bioinformatics transcriptome analyses at both the single-cell and bulk levels.
Methods
Data collection and preprocessing
The bulk transcriptome data and corresponding clinical data of BC patients receiving NAC were obtained from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) via GSE25055, GSE25065 and GSE22226. We chose GSE25055 with the most BC samples (N = 306) as the discovery cohort, then 198 BC samples from GSE25065 and 150 BC samples from GSE22226 were chosen as independent validation cohorts. Samples with incomplete survival information were excluded from the analysis.
The scRNA-seq data from 14 BC samples was obtained from the study of Qian et al [18], and downloaded from lambrechtslab - Laboratory of Translational Genetics (vib.be). The R package Seurat (version 4.0.0) was used to preprocess the scRNA-seq. Cell samples with more than 200 genes expressed and the mitochondrial gene expression rate less than 5% were retained. We used the “NormalizedData” function to standardize the scRNA-seq dataset and the “FindVariableFeatures” function to identify 2000 highly variable genes. We used R package Harmony to correct the batch effects. After data normalization, the principal component analysis (PCA),was performed and cells were grouped and visualized using uniform manifold approximation and projection (UMAP) [19]. The “DotPlot” function was then used to visualize the expression level of marker genes in a single cluster [20]. These clusters are assigned to known cell lineages by marker genes. Clusters of cells are identified using the K-nearest neighbor (KNN) algorithm and the “FindClusters” function with a resolution of 0.2.
Identification of candidate NAC marker genes and their cell expression activity
In the discovery cohort (GSE25055) with bulk transcriptome data, the differentially expressed genes (DEGs) between NAC resistant and sensitive groups were identified using the R package limma, with |log2FC |>0.585 and FDR < 0.05. For single-cell data, cell-specific genes were first identified using “FindAllMarkers” function in R package Seurat [18]. Then we obtained the intersection of NAC associated DEGs and cell-specific genes as the candidate NAC marker genes.
We used the R package AUCell (Version 1.12.0) to calculate the expression score of candidate NAC marker genes in each cell. The AUC value estimates the proportion of highly expressed genes in the gene set within each cell, and establishes a gene expression ranking for each cell. To calculate the threshold, we used the “AUCell_explore Thresholds” method.
Construction of NAC prognosis model
Univariate Cox regression analysis was utilised to screen the prognostic value of candidate NAC marker genes. Then the least absolute shrink-age and selection operator (LASSO) Cox regression model were used to construct a prognostic model to minimize the risk of over-fitting and reduce the redudant factors [21]. LASSO algorithm selects and contracts variables through R packet glmnet.
The risk score of patients was calculated according to the expression level of each prognostic related gene and its corresponding regression coefficient:
risk score =\(\sum _{i=1}^{n}{{exp}_{i}}^{*}{\beta }_{i}^{}\),
Where n is the number of prognostic genes, expi is the expression value of gene i, βi is the regression coefficient of gene i. According to the median risk score, the patients were divided into high-risk and low-risk group.
Assessment of the relevance with clinical variables
According to different clinicopathological characteristics, patients were divided into different subgroups, including age (> 50 and < = 50), grade (1/2 and grade 3/4), stage (I-II and III-IV), etc. Fisher’s exact test was used to compare the differences of each clinical variable between high- and low-risk groups, p < 0.05 was the significance threshold.
Estimation of immune cell infiltration in the TME
The single sample gene set enrichment analysis (ssGSEA) algorithm was used to quantify the relative abundance of each cell infiltration in the TME of BC patients [22]. We obtained the signature gene sets indicating a wide range of human immune cell subtypes from Charoentong’s research, including activated CD8 T cells, activated dendritic cells, macrophages, natural killer T cells, and regulatory T cells [23]. Also, the R package ESTIMATE was used to calculate the immune score and ESTIMATE score.
Chemotherapy sensitivity analysis
First, the IC50 values of drugs for each sample in the discovery cohort were calculated based on Genomics of Drug Sensitivity in Cancer (GDSC) (https://www.cancerrxgene.org) resource using calcPhenotype method from R package oncoPredict. To assess the correlation between small-molecule drug sensitivity and risk score, we calculated the Spearman correlation between risk score and drug IC50 values, and compared the differences of drug IC50 values between the high- and low-risk groups [24, 25].
Statistical analysis
All statistical analyses were performed using R software (version 4.0.0; https://www.R-project.org). The difference of immune cell infiltration was assessed by Wilcoxon rank-sum test. For survival analysis, univariate and multivariate Cox analyses were used to explore the prognostic value and independence. R package survminer was used. Kaplan-Meier (KM) curves were plotted to visualize differences in overall survival (OS) between groups, and log-rank tests were used to assess the significance of these differences. Time-dependent Receiver Operating Characteristic (ROC) curve analysis was employed to evaluate the sensitivity and specificity of the risk score in predicting prognosis, and using the R package timeROC. Functional enrichment analysis was performed using the R package cluster Profiler.
Result
Single-cell analysis revealed microenvironment heterogeneity of BC
Firstly, to investigate the heterogeneity of TME in BC patients, we obtained scRNA-seq data from 14 BC patients. After preprocessing and quality control, 44,024 cells were screened and UMAP analysis was performed to visualize the high-dimensional scRNA-seq data (Fig. 1A). Cell clustering revealed 15 subclusters (Fig. 1B), which were further annotated to 8 cell types based on marker genes expression (Fig. 1C, D). The top 5 highly expressed genes in each cell type were shown in Fig. 1E. It was worth noting that the cell composition of TME was highly heterogeneous, and the proportion of 8 cell types varied greatly among samples (Fig. 1F, G).

Analysis of cell subsets of single-cell RNA sequencing (scRNA-seq) from Breast Cancer (BC) patients. A. All the cell samples showed no significant batch effect; B. UMAP plot represents 15 cell clusters from 14 BC patients; C. The average expression of cell type marker genes in eight different cell types; D. UMAP plot represents the final identified eight cell types from (different colors represent different cell types) 14 BC patients; E. The expression of the top five highly expressed genes in each cell type; F. The barplot shows the total number of cell samples from each BC patient; G. The proportion of different cell types in each BC patient
Neoadjuvant chemotherapy signature identification through integrated single-cell and bulk RNA-seq data
Due to the lack of single-cell data from BC patients received NAC, we first identified NAC response-related genes based on bulk RNA-seq. Transcriptome data and clinical information from 306 BC patients underwent NAC were obtained from the GEO database (GSE25055), of which 57 patiences achieved pCR and 249 patiences were therapy-resistant. Then, 551 DEGs between the NAC sensitive group and resistant group were identified based on the R package limma (Fig. 2A, |log2FC| > 0.585, FDR < 0.05). As shown in Fig. 2B, among the top 50 DEGs, CA12 and TFF3 were found to be significantly increased in BC patiences who often showed worse NAC efficacy. The high and correlated expression of CA12 and TFF3 in estrogen receptor-positive BC may play a role in reducing the tumor’s sensitivity to chemotherapy drugs such as adriamycin and docetaxel, which in turn can negatively impact the efficacy of NAC [26].

Identification of NAC-related differentially expressed genes and enrichment analysis. A: Volcano plot compares the differentially expressed genes (DEGs), screened by the criteria of |log2FC|>0.585 and p < 0.05, the blue dots denotes the down-regulated DEGs and the red dots denotes the up-regulated DEGs; B: Heatmap of DEGs indicating the expression of the top 50 DEGs in NAC resistant and sensitive groups, each row represents one DEG and each column represents one sample. The red and blue colors represent up-regulated and down-regulated DEGs respectively; C: GO-Biological process (BP) enrichment analysis results of NAC-related DEGs; D: GO-Cellular component (CC) enrichment analysis results of NAC-related DEGs; E: GO-Molecular function (MF) enrichment analysis results of NAC-related DEGs; F: Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis results of NAC-related DEGs, the dot size represents the count of DEGs, and the color depth represents the p-value based significance; G: Expression of KRT7 in different cell clusters; H: Expression of AEBP1 in different cell clusters; I: Expression of BCL2A1 in different cell clusters; J: Expression of RGS1 in different cell clusters; K: AUC histogram for cell activity score of candidate NAC marker genes. The threshold was set as 0.16, and 9297 cells exceeded the threshold; L: The UMAP map is based on the candidate NAC marker genes score of each unit. Cell clusters with high ISG scores are highlighted
To further obtain NAC response-related genes that vary among different cell types, we identified190 intersecting key genes that were both NAC-response DEGs and highly variable genes of single-cell data. GO enrichment analysis revealed that these key genes are mainly distributed in the extracellular matrix and spindle and participate in the cell processes such as chromatid separation, cell cycle, mitosis, etc. By binding with chemokine receptors or regulating protein kinase (Fig. 2C-E). In addition, the results of KEGG [27–29] enrichment analysis showed that they were mainly involved in cell cycle, cellular senescence, and P53 signal pathways, among others (Fig. 2F).

Construction and verification of NAC prognostic risk model. A: Forest map shows the results of the univariate analysis of the top 10 NAC-related key genes; B: LASSO coefficient profiles of the prognostic genes. Change track of each independent variable in LASSO Cox regression analysis. The horizontal axis represents the log value of the independent variable lambda, and the vertical axis represents the coefficient of the independent variable; C: Cross-validation for turning parameter selection in the LASSO regression model. Two vertical dashed lines indicated the optimal values using the minimum criteria. Optimal genes with the best discriminative capability were selected for developing the Prognostic model; D: LASSO regression coefficients of the 9 optimized genes for constructing the prognostic model; E: Risk score distribution for each sample in the GSE25055 cohort; F: Kaplan-Meier curves comparing the OS of patients separated by risk groups, the red line represents line the high-risk score group, while the blue line represents the low-risk score group; G: Expression of the 9 prognostic model genes in the GSE25055 cohort between high- and low-risk group; H: Time-dependent receiver operating characteristic (ROC) curve of the prognostic model
In addition, we observed the cell-specific expression pattern of the 190 key genes of NAC response. Among them, KRT7 was highly expressed in cancer cells, AEBP1 was mainly expressed in fibroblasts, BCL2A1 and RGS1 were both highly expressed in myeloid cells (Fig. 2G-J). Specifically, RGS1 was also specifically expressed in T cell clusters, and a previous study has supported that RGS1 was associated with CD4 expression and was functionally associated with T cell activation [30]. The expression activity of the 190 key genes was calculated using the R package AUCell and showed a bimodal distribution among all cells (Fig. 2K). According to the bimodal distribution threshold of 0.16, all cells were divided into two groups of high and low activity. The results showed that 9297 cells (9297/44,024, 21%) had higher expression activity of NAC response-related genes, mainly including fibroblasts, cancer cells, and cycling cells (Fig. 2L). Further enrichment analysis of cell-specific genes showed that their functions were mainly related to oxidative phosphorylation, energy metabolism and DNA replication (Fig. S1).
Construction and validation of NAC prognostic model
To assess the prognostic value of key NAC response-related genes in patients with BC, univariate Cox analysis and log-rank test was performed in 306 BC patients underwent NAC (GSE25055) for 190 genes. The results showed that 126 genes were associated with OS in BC (P < 0.05), and the top 10 genes with the most significant p values were shown in Fig. 3A and S2. Among them, GATA3 is a key transcription factor involved in the development of breast tumors. Here, we found the higher expression of GATA3 indicated a better prognosis, which was supported by a previous analysis that GATA3 was required for homologous recombination repair and served as a tumor suppressor [31].
In order to construct a promising prognostic model based on the NAC response-related genes, we performed LASSO-Cox analysis to remove redundant prognostic factors (Fig. 3B, C), the resulting prognostic model, when applied to patients, can effectively predict OS outcomes. And nine prognostic genes related to NAC response were retained to construct the model, including NDRG1, CXCL14, HOXB2, NAT1, EVL, FBP1, MAGED2, AR and CIRBP (Fig. 3D). According to their expression levels, we measured the prognostic risk score for each patient and separated them into two groups based on the median score (Fig. 3E). K-Mcurve with the log-rank test further showed a significant reduction in OS in the high-risk score group (log-rank test p-value < 0.001, Fig. 3F). The nine prognostic genes related to NAC response had distinct expression patterns in different risk groups. NDRG1 was the only gene that up-regulated in the high-risk group, while the other eight genes were up-regulated in the low-risk group (Fig. 3G). ROC curve analysis was used to evaluate the prediction efficiency of the NAC prognostic model. And the area under the ROC curve (AUC) reached 0.804, 0.762 and 0.704 at 1, 3 and 5 years, respectively, indicating that the prediction effect of the model is reliable (Fig. 3H).

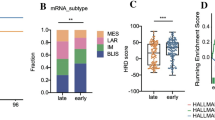
Clinicopathological significance of NAC response-related risk score in clinical variables.A: Differences in NAC response-related risk scores between age group; B: Differences in NAC response-related risk scores across different tumor grade; C: Differences in NAC response-related risk scores within different TNM stage; D: Differences in NAC response-related risk scores in different intrinsic subtypes; E: Differences in NAC response-related risk scores across ER status; F: Differences in NAC response-related risk scores across PR status; G: Differences in NAC response-related risk scores across HER2 status; H: Univariate cox analysis of risk score in the GSE25055 cohort; I: Multivariate cox analysis of risk score in the GSE25055 cohort; J: K-M curve of NAC prognosis model in different groups of age
In addition, we assessed the robustness and generalizability of the NAC prognostic model in two independent validation cohorts. Consistently, we calculated the risk score for each patient and separated them as the discovery cohort did. As expected, the high-risk group showed significantly reduced OS compared to the low-risk group (log-rank p-value < 0.001 for GSE25065 and GSE22226, respectively). It’s worth noting that the prognostic model also showed good predictive performance in external validation sets with the predictive AUC reached 0.87 and 0.753 respectively (Fig. S3).
Association of NAC risk scores with clinical variables
To further investigate the clinicopathologic significance of the NAC response-related risk score, we separated 306 patients by clinicopathologic variables and compared the difference in risk scores between groups. As expected, the risk score was significantly associated with tumor grade and TNM stage, indicating that as the disease progresses, the NAC response-related risk score increases (Wilcoxon rank-sum test p-value < 0.001, Fig. 4A, B). However, we didn’t observe its association with age (Fig. 4C). As for the three indicators of triple-negative breast cancer (TNBC), the risk score was significantly higher in ER and PR-negative groups (Wilcoxon rank-sum test p-value < 2.2e-16, Fig. 4D, E), while it didn’t show a relationship with Her-2 status, probably due to the limited sample size of Her-2+ patients (Fig. 4F). Simultaneously, among the intrinsic subtypes (PAM50) of BC, the basal-like (also considered as TNBC) subtype showed the highest risk score compared other groups (Fig. 4G). The two independent validation cohorts (GSE25065 and GSE22226) supported the clinicopathologic associations of NAC response-related risk model (Fig. S4). These results suggested that the NAC response-related risk score reflected a worse prognosis of TNBC [32].
In addition, we investigated the independence and predictive efficiency of the NAC response-related risk score in the 306 BC patients underwent NAC (GSE25055). Univariate Cox regression analysis revealed the prognosis associations (with OS) of the risk score, ER and PR status, AJCC stage, and tumor grade (HR = 4.98, 95% CI = 2.74–9.03, P < 0.001, Fig. 4H). Then, the multivariate Cox regression analysis showed that the risk score remained an independent predictor of OS after adjusting for other confounding factors(HR = 3.40, 95% CI = 1.34–8.65, P = 0.01, Fig. 4I). These results were also found in the two independent validation cohorts (Fig. S5A, B). To explore the applicability of the NAC prognostic model, we compared its prognosis associations in different clinical features groups. In particular, there was a strong prognostic effect of the NAC prognostic model regardless of whether the risk score was associated with this clinical variable (log-rank P < 0.05, Fig. 4J, S5C-F).
NAC risk score characterized differential tumor hallmarks and microenvironment features
In order to explore the underlying molecular mechanisms of this NAC response -related risk score, we first investigated the cancer hallmarks associated with the risk score. We calculated the activity score of 50 cancer hallmark pathways for 306 BC patients underwent NAC. The results indicated that 41 of the 50 pathways had significant differences between the high- and low- risk groups (Fig. 5A). For instance, the interferon-α/γ response, inflammatory response, Wnt-β catenin signaling, glycolysis, apoptosis, KRAS signaling, and hypoxia hallmarks were significantly activated in the high-risk group, whereas DNA repair, oxidative phosphorylation, and fatty acid metabolism hallmarks were significantly activated in the low-risk group.

The correlation between NAC risk score of patients and characterization of the immune microenvironment. A: Heat plot of the activity of cancer hallmarks in the GSE25055 cohort between high- and low-risk group; B: Spearman correlation between the NAC response-related risk score and the immune score (R = 0.27; p < 0.01); C: Spearman correlation between the NAC response-related risk score and the ESTIMATE score; D: The proportions of differentially infiltrated TME immune cells between the high- and low-risk score groups;
E: Top 6 drugs with the highest positive correlation with risk score; F: Comparison of log2 (IC50) values of the top 6 positively correlated drugs between the high- and low-risk score groups; G: Top 6 drugs with the highest negative correlation with risk score; H: Comparison of log2 (IC50) values of the top 6 negatively correlated drugs between the high- and low-risk score groups
As the risk score showed a relationship with inflammatory response, we further explored the association between the risk score with immune response. Using the ESTIMATE algorithm, we found that the NAC response-related risk score was slightly positively correlated with immune and ESTIMATE score of the TME (Spearman correlation analysis, Fig. 5B, C). Additionally, we analyzed the difference in immune cell infiltration between the high- and the low- risk groups. To our surprise, activated B cell, CD4 T cell, CD8 T cell, dendritic cell and effector memory CD8 T cell was significantly accumulated in the high-risk group (Fig. 5D). Furthermore, the immune-suppressive cell types including MDSC and Regulatory T cell were increased in the high-risk group. Previous studies have shown that tumors with immune-excluded phenotypes which associated with immune cells embedded in the surrounding tumor stroma away from tumor cells, also have higher immune infiltration, but whether the effector cells were surrounding the tumor or suppressed by the microenvironment is unclear [33].
NAC risk score and chemotherapy drug sensitivity
To further assess the potential impact of the risk score on drug response, we investigated drug response and potential therapeutic compounds based on the NAC response-related risk score. The spearman correlation between IC50 values and the risk score revealed the top 6 positive and top 6 negative correlated drugs (Fig. 5E, G). For the positively correlated drugs, the IC50 of Doramapimod, Elephantin, AZD2014, SB505124 and PRIMA-1MET differed significantly between the high and low groups (Fig. 5F). For negatively related drugs, there also was a substantial difference between the high- and low-risk groups (Fig. 5H). These results demonstrated that the risk score can be applied to drug resistance analysis.
Discussion
BC has the highest incidence rate among female malignant tumors, accounting for 25% of the total incidence rate [34]. NAC is a treatment strategy that involves administering chemotherapy before surgery. Its goal is to reduce tumor volume and aggressiveness, leading to improved surgical resection rates and therapeutic effects. In the case of BC, NAC offers multiple benefits [35, 36]. However, the lack of effective prognostic biomarkers for NAC presents a significant challenge to its clinical application in BC [37]. To address this challenge, we conducted a characterization of 190 key prognostic genes related to NAC. This characterization was achieved by integrating bulk RNA-seq data from 306 BC patients (including 57 with pathologic complete response and 249 resistant cases) and single-cell RNA-seq data from 14 BC patients. Through LASSO-Cox analysis, we identified nine NAC-related prognostic signatures. Our study then encompassed a comprehensive analysis of both the training cohort, GSE205055, and the validation cohorts, GSE25065 and GSE22226. This analysis enabled us to establish and validate, for the first time, a prognostic risk model specific to NAC response. Univariate and multivariate Cox regression analyses further confirmed that this prognostic risk model represents an independent risk factor associated with OS. Additionally, we evaluated the prognostic model’s association with various clinical variables. The establishment of this NAC response-related prognostic risk score will empower clinicians to make more informed decisions regarding personalized treatment strategies and follow-up care. Ultimately, this model has the potential to significantly improve survival rates and treatment outcomes for patients with BC.
The pathogenesis of BC is a complex process influenced by various genetic and environmental factors [38]. When integrating bulk-RNA seq and scRNA-Seq screening signatures, the key difference from other methods like whole-genome sequencing (WGS) or whole-exome sequencing (WES) is that it focuses solely on gene expression rather than genomic variations or mutations. While WGS and WES provide valuable insights into DNA sequence variations, they do not directly capture gene expression profiles, making it difficult to infer functional differences at the transcriptional level. By combining bulk-RNA seq and scRNA-Seq screening signatures, we can unravel the underlying regulatory mechanisms and gain a deeper understanding of how specific genes and pathways contribute to the observed cellular heterogeneity.Through our research, we have gained insights into the potential roles of NAC-related genes in regulating multiple cellular processes associated with BC development and progression. Our KEGG enrichment analysis further indicates that the differentially expressed genes identified in our study may be involved in regulating biological processes, such as cellular aging and the P53 signaling pathway. These pathways have been implicated in various types of cancer, suggesting that the differentially expressed genes identified in our study could play crucial roles in BC pathogenesis [39–41]. Notably, the observation of high expression of NAC-related genes in fibroblast, cancer cell, and cycling cell types is particularly significant, as these cell types are known to have important roles in BC pathogenesis [42–45].
Moreover, our scRNA-seq analysis revealed a highly heterogeneous cell composition within TME of BC, with significant variations in the proportions of eight distinct cell types across samples. Notably, sample sc5Rjuq064 exhibited a decreased proportion of T cells and an increased proportion of tumor cells compared to other samples, which correlated with the tumor stage. Through enrichment analysis, we found that the differentially expressed genes in these three active cell types primarily relate to oxidative phosphorylation, energy metabolism, and DNA replication. Interestingly, contrary to previous research, cancer-related fibroblasts appear to act as direct positive regulators of the adaptive immune system, suggesting the potential use of immune-stimulating CAF in cancer treatment [46]. Zheng et al.‘s research implies that combining chemotherapy with anti-cancer-related fibroblast therapy may enhance the effectiveness of T cell-based immunotherapy, providing a potential strategy for colon cancer treatment [47]. The role of fibroblasts in BC warrants further investigation. Overall, our research enhances the understanding of BC pathogenesis by shedding light on the specific genes and cellular processes involved in its development and progression. These findings have the potential to contribute to the development of novel diagnostic and therapeutic approaches for BC.
In this study, LASSO-Cox regression analysis identified nine prognostic genes associated with NAC response: NDRG1, CXCL14, HOXB2, NAT1, EVL, FBP1, MAGED2, AR, and CIRBP. Among these genes, AR has been found to stimulate breast tumor growth in the absence of the estrogen receptor, making it a promising molecular target in the treatment of TNBC [48]. Dong et al. designed a novel combination therapy using enzalutamide and ceritinib to target both androgen-dependent and androgen-independent AR signaling pathways in TNBC tumors [49]. CIRBP, known for its ability to bind and post-transcriptionally regulate mRNA, has been linked to cancer promotion and inflammation [50, 51]. Recent studies have identified CST3 as a downstream mediator for CIRBP functionality [52]. The melanoma-associated antigen (MAGE) family proteins are recognized tumor-specific antigens. MAGED2 exhibits distinct effects depending on the subtype of breast cancer. It has been identified as a potential prognostic factor for wild-type TP53 patients and breast cancer patients with varying pathological grades [53].
Our research also investigated the potential utility of the NAC response-related risk score in predicting drug response to chemotherapy in breast cancer (BC). SB505124, a small molecule inhibitor of TGF-β receptor I (ALK5), was examined in this context. We observed that BC patients in the low NAC risk group exhibited significantly lower IC50 values for SB505124 compared to those in the high NAC risk group. This suggests that patients in the low NAC risk group may be more responsive to SB505124 treatment. Notably, in conjunction with SMAD3, SB505124 can attenuate the activity of CD8 + T cells, which may contribute to the reduced efficacy of immunotherapy in malignant BC [54]. These findings emphasize the potential of risk scoring as a valuable tool for predicting drug response and optimizing therapy in BC patients, ultimately leading to more effective and personalized treatment approaches. However, the clinical classification of breast cancer has a great influence on the treatment effect of NAC. Although we validated the applicability of our model in different clinical classification of breast cancer, further studies are needed.
In conclusion, our study integrated single-cell and bulk RNA sequencing analyses to construct and validate a nine-gene signature associated with NAC prognosis. This signature serves as an independent prognostic indicator for BC patients. Additionally, our findings provide genomic evidence for future research directions in developing anti-BC treatment strategies, particularly for individuals who may not benefit from NAC.
Data Availability
The datasets GSE25055, GSE25065 and GSE22226 supporting the findings of this study are available in the from GEO (https://www.ncbi.nlm.nih.gov/geo/). For the discovery cohort databases. Further inquiries can be directed to the corresponding author.
References
Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J Clin. 2018;68(6):394–424. https://doi.org/10.3322/caac.21492.
Epidemiology of Breast Cancer
Harbeck N. Neoadjuvant and adjuvant treatment of patients with HER2-positive early Breast cancer. Breast (Edinburgh Scotland). 2022;62(Suppl 1):12–s16. https://doi.org/10.1016/j.breast.2022.01.006.
Petruolo O, Sevilimedu V, Montagna G, et al. How often does modern Neoadjuvant Chemotherapy Downstage patients to breast-conserving Surgery? Ann Surg Oncol. 2021;28(1):287–94. https://doi.org/10.1245/s10434-020-08593-5.
Denduluri N, Miller K. O’Regan RMJASoCOEB using a Neoadjuvant Approach for evaluating Novel therapies for patients with Breast Cancer. 2018(38):47–55.
Nicolini A, Ferrari P, Duffy MJ. Prognostic and predictive biomarkers in Breast cancer: past, present and future. Sem Cancer Biol. 2018;52(Pt 1):56–73. https://doi.org/10.1016/j.semcancer.2017.08.010.
Li J, Zhang B, Yang YF et al. Aldehyde dehydrogenase 1 as a predictor of the neoadjuvant chemotherapy response in Breast cancer a meta-analysis. 2019.
Wang RX, Chen S, Huang L, Shao ZMJBC. Predictive and prognostic value of Matrix metalloproteinase (MMP) – 9 in neoadjuvant chemotherapy for triple-negative Breast cancer patients. 2018, 18(1).
Mohammad N, Singh SV, Malvi P et al. Strategy to enhance efficacy of doxorubicin in solid Tumor cells by methyl-β-cyclodextrin: involvement of p53 and Fas receptor ligand complex. 2015, 5:11853.
A Metabolism-Related Gene Signature Predicts the Prognosis of Breast Cancer Patients: Combined Analysis of High-Throughput Sequencing and Gene Chip Data Sets
Pease AM, Riba LA, Gruner RA, Tung NM. James TA Oncotype DX(®) recurrence score as a predictor of response to Neoadjuvant Chemotherapy. Ann Surg Oncol. 2019;26(2):366–71. https://doi.org/10.1245/s10434-018-07107-8.
Luyendijk M, Jager A, Buijs SM, et al. Cost-effectiveness analysis of MammaPrint(®) to Guide the Use of Endocrine Therapy in patients with early-stage Breast Cancer. PharmacoEconomics. 2023;41(8):981–97. https://doi.org/10.1007/s40273-023-01277-4.
G-Protein-Coupled Estrogen Receptor Enhances the Stemness of Triple-Negative Breast Cancer Cells and Promotes Malignant Characteristics
PD-1 Relevant Genes Predict the Prognosis of Breast Cancer and Their Prediction Effect in Tumor Response to Immunotherapy
Papalexi E, Satija RJNRI. Single-cell RNA sequencing to explore immune cell heterogeneity. 2017.
Kim KT, Lee HW, Lee HO, et al. Single-cell mRNA sequencing identifies subclonal heterogeneity in anti-cancer drug responses of lung adenocarcinoma cells. Genome Biol. 2015;16(1):127. https://doi.org/10.1186/s13059-015-0692-3.
Kyu-Tae, Kim, Won H et al. Single-cell mRNA sequencing identifies subclonal heterogeneity in anti-cancer drug responses of lung adenocarcinoma cells. 2015.
Qian J, Olbrecht S, Boeckx B, et al. A pan-cancer blueprint of the heterogeneous Tumor microenvironment revealed by single-cell profiling. Cell Res. 2020;30(9):745–62. https://doi.org/10.1038/s41422-020-0355-0.
Melit Devassy B, George S, Nussbaum P. Unsupervised clustering of Hyperspectral Paper Data using t-SNE. J Imaging. 2020;6(5). https://doi.org/10.3390/jimaging6050029.
Aran D, Looney AP, Liu L, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. 2019;20(2):163–72. https://doi.org/10.1038/s41590-018-0276-y.
Simon N, Friedman J, Hastie T, Tibshirani R. Regularization paths for Cox’s proportional hazards Model via Coordinate Descent. J Stat Softw. 2011;39(5):1–13. https://doi.org/10.18637/jss.v039.i05.
Bindea G, Mlecnik B, Tosolini M, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39(4):782–95. https://doi.org/10.1016/j.immuni.2013.10.003.
Sturm G, Finotello F, List M. Immunedeconv: an R Package for Unified Access to computational methods for estimating Immune cell fractions from Bulk RNA-Sequencing data. Methods in Molecular Biology (Clifton NJ). 2020;2120:223–32. https://doi.org/10.1007/978-1-0716-0327-7_16.
Geeleher P, Cox NJ, Huang RS. Clinical drug response can be predicted using baseline gene expression levels and in vitro drug sensitivity in cell lines. Genome Biol. 2014;15(3):R47. https://doi.org/10.1186/gb-2014-15-3-r47.
Geeleher P, Cox N, Huang RS. pRRophetic: an R package for prediction of clinical chemotherapeutic response from Tumor gene expression levels. PLoS ONE. 2014;9(9):e107468. https://doi.org/10.1371/journal.pone.0107468.
Shen M, Yang L, Lei T, et al. Correlation between CA12 and TFF3 and their prediction value of neoadjuvant chemotherapy response in Breast cancer. J Clin Pharm Ther. 2022;47(5):609–18. https://doi.org/10.1111/jcpt.13580.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. https://doi.org/10.1093/nar/28.1.27.
Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Science: A Publication of the Protein Society. 2019;28(11):1947–51. https://doi.org/10.1002/pro.3715.
Kanehisa M, Furumichi M, Sato Y, Kawashima M, Ishiguro-Watanabe. M KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023;51(D1):D587–d592. https://doi.org/10.1093/nar/gkac963.
Zhang S, Wang H, Liu J, et al. RGS1 and related genes as potential targets for immunotherapy in Cervical cancer: computational biology and experimental validation. J Translational Med. 2022;20(1):334. https://doi.org/10.1186/s12967-022-03526-0.
Zhang F, Tang H, Jiang Y, Mao Z. The transcription factor GATA3 is required for homologous recombination repair by regulating CtIP expression. Oncogene. 2017;36(36):5168–76. https://doi.org/10.1038/onc.2017.127.
Gong Y, Ji P, Yang YS, et al. Metabolic-pathway-based subtyping of Triple-negative Breast Cancer reveals potential therapeutic targets. Cell Metabol. 2021;33(1):51–64e59. https://doi.org/10.1016/j.cmet.2020.10.012.
Hegde PS, Chen DS. Top 10 challenges in Cancer Immunotherapy. Immunity. 2020;52(1):17–35. https://doi.org/10.1016/j.immuni.2019.12.011.
Liang Y, Zhang H, Song X. Yang QJSicb metastatic heterogeneity of Breast cancer: molecular mechanism and potential therapeutic targets. 2020, 60:14–27 https://doi.org/10.1016/j.semcancer.2019.08.012.
Tadahiko S. Hiroji IJJJoCO Adjuvant and neoadjuvant therapy for Breast cancer. 2020(3):225–9.
Tracy-Ann M, Rachel S, Dang C, Monica MJPC. Overv Breast Cancer Therapy. 2018;13(3):339–54.
Zhang Z, Zhang H, Yu J et al. miRNAs as therapeutic predictors and prognostic biomarkers of neoadjuvant chemotherapy in Breast cancer: a systematic review and meta-analysis. Breast Cancer Res Treat 2022, 194(3):483–505 https://doi.org/10.1007/s10549-022-06642-z.
Wong GL, Manore SG, Doheny DL. Lo HW STAT family of transcription factors in Breast cancer: Pathogenesis and therapeutic opportunities and challenges. Sem Cancer Biol. 2022;86(Pt 3):84–106. https://doi.org/10.1016/j.semcancer.2022.08.003.
Baslan T, Morris JPt, Zhao Z, et al. Ordered and deterministic cancer genome evolution after p53 loss. Nature. 2022;608(7924):795–802. https://doi.org/10.1038/s41586-022-05082-5.
López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: an expanding universe. Cell. 2023;186(2):243–78. https://doi.org/10.1016/j.cell.2022.11.001.
Vaddavalli PL, Schumacher B. The p53 network: cellular and systemic DNA damage responses in cancer and aging. Trends Genet. 2022;38(6):598–612. https://doi.org/10.1016/j.tig.2022.02.010.
Tan Z, Kan C, Sun M, et al. Mapping Breast Cancer Microenvironment through single-cell omics. Front Immunol. 2022;13:868813. https://doi.org/10.3389/fimmu.2022.868813.
Avalle L, Raggi L, Monteleone E, et al. STAT3 induces Breast cancer growth via ANGPTL4, MMP13 and STC1 secretion by cancer associated fibroblasts. Oncogene. 2022;41(10):1456–67. https://doi.org/10.1038/s41388-021-02172-y.
Shan BQ, Wang XM, Zheng L, et al. DCAF13 promotes Breast cancer cell proliferation by ubiquitin inhibiting PERP expression. Cancer Sci. 2022;113(5):1587–600. https://doi.org/10.1111/cas.15300.
Chang CA, Jen J, Jiang S, et al. Ontogeny and vulnerabilities of Drug-Tolerant persisters in HER2 + Breast Cancer. Cancer Discov. 2022;12(4):1022–45. https://doi.org/10.1158/2159-8290.Cd-20-1265.
Tsoumakidou M. The advent of immune stimulating CAFs in cancer. Nat Reviews Cancer 2023 https://doi.org/10.1038/s41568-023-00549-7.
Zheng H, Liu H, Ge Y, Wang X. Integrated single-cell and bulk RNA sequencing analysis identifies a cancer associated fibroblast-related signature for predicting prognosis and therapeutic responses in Colorectal cancer. Cancer Cell Int. 2021;21(1):552. https://doi.org/10.1186/s12935-021-02252-9.
Mahtani R, Kittaneh M, Kalinsky K, et al. Advances in therapeutic approaches for triple-negative Breast Cancer. Clin Breast Cancer. 2021;21(5):383–90. https://doi.org/10.1016/j.clbc.2020.12.011.
Dong S, Yousefi H, Savage IV, et al. Ceritinib is a novel triple negative Breast cancer therapeutic agent. Mol Cancer. 2022;21(1):138. https://doi.org/10.1186/s12943-022-01601-0.
García-Cárdenas JM, Guerrero S, López-Cortés A, et al. Post-transcriptional regulation of Colorectal Cancer: a focus on RNA-Binding proteins. Front Mol Biosci. 2019;6:65. https://doi.org/10.3389/fmolb.2019.00065.
Moore S, Järvelin AI, Davis I, Bond GL, Castello A. Expanding horizons: new roles for non-canonical RNA-binding proteins in cancer. Curr Opin Genet Dev. 2018;48:112–20. https://doi.org/10.1016/j.gde.2017.11.006.
Indacochea A, Guerrero S, Ureña M, et al. Cold-inducible RNA binding protein promotes Breast cancer cell malignancy by regulating cystatin C levels. RNA (New York NY). 2021;27(2):190–201. https://doi.org/10.1261/rna.076422.120.
Jia B, Zhao X, Wang Y, et al. Prognostic roles of MAGE family members in Breast cancer based on KM-Plotter Data. Oncol Lett. 2019;18(4):3501–16. https://doi.org/10.3892/ol.2019.10722.
Xie F, Zhou X, Su P, et al. Breast cancer cell-derived extracellular vesicles promote CD8(+) T cell exhaustion via TGF-β type II receptor signaling. Nat Commun. 2022;13(1):4461. https://doi.org/10.1038/s41467-022-31250-2.
Acknowledgements
Not applicable.
Funding
This study was funded by Shanxi Province “136 Revitalization Medical Project Construction Funds”.
Author information
Authors and Affiliations
Contributions
Xiaojun Zhang, Ran Feng designed the research; Xiaojun Zhang, Junbin Guo and Ran Feng performed interpreted the results; Xiaojun Zhang, Lihui Pan and Yarong Yao performed the experiments and analyzed the results; Jinnan Gao drafted the manuscript; Xiaojun Zhang revised the manuscript and gave the final approval of the version to be published. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, X., Feng, R., Guo, J. et al. Integrated single-cell and bulk RNA sequencing analysis identifies a neoadjuvant chemotherapy-related gene signature for predicting survival and therapy in breast cancer. BMC Med Genomics 16, 300 (2023). https://doi.org/10.1186/s12920-023-01727-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-023-01727-0