
Abstract
Ichthyoses are a heterogeneous group of cornification disorders. The most common form of ichthyoses is ichthyosis vulgaris (IV) ([OMIM] #146,700), which can be inherited as autosomal semi-dominant mutation in the filaggrin gene (FLG). We present the findings of a study involving 35 Saudi patients with a clinical diagnosis of ichthyosis vulgaris. For identifying the pathogenic mutation of their disease, we used Sanger sequencing analysis of the extracted DNA samples. We also identified the underlying 22 FLG variants, which have been seen before. However, the detected mutations do not involve the common p.R501* c. 2282del4 mutations reported in European populations. Indeed, we did not identify any statistical influence of the homozygous or heterozygous genotypes on the phenotype severity of the disease.
Similar content being viewed by others


Background
Ichthyoses are a heterogeneous group of cornification disorders characterized by thickening of the stratum corneum, scaling, xerosis, and compromised perspiration. It can be classified into either a nonsyndromic or syndromic disease based on whether it is confined to the skin [1]. Several authors have developed classification systems based on genetic mutations, the mode of inheritance, clinical findings, the concept of retention versus hyperproliferative keratosis, or based on biochemical information [2,3,4,5].
The most common form of ichthyoses is ichthyosis vulgaris (IV) ([OMIM] #146,700), which is inherited in an autosomal semi-dominant manner [6, 7]. Indeed, the filaggrin (filament aggregating protein, FLG; #135,940) gene can be mutated in patients with IV. The gene FLG encodes a 400 kDa histidine-rich protein consisting of 10–12 repeats, each comprising 324 amino acids and short linkers, that is essentially involved in the cornification process of the cornified cell envelope [8,9,10,11]. Notably, the mutated gene is mapped to chromosome 1q21 within a cluster of markers in the epidermal differentiation complex region [12, 13]. The gene comprises three exons: exon one is noncoding, exon 2 contains the translation initiation codon, and exon 3 encodes a significant part of the profilaggrin protein [14].
In 2002, Compton et al. [12] mapped IV in a multi-generational family to the epidermal differentiation complex, including FLG. In 2006, Smith et al. identified a homozygous p. R501* mutation and p. R501*/c. 2282del4 compound heterozygous mutations in FLG in 15 families with a severe IV phenotype. The c. 2282del4 mutation they identified leads to a premature termination codon, stopping FLG translation. Screening IV families demonstrated high frequencies of these variants in patients of European ancestry [8].
Furthermore, in 2006, Palmer et al. showed that the two loss-of-function FLG variants, p. R501* and c. 2282del4, strongly predispose patients to atopic dermatitis. Atopic diathesis association with ichthyosis vulgaris is well established, as approximately 50% of IV patients develop atopic dermatitis [15,16,17].
Moreover, in 2006, Sandilands et al. described 15 variants of FLG to facilitate the genetic analysis, where European variants are either non-prevalent or rare. These variants they identified have resulted in a loss of FLG function [18].
In another study in 2007, Nomura et al. reported two other FLG variants, p. S2554* and c. 3321delA [19].
This study describes the clinical and genotypic data of 35 Saudi patients diagnosed clinically with ichthyosis vulgaris. Notably, we identified 22 common FLG variants mutated in our study population, all of which were missense variants that have been seen before.
Materials and methods
Ethical compliance
This descriptive-analytic study involving human participants obtained ethical approval from the Institutional Research Board of Imam Abdulrahman Bin Faisal University, Dammam. All study participants signed written informed consent forms.
Patients and phenotypes
Saliva samples were collected from 35 individuals diagnosed clinically with ichthyosis vulgaris by experienced dermatologists. The OrageneTM.DNA collection kit (OG-500 Disc format, DNA Genotek Inc, Ottawa, Ontario, Canada) was used for collecting saliva samples by trained nurses.The patients were asked to fast for 30 min before the collection. Fifteen minutes before sample collection, the patients were asked to rinse their mouths with water. The patients were then asked to rub the inside of their mouths with their tongues for 15 s and to spit the saliva into an empty container until the amount of liquid saliva (not the bubbles) reached the level shown on the collection container. After that, the container was sealed, labeled, and gently shaken for 10 s to mix the saliva with the Oragene solution. For the DNA extraction, a standard quantity of 2.0 ml Oragene/saliva mixture was used as the manufacturer’s protocol recommended.
A standard form was prepared for collecting patient data, including age, sex, ethnicity, parents’ consanguinity, coexisting atopy, degree of scaling and skin dryness, and inheritance pattern according to the family pedigree.
Genotyping
Genomic DNA was extracted using prepIP-L2P (DNA Genotek, Ottawa, ON, Canada) per the manufacturer’s protocol, and purity and quantity were checked using NanoDrop 8000 (Thermo Fisher Scientific Inc., Massachusetts, United States). PCR overlapping method was used to amplify target exons using conventional cycle settings and in-house developed primers. Regions of FLG gene were amplified individually in Bio-Rad MyCycler™ (Bio-Rad, Berkeley, CA, USA) (95 °C/4 minutes; 30 cycles of 95 °C/1 minute; annealing/1 minute; 72 °C/1.5 min; and final extension 72 °C/5 minutes) using amplification primers listed in Table 1, with AmpliTaq Gold DNA Polymerases (Thermo Fisher Scientific Inc., Massachusetts, United States). All the primers were synthesized from Applied Biosystems (Life Technologies Corporation, USA). The purified (QIAquick PCR Purification Kit, Qiagen, Germany) amplicons were cycle sequenced with forward and revere primers separately, as listed in Table 1, using a BigDye Terminator Cycle Sequencing Kit (Life Technologies Corporation, USA). The purified cycle sequenced products from forward and revere primers separately were sequenced using POP 7 in a Genetic Analyzer 3500 (Life Technologies Corporation, USA).
The DNA sequences obtained through Sanger Sequencing for all the patients were analyzed with SeqmanPro (DNASTAR Inc., Madison, WI, USA) [20]. Unknown missense variants were considered possibly pathogenic if they were anticipated to be harmful by at least two of the algorithms MutTaster [21], SIFT [22], and PolyPhen2 [23], impacted highly conserved amino acids and were not discovered as homozygous variants in control DNA sequences(www.internationalgenome.org). Human Splicing Finder 3.0 examined splice site variants [24]. PFAM was used to identify protein domains [25].
Statistical analysis
The collected data were entered into a computer database and analyzed statistically using Excel software [26]. We used the chi-square test for performing a logistic regression analysis to examine the influence of our descriptive genotype variables (homozygosity or heterozygosity of the FLG variant) on each of the phenotype variables severity (scale severity, palmer hyper linearity, and pruritus). The chi-square was calculated using the formula:
χ²= ∑ [(observed frequency – expected frequency)²/expected frequency].
Results
Patient characteristics
A total of 35 patients with ichthyosis vulgaris were included in this study; 22 (62.9%) were males, and 13 (37.1%) were females, giving a male-to-female ratio of 1.7:1. All patients were Saudis; their mean age was 21.1 ± 13.3. All patients’ symptoms presented after ten weeks of birth. There were 25 (68%) consanguineous marriages among the parents. Thirty-two (91.9%) patients had other affected family members with IV. (Figures 1 and 2)

Other affected family members

Parent consanguinity
Clinical findings
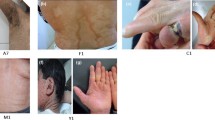
All patients had Fitzpatrick skin color type 4 and mild to moderate skin dryness. The majority of our patients had fine scales. The scales vary in severity and colors from delicate light brown in 84.5% of patients to coarse, large brown in 15.15%; most patients (84.8%) had scales on the neck, trunk, or extremities, while only a minority of them (5.4%) showed a generalized distribution of the scales. Flexures were spared in 20 (57%) patients. 40% of our patients had dental abnormalities, either in the form of caries (28.5%), abnormal occlusion (2.8%), or a combination of both (8.5%).
Plantar keratoderma was seen in 18 (51.2%) patients and palmoplantar keratoderma was seen in 4 (11.4%). Palmar hyperlinearity was encountered among 28 (80%) IV patients, while pruritis was discovered in 14 (40%). Their demographic data are outlined in Table 2.
Non-cutaneous coexisting disorders were identified in a minority of the patients, including sickle cell anemia (5.7%) or G6PD deficiency (2.8%). However, they might represent a confounding factor.
Most of our patients were treated with topical measures and were well-controlled; however, 8.5% required systemic treatment. We treated those patients with oral acitretin (neotigason capsules, Teva UK), whereas 66.6% of the treated patients required low-dose acitretin ranging between 5 and 10 mg/day, and one patient needed a higher dose of acitretin 20–30 mg/day for their disease control. The decision to use low-dose systemic retinoids versus higher doses is based on the symptomatology and severity of the clinical findings. Patients with milder, fine asymptomatic scales and palmoplantar keratosis were treated with low-dose retinoids, while those with pruritic larger, coarser scales were treated with higher doses. Fortunately, two patients responded well to systemic retinoids and tolerated continued use for over three years. The scale severity, hyperkeratosis thickness, and pruritus were improved with the treatment use. Topical treatments were given alone for minor cases or as an adjuvant to systemic therapies for severe patients. The topical choices were selected based on the involved areas, the type of scales, the degree of dryness, and the presence or absence of symptomatology. Patients with scalp involvement were treated with coal tar (5%)/salicylic acid (2%) shampoo, patients with dry, scaly skin were given Akerat cream (urea/salicylic acid), and patients with pruritus were advised to use LIPIKAR BAUME AP + from LA ROCHE for sensitive skin. The patients responded to the provided regimens, resulting in an improved quality of life and reasonable control of their clinical presentation.
FLG genetic diversity of the patients with IV
We comprehensively sequenced the gene FLG using the overlapping PCR method. No genetic alterations were identified in exons 1 and 2. We identified 22 different variants in exon 3; all have been reported previously with a known allele frequency according to the Genome Aggregation Database (gnomAD) [27]. (Table 3).
All variants found here were either missense or silent variants. We did not find the known loss-of-function variants p.R501*, and c. 2282del4 in our samples. The most prevalent allele size in our Saudi patient sample was 7–8 repeats. The two most frequent FLG variants were p.H2507Q and p.G2545R, presenting in 48.5% of our patients. Three identified variants were silent variants (#s 2, 3, 20) that might have no impact on the disease (Table 3). However, we do not have data about their allele frequencies in the general Saudi population.
Discussion
The gene FLG consists of 3 exons. Exon 3 is the largest and is the coding segment of all filaggrin repeats. In 2006, Smith et al. [8] detected the nonsense mutation p. R501* near repeat 1. They further sequenced the FLG leading to the identification of a second mutation, c. 2282del4, causing premature termination codon [8]. In 2007, Nomura et al. [19], on the other hand, screened Japanese families, however, only some Japanese individuals with IV carried the European mutations. Complete sequencing of FLG identified two other mutations in the Japanese IV families, namely, p.S2554* and c.3321delA. Similarly, in 2014, Polcari et al. [28] reported a low prevalence of European-specific mutations in the African American population. In parallel with the Japanese and the African American populations, ours does not have the common European mutations.
Near 60 FLG null mutations have been identified in European and Asian populations; some are unique to an ancestral population, and different ancestries share others [29, 30].
In 2018, Hassani et al. [31]. sequenced the entire FLG in Iranian patients and identified 45 variants with two previously unknown variants. None of the identified FLG variants were loss-of-function variants and were supposed to be non-pathogenic (nonsense). Considering the frequency of repeat numbers of FLG and the identified DNA variants between the patients and the control group, they concluded that other mechanisms, possibly inflammatory-driven or epigenetic FLG functional deficiency might be involved in the pathogenesis of IV rather than the FLG nonsense mutation [31].
In line with the Iranian study, sequencing the entire FLG coding repeats identified 22 FLG variants. In fact, most of the identified FLG variants were missense with a known general allele frequency; however, six were silent variants. Furthermore, we tested the hypothesis of correlating the FLG variants to the clinical severity of our IV patients. To correlate the genetic to the phenotype severity, we analyzed three clinical factors: scaling severity, palmer hyperlinearity, and the presence of pruritus. In contrast to studies of loss-of-function FLG variants, we did not identify any influence of the FLG variants on the clinical severity of the disease (Table 4).
Conclusion
In conclusion, we identified missense and silent FLG variants, but no loss-of-function variants in our Saudi patients with IV. This is in contrast to studies from several different ethnic groups. Moreover, there was no influence of the detected FLG genotype on the phenotype severity of the disease. This study may serve as a Saudi reference variant spectrum for the FLG gene mutation in ichthyosis vulgaris patients. In the future, we hope to perform more national cohort studies and further analyze the newly reported variants by comparing them with a control group.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author upon reasonable request.
References
Oji V, Preil ML, Kleinow B, Wehr G, Fischer J, Hennies HC, et al. S1 guidelines for the diagnosis and treatment of ichthyoses - update. JDDG: J Der Deutschen Dermatologischen Gesellschaft. 2017;15(10):1053–65.
Siemens HW. Studien über Vererbung Von Hautkrankheiten. Arch Dermatol Syph. 1929;158(1):111–27.
Wells RS, Kerr CB. Genetic classification of Ichthyosis. Arch Dermatol. 1965;92(1):1.
Frost P, Van Scott EJ. Ichthyosiform Dermatoses. Classification based on anatomic and biometric observations. Arch Dermatol. 1966;94(2):113.
Williams ML, Elias PM. Genetically transmitted, generalized disorders of cornification. The ichthyoses. Dermatol Clin. 1987;5(1):155–78.
Wells RS, Kerr CB. Clinical features of autosomal Dominant and Sex-linked ichthyosis in an English Population. BMJ. 1966;1(5493):947–50.
Majmundar VD, Baxi K. https://www.ncbi.nlm.nih.gov/books/NBK562318/. 2023. Hereditary and Acquired Ichthyosis Vulgaris.
Smith FJD, Irvine AD, Terron-Kwiatkowski A, Sandilands A, Campbell LE, Zhao Y, et al. Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat Genet. 2006;38(3):337–42.
Armengot-Carbo M, Hernández-Martín Á, Torrelo A. Filagrina: papel en la barrera cutánea y en El Desarrollo De patología. Actas Dermosifiliogr. 2015;106(2):86–95.
Thyssen JP, Godoy-Gijon E, Elias PM. Ichthyosis vulgaris: the filaggrin mutation Disease. Br J Dermatol. 2013;168(6):1155–66.
Sandilands A, Sutherland C, Irvine AD, McLean WHI. Filaggrin in the frontline: role in skin barrier function and Disease. J Cell Sci. 2009;122(9):1285–94.
Compton JG, DiGiovanna JJ, Johnston KA, Fleckman P, Bale SJ. Mapping of the associated phenotype of an absent granular layer in ichthyosis vulgaris to the epidermal differentiation complex on chromosome 1 *. Exp Dermatol. 2002;11(6):518–26.
Mischke D, Korge BP, Marenholz I, Volz A, Ziegler A. Genes Encoding Structural Proteins of Epidermal Cornification and S100 calcium-binding proteins Form a Gene Complex (epidermal differentiation complex) on human chromosome 1q21. J Invest Dermatology. 1996;106(5):989–92.
Presland RB, Haydock PV, Fleckman P, Nirunsuksiri W, Dale BA. Characterization of the human epidermal profilaggrin gene. Genomic organization and identification of an S-100-like calcium binding domain at the amino terminus. J Biol Chem. 1992;267(33):23772–81.
Palmer CNA, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 2006;38(4):441–6.
Enomoto H, Hirata K, Otsuka K, Kawai T, Takahashi T, Hirota T, et al. Filaggrin null mutations are associated with atopic dermatitis and elevated levels of IgE in the Japanese population: a family and case–control study. J Hum Genet. 2008;53(7):615–21.
Sandilands A, O’Regan GM, Liao H, Zhao Y, Terron-Kwiatkowski A, Watson RM, et al. Prevalent and rare mutations in the Gene Encoding Filaggrin cause Ichthyosis Vulgaris and predispose individuals to atopic dermatitis. J Invest Dermatology. 2006;126(8):1770–5.
Sandilands A, Terron-Kwiatkowski A, Hull PR, O’Regan GM, Clayton TH, Watson RM, et al. Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema. Nat Genet. 2007;39(5):650–4.
Nomura T, Sandilands A, Akiyama M, Liao H, Evans AT, Sakai K, et al. Unique mutations in the filaggrin gene in Japanese patients with ichthyosis vulgaris and atopic dermatitis. J Allergy Clin Immunol. 2007;119(2):434–40.
Blattner F, John Schroeder. Lasergene’s SeqMan Pro (RRID:SCR_000283). Madison. Wisconsin USA: DNASTAR, Inc.; 1984.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–2.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Desmet FO, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C. Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37(9):e67–7.
Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, et al. The pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016;44(D1):D279–85.
Microsoft Corporation. Microsoft Excel. https://office.microsoft.com/excel; 2018.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Sci. 2015;17(5):405–24.
Polcari I, Becker L, Stein SL, Smith MS, Paller AS. Filaggrin Gene mutations in African americans with both ichthyosis Vulgaris and atopic dermatitis. Pediatr Dermatol. 2014;31(4):489–92.
Chen H, Common JEA, Haines RL, Balakrishnan A, Brown SJ, Goh CSM, et al. Wide spectrum of filaggrin-null mutations in atopic dermatitis highlights differences between Singaporean Chinese and European populations. Br J Dermatol. 2011;165(1):106–14.
Li M, Cheng R, Shi M, Liu J, Zhang G, Liu Q, et al. Analyses of FLG mutation frequency and filaggrin expression in isolated ichthyosis vulgaris (IV) and atopic dermatitis-associated IV. Br J Dermatol. 2013;168(6):1335–8.
Hassani B, Isaian A, Shariat M, Mollanoori H, Sotoudeh S, Babaei V, et al. Filaggrin gene polymorphisms in Iranian ichthyosis vulgaris and atopic dermatitis patients. Int J Dermatol. 2018;57(12):1485–91.
Acknowledgements
This work is supported by King Abdulaziz City for Science and Technology, Riyadh, Saudi Arabia, grant numbers 10-BIO1344-46. This is a government agency. However, they have no rule in designing, collecting, analyzing, or interpreting the data and writing the manuscript.
Funding
This work is supported by King Abdulaziz City for Science and Technology, Riyadh, Saudi Arabia, grant numbers 10-BIO1344-46. This is a government agency. However, they have no rule in designing, collecting, analyzing, or interpreting the data and writing the manuscript.
Author information
Authors and Affiliations
Contributions
Omar Alakloby wrote the main manuscript, analyzed the data, reviewed the literature, generated the idea, prepared the proposal, collected the data, and prepared the tables. Fatimah Almuqarrab wrote the main manuscript, analyzed and interpreted the data, and reviewed the literature. Johannes Zschocke: generated the idea, prepared the proposal, collected the data, and prepared the tables. Mathias Schmuth: generated the idea, prepared the proposal, collected the data, and prepared the tables. Adnan Abdulkareem: generated the idea, prepared the proposal, collected the data, and prepared the tables. Kholood Alnutaifi: generated the idea, prepared the proposal, collected the data, and prepared the tables. Francis Borgio: generated the idea, prepared the proposal, collected the data, and helped in writing the manuscript. Robert Gruber: Collected the data, interpreted the results, and analyzed the genetic variants. Hans Hennies: Collected the data, interpreted the results, and analyzed the genetic variants.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This is a descriptive-analytic study involving human participants; ethical permission was obtained from the Institutional Research Board of Imam Abdulrahman Bin Faisal University, Dammam. All study participants signed written informed consent forms. All experiments were performed in accordance with the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
None.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Alakloby, O., Almuqarrab, F., Zschocke, J. et al. Filaggrin gene variants among Saudi patients with ichthyosis vulgaris. BMC Med Genomics 16, 256 (2023). https://doi.org/10.1186/s12920-023-01700-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-023-01700-x