
Abstract
Background
Epidemiological studies have indicated a potential link between the gut microbiome and autoimmune liver disease (AILD) such as autoimmune hepatitis (AIH), primary biliary cholangitis (PBC), and primary sclerosing cholangitis (PSC). The relationship between the gut microbiome and autoimmune liver disease is still uncertain due to confounding variables. In our study, we aim to shed light on this relationship by employing a two-sample Mendelian randomization approach.
Methods
We conducted a two-sample Mendelian randomization (MR) study using the R package "TwoSampleMR". The exposure data consisted of genetic variants associated with 194 bacterial traits obtained from the MiBioGen consortium. Summary statistics for AILD were obtained from the GWAS Catalog website. Furthermore, a series of sensitivity analyses were performed to validate the initial MR results.
Results
There were two, four and three bacteria traits associated with an increased risk of AIH. PBC, and PSC respectively. In contrast, there were five, two and five bacteria traits associated with a decreased risk for AIH, PBC and PSC. Notably, the genus_Clostridium_innocuum_group showed a negative association with AIH (OR = 0.67, 95% CI: 0.49–0.93), and the genus_Actinomyces was found to be genetically associated with a decreased risk of PSC (OR = 0.62, 95% CI: 0.42–0.90).
Conclusions
Our study identified the causal impact of specific bacterial features on the risk of AILD subtypes. Particularly, the genus_Clostridium_innocuum_group and the genus_Actinomyces demonstrated significant protective effects against AIH and PSC respectively. These findings provide further support for the potential use of targeted probiotics in the management of AILD.
Similar content being viewed by others
Background
Autoimmune liver disease (AILD) is a rare chronic liver disorder characterized by autoimmune abnormalities. It encompasses three main types: autoimmune hepatitis (AIH), primary biliary cholangitis (PBC), and primary sclerosing cholangitis (PSC). The diagnosis of AILD typically involves a combination of specific autoantibody testing, serum biochemistry, and liver histology [1, 2]. Notably, specific autoantibodies associated with PSC have not been identified, and non-invasive imaging techniques are recommended for evaluating liver and bile duct fibrosis [3]. Despite the low incidence and prevalence of autoimmune liver diseases, they impose a disproportionate clinical burden on affected individuals. The global incidence rates per 100,000 population vary, ranging from 0.4 to 2.39 for AIH, 0.84 to 2.75 for PBC, and 0.1 to 4.39 for PSC [4]. Treatment typically involves the use of immunosuppressants, which are generally effective but often require long-term administration, raising concerns regarding potential side effects and patient adherence to therapy [5].
The gut microbiota, considered a virtual metabolic organ, is increasingly recognized for its role in various extraintestinal systems [6]. Recent research has emphasized the importance of the gut-liver axis in liver disease pathogenesis, involving intestinal barrier homeostasis, bile-mediated liver communication, and the composition and function of the gut microbiota [7]. While the crucial involvement of the gut microbiota in alcohol-associated liver disease and non-alcoholic fatty liver disease has been extensively investigated, studies focusing on the relationship between AILD and the gut microbiota are limited [8]. Over the past decades, Genome-Wide Association Study (GWAS) has successfully identified genetic factors associated with AIH, PBC, and PSC [9]. These GWAS data can be repurposed to explore the causal relationship between the gut microbiota and AILD. Mendelian randomization (MR), a statistical method, offers an opportunity to infer causal effects by using single nucleotide polymorphisms (SNPs) as instrumental variables (IVs) [10]. Through MR analysis, the causal relationship between the gut microbiome and the occurrence of AIH, PBC, and PSC can be explored, providing valuable insights for clinical practice. Additionally, the results of MR analysis may also support the potential development of probiotic therapies targeting the gut microbiota for AILD.
Methods
Study design
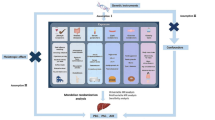
We conducted a two-sample MR study to investigate the causal association between the gut microbiome and autoimmune liver disease, and a workflow of the study is shown in Fig. 1. To ensure valid instrumental variables (IVs), three fundamental assumptions of the MR design were satisfied: (I) genetic variation as an IV must be significantly associated with the gut microbiome; (ii) genetic variation must be independent of confounders; and (iii) variation must be associated with autoimmune liver disease only through the gut microbiome [11]. The summary data were primarily based on independent GWAS and MR utilized SNPs to assess causality.

Study design of the two-sample Mendelian randomization for the effect of genetically predicted gut microbiome on AILD subtypes. AIH, autoimmune hepatitis; PBC, primary biliary cholangitis; PSC, primary sclerosing cholangitis; N: number of discovery cases; SNPs, single nucleotide polymorphisms
GWAS data source
The full GWAS summary statistics for the microbiota were primarily derived from a large-scale multi-ethnic GWAS meta-analysis conducted by the international MiBioGen consortium [12], which was established to study the influence of human genetics on the gut microbiome. This meta-analysis included 211 gut microbiota and 122,110 related SNPs. To exclude the influence of ethnicity, GWAS data for participants of European ancestry were selected.
The summary statistics data for AIH, PBC, and PSC were acquired from the corresponding studies in the GWAS Catalog. For AIH (GCST90018785), there were 821 cases and 484,413 controls of European ancestry [13]. For PBC (GCST90061440), there were 8,021 cases and 16,489 controls of European ancestry [14]. For PSC (GCST004030), there were 2,871 cases and 12,019 controls of European ancestry [15]. Detailed descriptions of the study procedures, ethical approvals, and consent to participate can be found in the original studies.
The selection of instrumental variables
To ensure the reliability of the results, instrumental variables were carefully selected. First, 17 bacterial traits with unknown classifications were excluded. Then SNPs with a p-value less than 10–5 were chosen, while those with weak associations or low minor allele frequencies were excluded. Independent SNPs were identified by clumping SNPs based on the European 1000 Genomes Project reference panel (r2 < 0.01 and clump distance > 10,000 kb). The instrumental strength of each SNP was assessed using the F statistics = (β/SE)2, and variables with F statistics values > 10 were excluded [16]. The corresponding data of the selected SNPs was extracted from the GWAS outcome data, and proxy SNPs were not allowed. The selection process was carried out using the “TwoSampleMR” R package (version 0.5.6) [17]. It's worth noting that due to the absence of the rsID column in the GWAS summary data of AIH, a set of unique identifiers for distinguishing similar SNPs was unavailable. To address this, we utilized the SNP Annotation Tool [18] to derive rsID information based on the available chromosome number and position details. As the GWAS summary data were annotated in the GRCh37 version, we ensured the use of the same version for consistency during the querying process. This enabled us to obtain the necessary rsID information for the SNPs. This information was then matched with the outcome data and processed using the “format_data” function in the “TwoSampleMR” package to obtain standardized data. During the harmonizing process, positive strand alleles were inferred using allele frequencies for palindromes to ensure that the effects of SNPs on exposure corresponded to the same allele as the effects of SNPs on the outcome. Finally, as a quality control measure, exposure traits with less than 3 SNPs were excluded.
Conduction of MR analyses
MR analyses were performed using inverse variance-weighted (IVW), weighted median, MR–Egger, and maximum likelihood methods to identify gut microbiome related to three subtypes of autoimmune liver diseases. IVW was used as the primary method assuming all SNPs are valid variables. The weighted median approach yields consistent estimates assuming more than half of the weights are from valid SNPs [19]. MR–Egger analysis can calibrate for pleiotropy and calculate causal inferences even when all genetic variants are pleiotropic [20]. The maximum likelihood-based approach can generate appropriate confidence interval (CI) estimation when weak IV is observed. Guidance on interpreting the outcomes of these methods can be found elsewhere [21]. We further visualized the results of IVW method with a heatmap for more intuitive interpretation. Finally, several essential sensitivity analyses were performed to verify the robustness of the MR analysis results. A test for heterogeneity was conducted using Cochran’s test. MR-Pleiotropy Residual Sum and Outlier (MR-PRESSO) was performed to examine horizontal pleiotropy if available in order to eliminate SNPs with horizontal pleiotropic outliers [22]. The MR–Egger regression intercept was used to estimate potential pleiotropy of SNP, with a P-value > 0.05 indicating no horizontal pleiotropy. A leave-one-out analysis was also used to detect pleiotropy caused by each SNP. Sensitivity analyses were conducted using the “TwoSampleMR” R package. Results were presented as odds ratios (OR) with respective 95% CI. P-values were two-sided and statistical significance was set at the 5% level.
Results
Instrument variables for gut microbiome
SNPs from 194 bacterial traits containing five biological levels (i.e. phylum, class, order, family, and genus) were included in our study. Detailed information (i.e. effect allele, other allele, beta, standard error, p-value, and F statistics) of the final SNPs for each bacterial trait is shown in Supplementary Table 1.
Causal effects of the gut microbiome on autoimmune liver diseases
Using the IVW method as the primary MR detection method, a total of 21 gut microbiome traits were found to have a potential causal relationship with AILD (7 traits for AIH, 6 traits for PBC, and 8 traits for PSC). A heatmap was created to display these results, with red representing risk factors and blue representing protective factors (Fig. 2). These taxonomic groups demonstrate the hierarchical relationship between phyla, classes, orders, families, and genera of bacteria, with groups related at the highest level (phylum) and branching into more specific categories. In detail, the phylum Tenericutes includes the class Mollicutes. The class Clostridia contains two families: Clostridiales vadin BB60 group and Victivallaceae. The class Coriobacteriia includes the order Coriobacteriales and the family Coriobacteriaceae. The order Actinomycetales contains the family Actinomycetaceae and the genus Actinomyces. Genera in the results do not have a hierarchical relationship with each other. These results provide insight into the potential associations between certain bacterial features and the risk of AILD.

Heatmap illustrating positive IVW-MR results, with red indicating risk factors and blue indicating protective factors. Asterisks (*) denote statistical significance, with * indicating P < 0.05 and ** indicating P < 0.01
AIH
According to our IVW-MR analysis, we found that genetically predicted levels of two bacterial features (family_Victivallaceae: OR 1.42, 95% CI 1.03–1.92, P = 0.004; genus_Ruminococcaceae NK4A214 group: OR 1.64, 95% CI 1.03–2.64, P = 0.039) were potentially associated with an decreased risk of AIH. Additionally, we observed that genetically predicted levels of five bacterial features were associated with a lower risk of AIH: class_Clostridia (OR 0.58, 95% CI 0.35–0.97, P = 0.039), genus_Ruminiclostridium9 (OR 0.39, 95% CI 0.20–0.76, P = 0.005), genus_Holdemania (OR 0.66, 95% CI 0.46 to 0.95, P = 0.024). genus_Clostridium innocuum group (OR 0.67, 95% CI 0.49 to 0.93, P = 0.016) and genus_Anaerostipes (OR 0.35, 95% CI 0.17 to 0.74, P = 0.006) (Table 1). The aggressive role of family_Victivallaceae and the protectiove role of genus_Anaerostipes were supported by the Weighted Median method, whilst others were not (see Supplementary Table 2).
PBC
The same approaches were utilized to explore the causal effect of gut microbiome on PBC. There were four bacterial traits(class_Coriobacteriia: OR 2.18, 95% CI 1.30 to 3.66, P = 0.003; family_Coriobacteriaceae: OR 2.18, 95% CI 1.30 to 3.66, P = 0.003; order_Coriobacteriales: OR 2.18, 95% CI 1.30 to 3.66, P = 0.003; genus_Ruminiclostridium5, OR 1.47, 95% CI 1.03 to 2.09, P = 0.031) potentially related to an increased risk of PBC utilizing the IVW method, while two bacterial traits(class_Deltaproteobacteria, OR 0.52, 95% CI 0.36 to 0.74, P < 0.001; family_Desulfovibrionaceae, OR 0.53, 95% CI 0.34 to 0.81, P = 0.003) were associated with a lower risk of PBC in the IVW–MR analysis (Table 1). Meanwhile, all the results above were supported by the Weighted Median method (P < 0.05, Supplementary Table 2).
PSC
Similarly, on the causal effect of gut microbiome on PSC, the estimates of the IVW test indicated that certain bacterial traits was associated with either an increased or reduced risk of PSC. Specifically, three bacterial features (phylum_Tenericutes: OR 1.66, 95% CI 1.06 to 2.61, P = 0.026; class_Mollicutes: OR 1.66, 95% CI 1.06 to 2.61, P = 0.026; family_Oxalobacteraceae, OR 1.44, 95% CI 1.08 to 1.91, P = 0.013) were associated with an increased risk of PSC according to IVW analysis. In the contrast, five bacterial features (order_Actinomycetales, OR 0.59, 95% CI 0.36 to 0.98, P = 0.041; family_Clostridiales vadin BB60 group, OR 0.73, 95% CI 0.54 to 0.98, P = 0.035; family_Actinomycetaceae, OR 0.60, 95% CI 0.36 to 0.98, P = 0.042; genus_Alloprevotella, OR 0.68, 95% CI 0.50 to 0.94, P = 0.018; genus_Actinomyces OR 0.62, 95% CI 0.42 to 0.90, P = 0.012) were associated with an lower risk of PSC (Table 1). The Weighted Median method provided support for the aggressive role of family_Oxalobacteraceae and the protective role of genus_Alloprevotella, while other associations did not reach statistical significance (Supplementary Table 2).
In summary, Supplementary Table 2 presents a comprehensive overview of all positive results, providing in-depth details and information. On the other hand, Supplementary Table 3 includes all results, including negative ones, in a tabulated format. These supplementary tables are intended to complement and provide further context to the main findings discussed in the sections above.
Sensitivity analyses
When the exposure is the genus_Anaerostipes and the outcome is AIH, the MR result did not pass the heterogeneity test (Q p-value = 0.02, Supplementary Table 4). Since there is only heterogeneity and no pleiotropy for this exposure trait, the result of the Weighted Median method are preferred [23]. The value of the Weighted Median method is P < 0.001, indicating that causality exists. To increase the credibility of the result, the random effects model of IVW was further performed and the result was P = 0.006 < 0.05, which was statistically significant. For other exposure-outcome pairs, no heterogeneity or outliers were found using Cochran’s Q and MR-PRESSO tests (P > 0.05, Supplementary Table 4–5). All P-values of MR–Egger interpret were > 0.05, indicating no horizontal pleiotropy (Supplementary Table 4). Moreover, Supplementary figures S1-3 show the results of sensitivity analyses in scatter plots. Furthermore, we conducted leave-one-out analyses to evaluate the potential influence of individual SNPs on the observed associations. Supplementary figures S4-6 present the leave-one-out analysis, evaluating the influence of individual SNPs on the associations.
Discussion
We conducted an MR study using the most comprehensive GWAS data available to overcome a common limitation in epidemiological studies. This could provide important insights into the genetic correlations between the gut microbiome and AILD subtypes. Our results highlighted a causal effect of the abundance of specific bacterial features on the risk of AILD subtypes. To the best of our knowledge, our study is the first to employ the MR framework to investigate the causal relationship between the gut microbiome and AILD. Our discussion primarily focuses on the findings at the genus level, as such an approach is more clinically oriented. Notably, the genera Anaerostipes, Clostridium_innocuum_group, Holdemania, and Ruminiclostridium9 played a role in protection against AIH, while Ruminococcaceae_NK4A214_group increased the risk of AIH. Ruminiclostridium5 increased the risk of PBC. Alloprevotella and Actinomyces protected against PSC.
The microbiome's implication in the etiology of autoimmune disorders has garnered substantial attention. The primary mechanism involves immune system deviations mediated through microbial signaling, predominantly via the gut-liver axis [24]. While the implication of microbiota in the pathogenesis of disorders like Type 1 diabetes, rheumatoid arthritis, and coeliac disease has been extensively explored, the literature concerning AILDs remains comparatively limited [25]. However, evidence from animal models underscores a causal connection between dysbiosis of gut microbiota or specific pathobionts and AILDs. For example, gnotobiotic mice administered with microbiota from PSC patients exhibited heightened Th17 cell responses within the liver, rendering them more susceptible to hepatobiliary injuries [26]. This suggests a potential role of gut microbiota in driving PSC pathogenesis. Other research indicates that gut pathobiont translocation, stemming from compromised gut barriers, infiltrates systemic organs in hosts prone to autoimmunity, instigating autoimmune pathogenesis [27].
Several population-based observational studies have been conducted to examine the gut microbiome in patients with AILD [28,29,30]. Comparing the results to observational studies, we have observed both consistencies and inconsistencies in the association of bacterial traits with AILD. These variations may stem from differences in genetic backgrounds and synergistic activities among populations from different regions. For instance, the genus_Veillonella is frequently reported to be enriched in AIH, PBC, and PSC in Asian cohorts [31]. However, our study did not find a causal effect between the genus_Veillonella and AILD in the MR analysis. Here, we will specifically examine the potential impact of two traits, namely genus_Clostridium_innocuum_group and genus_Actinomyces as they exhibited protective role for AIH and PSC respectively. Meanwhile, these two traits were previously reported to be associated with AILD, and the details of the MR analysis pertaining to their effects were provided in Table 2.
So far, there are relatively few studies about the relationship between gut microbiome and AIH. In the current study, the genus_Clostridium_innocuum_group could mitigate the risk of AIH. It belongs to the order_Clostridiales and the latter has been adapted as a biomarker to distinguish AIH from controls in a microbial diagnostic model [28]. It is also reported that the genus_Clostridium was more abundant in all subgroups of PBC and PSC [32, 33], however, in our study, no causal effect has been revealed between the genus_Clostridium and these subtypes, though a group of family_Clostridiales exhibited protective role in our MR analysis for PSC. Moreover, the genus_Clostridium has been found to modulate the induction of T regulatory cells through the provision of bacterial antigens and short-chain fatty acids [34]. These factors influence the activity of T regulatory cells and contribute to the reduction of pro-inflammatory cytokine levels [35].
Evidence indicates an elevated relative abundance of genus_Actinomyces in both saliva and fecal samples of PSC patients [36, 37]. Our findings suggest that the genus_Actinomyces may play a protective role in PSC patients, further supporting these findings. In contrast, the genus_Actinomyces was observed to be lower in AIH patients than in healthy controls [38]. Therefore, the role of the genus_Actinomyces in different subtypes of AILD may differ, and further investigation is required.
Notably, our MR analysis revealed contrasting findings regarding the genus_Ruminiclostridium in relation to AIH and PBC. The presence of genus_Ruminiclostridium5 was associated with an increased risk of PBC, while the genus_Ruminiclostridium9, another unidentified group within the genus_Ruminiclostridium, exhibited a protective effect with greater statistical significance in AIH. The genus_Ruminiclostridium is known to be involved in glucose and bile acid metabolism [39]. Considering that a subset of patients (2–19%) may exhibit overlapping features of both PBC and AIH, known as PBC-AIH overlap syndrome [40], caution should be exercised in interpreting these results. Further investigation is warranted to elucidate the role of this microbial trait in the pathogenesis of these subtypes.
The investigation of alterations in the gut microbiome holds significant clinical implications for AILD. First, changes in the gut microbiome can serve as a biomarker for disease screening, diagnosis, and prognosis throughout the course of AILD. The human microbiome has been successfully utilized to develop diagnostic biomarkers for various diseases, including hepatocellular carcinoma [41]. In the context of AILD, diagnostic models based on the microbiome have been established for AIH [28] and PSC [42], but there is currently no reported model specifically for PBC. In the future, there is a need for more longitudinal data on the gut microbiome to support the development of screening, diagnosis, and prognosis models for AILD.
Second, current clinical approaches for treating AILD are limited. Standard therapy for AIH involves a combination of prednisone and azathioprine [43], while ursodeoxycholic acid (UDCA) is commonly used for PBC and PSC. However, the efficacy of UDCA in improving survival in PSC is uncertain, and higher doses are associated with increased adverse events [44]. Therefore, understanding the causal relationship between the gut microbiome and the development and progression of AILD is of great significance in identifying new therapeutic targets and drugs. Probiotics have shown potential as a promising adjunctive therapeutic option in the management of AILD [45]. In the routine management of patients, incorporating food products containing beneficial bacterial components into their daily diet is a relatively easy to implement, cost-effective, and efficient approach. Probiotics were found to increase the population of T regulatory cells in AIH mouse model, indicating their immunomodulatory role in alleviating autoimmune hepatitis [46]. The therapeutic potential of Lactobacillus in combination with prednisone for the treatment of AIH has been suggested from a clinical trait [47]. However, a randomized, placebo-controlled study of probiotics in patients with PSC did not show any benefits in relieving PSC symptoms, indicating that probiotics alone may not be effective in treating PSC [48].
Limitations of our research include the potential influence of various factors on the abundance of the gut microbiome, such as diet, sex, medication, and sampling time. To obtain more comprehensive results, we refrained from applying a strict false discovery rate correction to re-evaluate positive outcomes. Future research demands more rigorous experimental and clinical validation of our findings. Furthermore, it underscores the necessity for comprehensive GWAS tailored to Asian populations to delve into host genetic variants associated with the gut microbiome [49]. Additionally, the overlapping symptoms among different subtypes of AILD can complicate the diagnosis process, making it more relevant to compare results across subtypes rather than relying solely on subtype-specific conclusions to draw definitive causal inferences regarding the relationship between the gut microbiome and AILD.
Conclusion
Our study provides evidence supporting the causal effect of specific bacterial features on the risk of AILD subtypes. Specifically, we found that the genus_Clostridium_innocuum_group displayed a significant protective effect against AIH, while the genus_Actinomyces showed a significant protective effect against PSC. Further longitudinal studies and clinical trials are needed to validate these findings and explore the potential of targeted probiotics for the management of AILD.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary files. The accession number for GWAS data of AIH is GCST90018785, available at http://ftp.ebi.ac.uk/pub/databases/gwas/summary_statistics/GCST90018001-GCST90019000/GCST90018785/GCST90018785_buildGRCh37.tsv.gz). The accession number for PBC is GCST90061440, available at http://ftp.ebi.ac.uk/pub/databases/gwas/summary_statistics/GCST90061001-GCST90062000/GCST90061440/GCST90061440_buildGRCh37.tsv, and accession number for PSC is GCST004030, available at http://ftp.ebi.ac.uk/pub/databases/gwas/summary_statistics/GCST004001-GCST005000/GCST004030/harmonised/27992413-GCST004030-EFO_0004268-Build37.f.tsv.gz.
Abbreviations
- AIH:
-
Autoimmune hepatitis
- AILD:
-
Autoimmune liver disease
- CI:
-
Confidence interval
- GWAS:
-
Genome-Wide Association Study
- IVW:
-
Inverse variance-weighted
- IV:
-
Instrumental variable
- MR:
-
Mendelian randomization
- MR-PRESSO:
-
MR-Pleiotropy Residual Sum and Outlier
- OR:
-
Odds ratio
- PBC:
-
Primary biliary cholangitis
- PSC:
-
Primary biliary cholangitis
- SNP:
-
Single nucleotide polymorphism
- UDCA:
-
Ursodeoxycholic acid
References
Mieli-Vergani G, Vergani D, Czaja AJ, Manns MP, Krawitt EL, Vierling JM, et al. Autoimmune hepatitis. Nat Rev Dis Primers. 2018;4:18017.
Komori A. Recent updates on the management of autoimmune hepatitis. Clin Mol Hepatol. 2021;27:58–69.
Chapman MH, Thorburn D, Hirschfield GM, Webster GGJ, Rushbrook SM, Alexander G, et al. British Society of Gastroenterology and UK-PSC guidelines for the diagnosis and management of primary sclerosing cholangitis. Gut. 2019;68:1356–78.
Trivedi PJ, Hirschfield GM. Recent advances in clinical practice: epidemiology of autoimmune liver diseases. Gut. 2021;70:1989–2003.
Hadzic N, Hierro L. Autoimmune liver disease: Novelties in management. Clin Res Hepatol Gastroenterol. 2014;38:273–6.
Chen Y, Zhou J, Wang L. Role and Mechanism of Gut Microbiota in Human Disease. Front Cell Infect Microbiol. 2021;11:625913.
Albillos A, de Gottardi A, Rescigno M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J Hepatol. 2020;72:558–77.
Cheng Z, Yang L, Chu H. The gut microbiota: a novel player in autoimmune hepatitis. Front Cellular Infect Microbiol. 2022:977.
Ellinghaus D. How genetic risk contributes to autoimmune liver disease. Semin Immunopathol. 2022;44:397–410.
Emdin CA, Khera AV, Kathiresan S. Mendelian randomization. Jama. 2017;318:1925–6.
Boef AGC, Dekkers OM, le Cessie S. Mendelian randomization studies: a review of the approaches used and the quality of reporting. Int J Epidemiol. 2015;44:496–511.
Kurilshikov A, Medina-Gomez C, Bacigalupe R, Radjabzadeh D, Wang J, Demirkan A, et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat Genet. 2021;53:156–65.
Sakaue S, Kanai M, Tanigawa Y, Karjalainen J, Kurki M, Koshiba S, et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat Genet. 2021;53:1415–24.
Cordell HJ, Fryett JJ, Ueno K, Darlay R, Aiba Y, Hitomi Y, et al. An international genome-wide meta-analysis of primary biliary cholangitis: Novel risk loci and candidate drugs. J Hepatol. 2021;75:572–81.
Ji S-G, Juran BD, Mucha S, Folseraas T, Jostins L, Melum E, et al. Genome-wide association study of primary sclerosing cholangitis identifies new risk loci and quantifies the genetic relationship with inflammatory bowel disease. Nat Genet. 2017;49:269–73.
Brion M-JA, Shakhbazov K, Visscher PM. Calculating statistical power in Mendelian randomization studies. Int J Epidemiol. 2013;42:1497–501.
Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife. 2018;7:e34408.
SNP Annotation Tool. https://www.snp-nexus.org/v4/. Accessed 2 May 2023.
Boehm FJ, Zhou X. Statistical methods for Mendelian randomization in genome-wide association studies: a review. Comput Struct Biotechnol J. 2022;20:2338–51.
Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44:512–25.
Davies NM, Holmes MV, Davey SG. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362:k601.
Verbanck M, Chen C-Y, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50:693–8.
Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol. 2017;32:377–89.
Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, Schnabl B, et al. The gut–liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol. 2018;15:397–411.
McLean MH, Dieguez D, Miller LM, Young HA. Does the microbiota play a role in the pathogenesis of autoimmune diseases? Gut. 2015;64:332–41.
Nakamoto N, Sasaki N, Aoki R, Miyamoto K, Suda W, Teratani T, et al. Gut pathobionts underlie intestinal barrier dysfunction and liver T helper 17 cell immune response in primary sclerosing cholangitis. Nat Microbiol. 2019;4:492–503.
Manfredo Vieira S, Hiltensperger M, Kumar V, Zegarra-Ruiz D, Dehner C, Khan N, et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science. 2018;359:1156–61.
Wei Y, Li Y, Yan LI, Sun C, Miao Q, Wang Q, et al. Alterations of gut microbiome in autoimmune hepatitis. Gut. 2020;69:569–77.
Furukawa M, Moriya K, Nakayama J, Inoue T, Momoda R, Kawaratani H, et al. Gut dysbiosis associated with clinical prognosis of patients with primary biliary cholangitis. Hepatol Res. 2020;50:840–52.
Kummen M, Thingholm LB, Rühlemann MC, Holm K, Hansen SH, Moitinho-Silva L, et al. Altered Gut Microbial Metabolism of Essential Nutrients in Primary Sclerosing Cholangitis. Gastroenterology. 2021;160:1784-1798.e0.
Zheng Y, Ran Y, Zhang H, Wang B, Zhou L. The microbiome in autoimmune liver diseases: Metagenomic and metabolomic changes. Front Physiol. 2021;12:715852.
Bajer L, Kverka M, Kostovcik M, Macinga P, Dvorak J, Stehlikova Z, et al. Distinct gut microbiota profiles in patients with primary sclerosing cholangitis and ulcerative colitis. WJG. 2017;23:4548.
Tang R, Wei Y, Li Y, Chen W, Chen H, Wang Q, et al. Gut microbial profile is altered in primary biliary cholangitis and partially restored after UDCA therapy. Gut. 2018;67:534–41.
Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–6.
Bhaskaran N, Quigley C, Paw C, Butala S, Schneider E, Pandiyan P. Role of short chain fatty acids in controlling tregs and immunopathology during mucosal infection. Front Microbiol. 2018;9:1995.
Lapidot Y, Amir A, Ben-Simon S, Veitsman E, Cohen-Ezra O, Davidov Y, et al. Alterations of the salivary and fecal microbiome in patients with primary sclerosing cholangitis. Hepatol Int. 2021;15:191–201.
Quraishi MN, Acharjee A, Beggs AD, Horniblow R, Tselepis C, Gkoutos G, et al. A Pilot Integrative analysis of colonic gene expression, gut microbiota, and immune infiltration in primary sclerosing cholangitis-inflammatory bowel disease: association of disease with bile acid pathways. J Crohns Colitis. 2020;14:935–47.
Rao B, Lou J, Lu H, Liang H, Li J, Zhou H, et al. Oral Microbiome Characteristics in Patients With Autoimmune Hepatitis. Front Cell Infect Microbiol. 2021;11:656674.
La Reau AJ, Suen G. The Ruminococci: key symbionts of the gut ecosystem. J Microbiol. 2018;56:199–208.
Chazouillères O. Overlap syndromes. Dig Dis. 2015;33(Suppl 2):181–7.
Rao B-C, Lou J-M, Wang W-J, Li A, Cui G-Y, Yu Z-J, et al. Human microbiome is a diagnostic biomarker in hepatocellular carcinoma. Hepatobiliary Pancreat Dis Int. 2020;19:109–15.
Rühlemann MC, Heinsen F-A, Zenouzi R, Lieb W, Franke A, Schramm C. Faecal microbiota profiles as diagnostic biomarkers in primary sclerosing cholangitis. Gut. 2017;66:753–4.
Mack CL, Adams D, Assis DN, Kerkar N, Manns MP, Mayo MJ, et al. Diagnosis and management of autoimmune hepatitis in adults and children: 2019 practice guidance and guidelines from the American Association for the study of liver diseases. Hepatology. 2020;72:671–722.
Carbone M, Neuberger JM. Autoimmune liver disease, autoimmunity and liver transplantation. J Hepatol. 2014;60:210–23.
Maslennikov R, Ivashkin V, Efremova I, Poluektova E, Shirokova E. Probiotics in hepatology: An update. World J Hepatol. 2021;13:1154.
Liu Q, Tian H, Kang Y, Tian Y, Li L, Kang X, et al. Probiotics alleviate autoimmune hepatitis in mice through modulation of gut microbiota and intestinal permeability. J Nutr Biochem. 2021;98:108863.
Ma L, Zhang L, Zhuang Y, Ding Y, Chen J. Lactobacillus improves the effects of prednisone on autoimmune hepatitis via gut microbiota-mediated follicular helper T cells. Cell Commun Signal. 2022;20:83.
Vleggaar FP, Monkelbaan JF, Van Erpecum KJ. Probiotics in primary sclerosing cholangitis: a randomized placebo-controlled crossover pilot study. Eur J Gastroenterol Hepatol. 2008;20:688–92.
Ishida S, Kato K, Tanaka M, Odamaki T, Kubo R, Mitsuyama E, et al. Genome-wide association studies and heritability analysis reveal the involvement of host genetics in the Japanese gut microbiota. Commun Biol. 2020;3:686.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from National Natural Science Foundations of China (82104549), Shanghai Natural Science Foundation of China (23ZR1461200; 22ZR1459400), Future Plan of Shanghai Medical Innovation and Development Foundation (WL-YBXM-2022002 K).
Author information
Authors and Affiliations
Contributions
The study was supervised by Y. Li. Y. Fu was responsible for conceiving and conducting the statistical analysis. Y. Fu and J. Li drafted the manuscript. Y. Zhu, C. Chen, J. Liu, S. Gu, Y. Zheng, Y. Li provided suggestions on figures and tables. The final version of the manuscript was approved by all authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval is presented in the mentioned GWAS studies.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Supplementary Figure S1-3. Results of sensitivity analyses displayed in scatter plots for AIH (S1), PBC (S2) and PSC (S3). Supplementary Figure S4-6. Results of leave-one-out analyses, evaluating the influence of individual SNPs on the associations for AIH (S4), PBC (S5) and PSC (S6). Supplementary Table S1. Instrumental Variables for each baterial triats. Supplementary Table S2. Positive results of MR analyses. Supplementary Table S3. All MR results for 194 traits. Supplementary Table S4. Results of sensitivity analyses for IVW positive MR analyses. Supplementary Table S5. Results of MR-PRESSO.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fu, Y., Li, J., Zhu, Y. et al. Causal effects of gut microbiome on autoimmune liver disease: a two-sample Mendelian randomization study. BMC Med Genomics 16, 232 (2023). https://doi.org/10.1186/s12920-023-01670-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-023-01670-0