
Abstract
Background
Hereditary hemolytic anemia (HHA) refers to a heterogeneous group of genetic disorders that share one common feature: destruction of circulating red blood cells (RBCs). The destruction of RBCs may be due to membranopathies, enzymopathies, or hemoglobinopathies. Because these are genetic disorders, incorporation of next-generation sequencing (NGS) has facilitated the diagnostic process of HHA.
Method
Genetic data from 29 patients with suspected hereditary anemia in a tertiary hospital were retrospectively reviewed to evaluate the efficacy of NGS on hereditary anemia diagnosis. Targeted NGS was performed with custom probes for 497 genes associated with hematologic disorders. After genomic DNA was extracted from peripheral blood, prepared libraries were hybridized with capture probes and sequenced using NextSeq 550Dx (Illumina, San Diego, CA, USA).
Result
Among the 29 patients, ANK1 variants were detected in five, four of which were pathogenic or likely pathogenic variants. SPTB variants were detected in six patients, five of which were classified as pathogenic or likely pathogenic variants. We detected g6pd pathogenic and spta1 likely pathogenic variants in two patients and one patient, respectively. Whole-gene deletions in both HBA1 and HBA2 were detected in two patients, while only HBA2 deletion was detected in one patient. One likely pathogenic variant in PLKR was detected in one patient, and one likely pathogenic variant in ALAS2 was detected in another.
Conclusion
Here, NGS played a critical role in definitive diagnosis in 18 out of 29 patients (62.07%) with suspected HHA. Thus, its incorporation into the diagnostic workflow is crucial.
Similar content being viewed by others

Background
Hereditary hemolytic anemia (HHA) refers to a heterogeneous group of genetic disorders that share one common feature: destruction of circulating red blood cells (RBCs). The destruction of RBCs may be due to membranopathies, enzymopathies, or hemoglobinopathies [1].
Membranopathies arise from mutations in genes coding for RBC membrane proteins such as spectrin (SPTA1 and SPTB) and ankyrin 1 (ANK1) and account for more than half of HHAs [2]. Examples include hereditary spherocytosis (HS), hereditary elliptocytosis (HE), and hereditary stomatocytosis (HSt), among which HS is the most common [3]. Enzymopathies affect the cellular metabolism of RBCs, which consists of anaerobic glycolysis, hexose monophosphate shunt, glutathione metabolism, and nucleotide metabolism [4, 5]. Enzymes involved in these pathways, such as glucose-6-phosphate dehydrogenase (G6PD) and pyruvate kinase (PKLR), are the most common enzyme disorders in HHAs [6]. Hemoglobinopathies include thalassemias (abnormal hemoglobin proportion) and structurally abnormal hemoglobin. Abnormal hemoglobin molecules produced in these conditions alter the RBC membrane, leading to cell destruction [7].
HHA prevalence is low in Korea because HS is less frequent in Asians compared with Caucasians, and Korea is not located in the thalassemia belt [8]. However, prevalence has increased over time due to the increased number of immigrants from Southeast Asia through international marriages and the growing awareness of the disease entity [9,10,11,12].
Moreover, no high-sensitivity, high-specificity test was available in the past; to rule out all the possible causes of HHA-like symptoms, patients had to go through an extensive battery of tests including osmotic fragility test, flow cytometry, mass spectrometry, direct sequencing, and multiplex ligation-dependent probe amplification (MLPA). Not only was this way of diagnosing cost-intensive but not possible in most facilities because of lack of required equipment and highly trained personnel. Next-generation sequencing (NGS) can sequence multiple genes simultaneously and can offer information about copy number variations as well. Therefore, incorporation of NGS has greatly facilitated the HHA diagnostic process. In this study, we report data for patients who underwent hemolytic anemia panel by NGS at a single tertiary hospital.
Methods
Patients
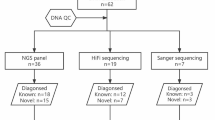
The study included 29 unrelated pediatric and young adult patients (age ≤ 25 years) who underwent NGS testing at our center for suspected hereditary anemia from March 2017 to December 2022. Clinical information and laboratory test results, including NGS, were retrospectively obtained from electronic medical records. The primary diagnosis at the time of NGS referral was regarded as the “diagnosis at referral”. In contrast, the main diagnosis after NGS test results was denoted as the “final diagnosis”.
Research involving human specimens complied with all relevant national regulations, institutional policies and the tenets of the Helsinki Declaration (as revised in 2013). This study was approved, and the requirement for informed consent was waived by the Institutional Review Board of Severance Hospital, Seoul, Republic of Korea (4-2023-0138).
Next-generation sequencing analysis
Genomic DNA was extracted from peripheral blood using a QIAsymphony DNA Mini Kit (Qiagen, Hilden, Germany). A custom capture panel (Dxome, Seoul, Republic of Korea) targeting coding exons and intron-exon boundaries of 497 genes related to hematologic disorders (Supplementary Table S1) was used. Prepared libraries were hybridized with capture probes and sequenced as paired-end reads (2 × 150 bp) using NextSeq 550Dx (Illumina, San Diego, CA, USA). NGS data analysis was performed with DxSeq Analyzer (Dxome). Single-nucleotide variants, small insertion and deletions, and copy number variants were identified [13, 14]. All variants were classified and reported as a 5-tier system according to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology guidelines [15]. Genes included in our panel that are associated with HHAs are classified according to their relevant phenotypes in Table 1.
Statistical analysis
Microsoft Excel 2013 (Seattle, WA, USA), Statistical Package for the Social Sciences (SPSS) v.23 (SPSS Inc., Chicago, IL, USA) were used for statistical analyses. Continuous variables were evaluated for normality using the Shapiro-Wilk test. The Mann-Whitney U test or independent two-sample t-test was employed according to the normality of independent variables. Fisher’s exact test was used to compare categorical variables. All parameters without normal distributions were presented as median with first and third quartiles. A P-value < 0.05 was interpreted as being statistically significant. Diagnostic yield was calculated by dividing the number of patients in whom at least one likely pathogenic or pathogenic (LP/P) variant was detected by the total number of patients. If only one LP/P variant was detected in an autosomal recessive gene, it was excluded from the calculation.
Results
Patient demographics are summarized in Table 2. Patients were divided into those with LP/P variants versus those with only variants of uncertain significance (VOUS) or no variants (none). The LP/P group was younger; showed higher reticulocyte indices and lactate dehydrogenase (LDH) and total bilirubin levels; and was more likely to have poikilocytosis. However, no statistically significant difference was observed between the two groups in terms of age, sex, hemoglobin level, reticulocyte count, red cell distribution width (RDW), LDH, and total bilirubin.
Among the 29 patients, ANK1 variants were detected in five, four of which were LP/P variants (Table 3). SPTB variants were found in six patients, five of which were classified as LP/P variants. Of three variants identified in the SPTA1 gene, one was classified as LP, while the other two were classified as VOUS. VOUS variants in the SLC4A1 gene were found in two cases, while four VOUS and one LP variant of PIEZO1 were detected in five patients. Pathogenic G6PD mutations were each identified as hemizygotes in two male patients. One likely pathogenic variant and one VOUS variant in the PKLR gene were detected in two patients, and one LP variant of ALAS2 was detected in another. Other enzyme-related genes with variants included GSR and GPI, but the variant was VOUS or the patient was a heterozygote carrier. Whole-gene deletions in both HBA1 and HBA2 were detected in two patients while lone HBA2 deletion was detected in one patient. One patient had a mutation at the start codon of HBB.
Discussion
In our study, NGS served as a critical diagnostic tool in 18 of 29 patients with suspected HHA (diagnostic yield: 62.07%). This LP/P variant detection rate was similar to that in previous studies using targeted NGS (54% by Fermo et al. [16], 64.9% by Russo et al. [17], and 67.3% by Nieto et al. [18], and 64.29% by Kim et al. [19] (Table 4)). Poikilocytosis was the only significantly different demographic factor between the LP/P and VOUS/none groups (Table 1). Although not statistically significant, the group with LP/P variants tended to be younger than the VOUS/none group. Newborns included due to hemolytic disease of the fetus and newborn (HDFN) included in the VOUS/none group might have masked some of the difference. Also, reticulocyte indices were noticeably elevated in the LP/P group compared with the VOUS/none group despite their differences not being significant.
Through the NGS HHA panel test, LP/P variants in membranopathy genes (ANK1, SPTB, SPTA1, PIEZO1, and SLC4A1) were found in 11 patients, accounting for 61.11% of the group. This finding was in line with the previous report that membranopathies accounted for more than half of HHAs [2] and other studies reported a similar proportion of membranopathies as well (Table 4). LP/P variants in enzymopathy genes, which include G6PD, PKLR, ALAS2, GPI, and GSR, were found in four cases (22.22% of confirmed HHA patients). Among these, G6PD and PKLR were the most common, which was also consistent with prior findings [6]. Last, whole-gene deletion of HBA1 and HBA2 and loss of function variants in HBB were found in 22.22% (4/18) of HHA patients.
A family history of hemolytic anemia or a history suggestive of hemolytic anemia such as cholecystectomy and splenectomy were found in seven patients with LP/P variants and one patient with two VOUS variants on SLC4A1 (Case 20). The parents of case 15 were asymptomatic carriers of PKLR gene variants. A more thorough history-taking that included specific questions about whether a relative had undergone either cholecystectomy or splenectomy would have increased the detection rate of an affected family member. Moreover, considering the low awareness of this disease entity in prior generations, it seems reasonable to assume the prevalence of a positive family history was underestimated.
Upon review, the SPTA1 R28H variant (case 11) was reclassified because we found a functional study that demonstrated no detectable affinity of the α-spectrin harboring R28H mutation to the β-subunit [20]. Thus, with the addition of a PS3, the then VOUS variant was reclassified as LP and reported to the clinician. Another variant, PIEZO1 R2456H (case 13), was also reclassified from VOUS to LP based on a functional study showing prolonged cation channel activity resulting in reduced osmotic fragility [21,22,23,24].
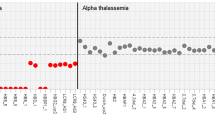
The clinical history of Case 13 is notable because it demonstrates the value of NGS in diagnosing HHA. When the patient was seven years old, his Hb dropped to 5.6 g/dL and he received a blood transfusion. Since he had no known family history of anemia, the clinician suspected leukemia and performed a bone marrow examination, with a negative result. Results of the Coomb’s test, paroxysmal nocturnal hemoglobinuria (PNH) test, G6PD enzyme activity test, osmotic fragility test and Hb electrophoresis test were all within normal limits. Two years prior to NGS testing, by the age of 21, he had undergone cholecystectomy and splenectomy due to gallbladder stone. In 2017, the patient was referred to our hospital due to hemolytic anemia. Subsequent NGS analysis uncovered a whole-gene deletion of HBA1 and HBA2, alongside an LP/P variant in PIEZO1, leading to a conclusive diagnosis of alpha thalassemia minor combined with dehydrated hereditary stomatocytosis. While each of these diseases individually is typically associated with mild anemia and infrequent symptom manifestation, the combined effect of these genetic aberrations accounted for the severity observed in this case. Additionally, this genetic combination shed light on the absence of any notable family history pertaining to these diseases. If NGS had been available when he was seven years old, he would not have had to do all those tests, especially a bone marrow biopsy. This case is representative of the limitations of conventional HHA tests and how NGS can overcome them.
The biggest limitation of this study is the small number of patients; if we had compared larger groups of patients, we might have obtained a significant difference between the LP/P and VOUS/none groups and been able to guide clinicians on the demographic factors and biochemical results most suggestive of HHA. Also, because this was a retrospective study, some test results were missing, and samples from other potentially affected family members could not be obtained. Nevertheless, we demonstrated the value of NGS for a confirmatory diagnosis of HHA. Moreover, NGS test results can aid patients in genetic counseling and family planning. Further studies with larger HHA cohorts and genotype-phenotype correlation information are needed.
Conclusion
NGS played a critical role in definitive diagnosis in 18 of 29 patients (62.07%) with suspected HHA because the disease comprises overlapping phenotypes and ample genetic heterogeneity, hindering an accurate diagnosis. Through our review of patient data, we demonstrated the invaluable role of NGS in HHA diagnosis and the time and resources that can be saved if such analysis is performed at an early stage of suspicion. Thus, incorporation of NGS into the diagnostic workflow is crucial.
Data Availability
The datasets generated and/or analysed during the current study are available in ClinVar, SCV003932458, SCV003932459, SCV003932460, SCV003932461, SCV003932462.
Abbreviations
- G6PD:
-
glucose-6-phosphate dehydrogenase
- Hb:
-
hemoglobin
- HDFN:
-
hemolytic disease of the fetus and newborn
- HE:
-
hereditary elliptocytosis
- HHA:
-
hereditary hemolytic anemia
- HS:
-
hereditary spherocytosis
- HSt:
-
hereditary stomatocytosis
- LDH:
-
lactate dehydrogenase
- LP:
-
likely pathogenic
- MLPA:
-
multiple ligation-dependent probe amplification
- NGS:
-
next-generation sequencing
- P:
-
pathogenic
- RBC:
-
red blood cell
- RDW:
-
red cell distribution width
- VOUS:
-
variants of uncertain significance
References
Kedar PS, Colah RB, Kulkarni S, Ghosh K, Mohanty D. Experience with eosin-5’-maleimide as a diagnostic tool for red cell membrane cytoskeleton disorders. Clin Lab Haematol. 2003;25:373–6.
Yawata Y, Kanzaki A, Yawata A, Nakanishi H, Kaku M. Hereditary Red Cell membrane Disorders in Japan: their genotypic and phenotypic features in 1014 cases studied. Hematology. 2001;6:399–422.
King MJ, Garçon L, Hoyer JD, Iolascon A, Picard V, Stewart G, et al. ICSH guidelines for the laboratory diagnosis of nonimmune hereditary red cell membrane disorders. Int J Lab Hematol. 2015;37:304–25.
Arya R, Layton DM, Bellingham AJ. Hereditary red cell enzymopathies. Blood Rev. 1995;9:165–75.
Jacobasch G, Rapoport SM. Hemolytic anemias due to erythrocyte enzyme deficiencies. Mol Aspects Med. 1996;17:143–70.
Prchal JT, Gregg XT. Red cell enzymes. Hematol Am Soc Hematol Educ Program 2005:19–23.
Poyart C, Wajcman H. Hemolytic anemias due to hemoglobinopathies. Mol Aspects Med. 1996;17:129–42.
Ahn DH, Sohn K, Kang I, Kang J, Ko Y, Ko Y, et al. Statistical analysis of hemolytic anemia in Korea. Korean J Hematol. 1991;26:445–61.
Kim Y-P, Joh J-Y, Shin I-S. Family function of the families consisting of asian immigrant women living in South Korea: a 3-year longitudinal study. Asia Pac J Public Health. 2015;27:NP2702–NP11.
Lee YK, Kim H-J, Lee K, Park SH, Song SH, Seong M-W, et al. Recent progress in laboratory diagnosis of thalassemia and hemoglobinopathy: a study by the Korean Red Blood Cell disorder Working Party of the korean Society of Hematology. Blood Res. 2019;54:17–22.
Risinger M, Emberesh M, Kalfa TA. Rare hereditary hemolytic anemias: diagnostic approach and considerations in management. Hematology/Oncology Clin. 2019;33:373–92.
Shim YJ, Jung HL, Shin HY, Kang HJ, Choi JY, Hah JO, et al. Epidemiological study of Hereditary hemolytic Anemia in the korean Pediatric Population during 1997–2016: a Nationwide Retrospective Cohort Study. J Korean Med Sci. 2020;35:e279.
Kim H, Shim Y, Lee TG, Won D, Choi JR, Shin S, et al. Copy-number analysis by base-level normalization: an intuitive visualization tool for evaluating copy number variations. Clin Genet. 2023;103:35–44.
Kook HW, Kim JJ, Park MR, Jang JE, Min YH, Lee ST, et al. Therapy-related Acute Lymphoblastic Leukaemia has a Unique Genetic Profile compared to De Novo Acute Lymphoblastic Leukaemia. J Cancer. 2022;13:3326–32.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Fermo E, Vercellati C, Marcello AP, Keskin EY, Perrotta S, Zaninoni A et al. Targeted next generation sequencing and diagnosis of congenital hemolytic anemias: a three years experience monocentric study. Front Physiol 2021:724.
Russo R, Andolfo I, Manna F, Gambale A, Marra R, Rosato BE, et al. Multi-gene panel testing improves diagnosis and management of patients with hereditary anemias. Am J Hematol. 2018;93:672–82.
Nieto JM, Rochas-López S, González-Fernández FA, Villegas-Martínez A, Bolaños-Calderón E, Salido-Fiérrez E, et al. Next generation sequencing for diagnosis of hereditary anemia: experience in a spanish reference center. Clin Chim Acta. 2022;531:112–9.
Kim N, Kim TY, Han JY, Park J. Five years’ experience with gene panel sequencing in Hereditary hemolytic Anemia screened by routine peripheral blood smear examination. Diagnostics (Basel) 2023;13.
Gaetani M, Mootien S, Harper S, Gallagher PG, Speicher DW. Structural and functional effects of hereditary hemolytic anemia-associated point mutations in the alpha spectrin tetramer site. Blood. 2008;111:5712–20.
Albuisson J, Murthy SE, Bandell M, Coste B, Louis-Dit-Picard H, Mathur J, et al. Dehydrated hereditary stomatocytosis linked to gain-of-function mutations in mechanically activated PIEZO1 ion channels. Nat Commun. 2013;4:1884.
Andolfo I, Alper SL, De Franceschi L, Auriemma C, Russo R, De Falco L, et al. Multiple clinical forms of dehydrated hereditary stomatocytosis arise from mutations in PIEZO1. Blood. 2013;121:3925–35. s1-12.
Bae C, Gnanasambandam R, Nicolai C, Sachs F, Gottlieb PA. Xerocytosis is caused by mutations that alter the kinetics of the mechanosensitive channel PIEZO1. Proc Natl Acad Sci U S A. 2013;110:E1162–8.
Glogowska E, Schneider ER, Maksimova Y, Schulz VP, Lezon-Geyda K, Wu J, et al. Novel mechanisms of PIEZO1 dysfunction in hereditary xerocytosis. Blood. 2017;130:1845–56.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Research Foundation of Korea (NRF-2021R1I1A1A01045980).
Author information
Authors and Affiliations
Contributions
YJC analyzed patient data and wrote the manuscript. HK provided insight into the statistical analysis. WKA, JWH, and CJL provided clinical insight about HHA. STL and JRC provided the NGS data and information on its methodology. SH and SS designed the study, and reviewed/edited the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Research involving human specimens complied with all relevant national regulations, institutional policies and the tenets of the Helsinki Declaration (as revised in 2013). This study was approved, and the requirement for informed consent waived by the Institutional Review Board of Severance Hospital, Seoul, Korea (4-2023-0138).
Consent for publication
Not applicable.
Competing interests
STL and JRC are employees of Dxome co., Ltd. Other authors declare no potential conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Choi, Y.J., Kim, H., Ahn, W.K. et al. Diagnostic yield of targeted next-generation sequencing for pediatric hereditary hemolytic anemia. BMC Med Genomics 16, 215 (2023). https://doi.org/10.1186/s12920-023-01648-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-023-01648-y