
Abstract
Background
Concurrent variation in adiposity and inflammation suggests potential shared functional pathways and pleiotropic disease underpinning. Yet, exploration of pleiotropy in the context of adiposity-inflammation has been scarce, and none has included self-identified Hispanic/Latino populations. Given the high level of ancestral diversity in Hispanic American population, genetic studies may reveal variants that are infrequent/monomorphic in more homogeneous populations.
Methods
Using multi-trait Adaptive Sum of Powered Score (aSPU) method, we examined individual and shared genetic effects underlying inflammatory (CRP) and adiposity-related traits (Body Mass Index [BMI]), and central adiposity (Waist to Hip Ratio [WHR]) in HLA participating in the Population Architecture Using Genomics and Epidemiology (PAGE) cohort (N = 35,871) with replication of effects in the Cameron County Hispanic Cohort (CCHC) which consists of Mexican American individuals.
Results
Of the > 16 million SNPs tested, variants representing 7 independent loci were found to illustrate significant association with multiple traits. Two out of 7 variants were replicated at statistically significant level in multi-trait analyses in CCHC. The lead variant on APOE (rs439401) and rs11208712 were found to harbor multi-trait associations with adiposity and inflammation.
Conclusions
Results from this study demonstrate the importance of considering pleiotropy for improving our understanding of the etiology of the various metabolic pathways that regulate cardiovascular disease development.
Similar content being viewed by others

Introduction
Self-identified Hispanic/Latino Americans (hereafter Hispanics) harbor an elevated burden of obesity (> 40%) [1]. Excess body weight is a known risk factor for a range of metabolic abnormalities, including insulin resistance [2], high blood pressure [3], dyslipidemia [4], and metabolic hormones disorders [5].
The associations between obesity and metabolic irregularities are often attributed to induction of inflammatory responses by adipose tissue [6, 7]. Fat accumulation leads to enlargement (hypertrophy) of adipocytes with impaired capacity for fat oxidation, glucose regulation and amelioration of inflammatory activities [8, 9]. Hypertrophied adipocytes exhibit a distorted cytokine secretion profile [7, 10]; cytokines are key drivers of C-reactive protein (CRP) production [11], which is associated with cardiovascular diseases (CVD) [12], and a significant predictor of CVDs’ clinical course [13]. Clinical studies suggest lowering serum CRP levels significantly reduce CVD incidences [14].
Although this process has been highlighted as the presumptive pathophysiological direction of adiposity-CVD risk factors association, compelling evidence also suggests that inflammation could induce adiposity. For example, inflammation-induced insulin resistance may trigger hyperplastic and hypertrophic changes in adipose tissues to alleviate glucose accumulation [15]. Furthermore, results from a longitudinal study illustrated steeper weight gain among those with higher baseline CRP levels in a 3-year follow-up period [16]. More corroborating evidence for adiposity-inflammation interrelatedness has emerged with the increased availability of genetic studies. High levels of inflammatory markers in mice carrying genes that increase obesity [17], or concurrent inflammatory effects where the expression levels of adipose tissue genes were regulated [18, 19], all support common molecular pathways for inflammation and obesity.
These common pathways [20] suggest pleiotropic disease underpinnings [21], which may inform classification and treatment of disease. Yet, exploration of pleiotropic variants in the context of adiposity-inflammation has been scarce [22], and none have included Hispanics populations. Given the high level of ancestral diversity in Hispanics, genetic studies may reveal variants that are infrequent/monomorphic in more homogeneous populations [23, 24]. Furthermore, the differential distribution of inflammatory markers associated with comparative adiposity configurations [25, 26], patterns of fat accumulation [27, 28], and the prevalence of cardiometabolic abnormalities in Hispanics [29] highlights the need for adiposity-inflammation pleiotropy study in this underserved group.
We, therefore, examined individual and shared genetic effects underlying inflammatory (CRP) and adiposity-related traits (overall obesity: Body Mass Index [BMI], and central adiposity: Waist to Hip Ratio [WHR]) in self-identified Hispanics participating in the Population Architecture Using Genomics and Epidemiology (PAGE) study with replication of effects in the Cameron County Hispanic Cohort. For multi-trait analysis, we used adaptive Sum of Powered Score (aSPU) [30] method. Advantages of aSPU over similar methods include its ability to accommodate differences in sample size between traits and phenotypic distributions, preserved type 1 error in select scenarios, where some other methods did not [30], performance in the context of heterogeneity in direction of phenotypic effect, and scalability and computational efficiency which enabled examination of millions of variants in a reasonable time.
We also assessed whether replicated signals were directly associated with relevant phenotypes (i.e., biologic pleiotropy) or indirectly associated as mediators (i.e., mediated pleiotropy). Assuming that adiposity and inflammation are interrelated manifestations of biological mechanisms, pleiotropy assessment may provide insights on the development of effective preventive strategies designed to improve the cardiovascular health of Hispanics. Our results illustrate the broad utility of pleiotropy assessment to illuminate the biology of complex diseases and traits.
Materials and methods
Study populations
The conceptual framework for the adiposity-inflammation pleiotropy study is displayed in Additional file 2: Fig. S1. We chose a discovery and replication study design of self-identified Hispanics study participants from the two studies. The PAGE study population includes several studies at different sites [31, 32] as described previously [33]. In brief, PAGE consists of multiple populations grouped by self-identified ethnicities: European Americans (Non-Hispanic whites [NHW]), African Americans (AA), Hispanics, American Indians (AI), East Asians (ASN), and Native Hawaiians/Pacific Islander (HAW). All participating sites in PAGE ascertained both men and women except for the women only Women’s Health Initiative (WHI). We studied the Hispanics sub-cohort as our discovery set (N = 35,871) from four studies within the PAGE: Hispanic Community Health Study/ Study of Latinos (HCHS/SOL), the Women’s Health Initiative (WHI), BioMe Biobank (BioMe), and MultiEthnic Cohort (MEC). Principal component analyses suggest substantial ancestral variability among self-identified Hispanics/Latinos in PAGE (Additional file 1: Appendix, Section 1. Fig. 1.). All study participants provided written informed consent and each study was approved by relevant institutional review board. The replication study included self-identified Hispanics study participants recruited at the US-Mexico border, the Cameron County Hispanic Cohort (CCHC). This cohort was established in 2004 and now numbers 5,000 individuals randomly selected from a population with severe health disparities. Many of the participants are originally from Mexico, and we have collected detailed information on participant place of birth, how recently they arrived in the US, income, marital status, and employment [34]. Out of 5000 individuals recruited for the cohort, a sub sample of 3,313 genotyped individuals are included in this study. The CCHC is almost exclusively self-report Mexican American, with substantial patterns of admixture from Native American and European ancestry, with limited African admixture (Additional file 1: Appendix, Section 1. Fig. 2.)
Phenotype measurements and quality control
We studied two anthropometric phenotypes: BMI (kg/m2; a measure of overall adiposity) and WHR (proxy measure of central obesity). For all studies, except in MEC, height and weight were measured by study staff at study enrollment, to calculate BMI (weight/height2). In MEC, BMI is based on self-reported height and weight at enrollment. Pilot analyses of BMI in MEC illustrated a comparable distribution to national surveys [35]. Waist circumference was measured at the level of natural waist in horizontal plane to the nearest 0.5 cm [36]; no waist or hip measurement was available for BioMe sub-cohort; WHR measures were multiplied to 100 for ease of interpretation. Analyses of WHR measures were stratified by sex to account for well-established sexual dimorphism. For inflammation, we used high sensitivity CRP (hsCRP) as robust marker of systemic inflammation with strong association with central obesity [37]; CRP was measured at enrollment separately in each contributing cohort and subsequently harmonized for the entire set [38].
Genotype and quality control
Most individuals (~ 20,000) were genotyped using the MEGA array panel; details on the genotyping were published previously [39]. The remaining PAGE samples were genotyped with Affymetrix and Illumina arrays. We used the genome-wide variants’ set that was imputed to 1000 Genome Phase 3 reference population after quality control (QC), and details are accessible [40]. For this study analyses, we next excluded genetic variants with poor imputation (R2 < 0.4), effective sample size of < 30, and minor allele frequency (MAF) of < 0.05; criteria used for calculating effective sample size for each single nucleotide polymorphisms (SNP) is defined in Additional file 1: Appendix, Section 2. Out of > 60 million SNPs, approximately 32 million SNPs were removed because of low MAF. The replication cohort from the CCHC was genotyped at the Vanderbilt University Medical Center genotyping core facility, VANTAGE, using MEGA-EX Array panel. After standard QC, imputation was completed using the Trans-Omics for Precision Medicine (TOPMed) [41] freeze 8 panel available on the NHLBI Imputation Server (https://imputation.biodatacatalyst.nhlbi.nih.gov), by having American ancestry as reference group. We used same QC criteria to exclude SNPs in this replication cohort as substudies from PAGE.
Statistical analyses
Association tests
The skewed distribution of CRP necessitated log-transformation before regression analyses. Extreme observations (defined as values outside three standard deviations (SD) from the mean in the log transformed distributions (CRP), or 4SD for BMI and WHR) were flagged as outliers and excluded from all analyses. WHR for women and men were treated as separate phenotypes, so that the phenotypes included were CRP, BMI, WHR-men, and WHR-women. Residuals for each phenotype were estimated from linear regression models adjusted for age, age2, sex (when applicable), BMI (when applicable), center, cohort, and age-by-sex interaction, performed separately in each study. Residuals were then inverse rank normalized and used as the outcome for univariate genome-wide association tests (GWAS). All MEGA-genotyped samples were pooled together for genetic association testing while the remaining non-MEGA data were analyzed by study. GWAS were performed using SUGEN, adjusted for 10 genetic principal components (PC). SUGEN employs generalized estimating equations (GEE) to adjust for family relationships (first or second degree), with independent error distributions by self-identified group [42]. GWAS results from MEGA samples and other studies were then meta-analyzed with METAL [43] assuming fixed-effect inverse-variance weighting.
Multi-trait association test
Various multi-trait methods have recently been proposed [44]. Most of these methods offer superior statistical power compared with multivariate analysis [45]. To identify loci with evidence for associations with one or more of the adiposity and inflammation traits, we combined Z-score statistics from meta-analysis results from each phenotype using aSPU [30] test.
Briefly, aSPU aggregates information across n phenotypes for a given SNP by taking the sum of its univariate GWAS Z-scores Sp, each raised to some power γ, so that a higher γ increases the influence of strongly-associated phenotypes on the score \(SPU\left(\gamma \right)={\sum_{n}{(S}_{p})}^{\gamma }\) [30]. With γ taking one of many competing values (1, 2, …, 8), aSPU selects a maximally-efficient scheme to detect combined phenotype effects on the entire group of phenotypes. Monte-Carlo methods applied to univariate GWAS Z-scores estimated by inverse-variance-weighted meta-analysis are then used to generate aSPU P-values, which are interpreted as evidence for an association of the SNP with at least one phenotype. SNP-specific γ scores also provide a mechanism to group loci from most to least likely to be pleiotropic.
A set containing trait-specific GWAS-inferred Z-scores (from all contributing phenotypes) was constructed. Multi-trait analysis was performed using the JaSPU (github.com/kaskarn/JaSPU) package, with the number of iterations set to 100 billion. This program relies on a Markov Chain Monte-Carlo (MCMC) [46] iterative process to estimate p-values. The p-values generated from this test were used to assess statistical significance of whether SNPs were associated with one or more phenotypes.
We considered a SNP as a candidate for pleiotropy analyses if a): exceeded significance threshold for multi-trait test at PaSPU < 1.25 × 10–8, a conservative threshold [47] which accounted for multiple testing on likely low frequency SNPs and the number of traits tested, and b) nominally significant for inflammation and at least one adiposity trait in univariate GWAS results. We subsequently identified significantly associated loci where: i) variants in the region met the criteria above, and (ii) were present in pooled MEGA results; latter criterion was used to ensure understudied SNPs existed in both PAGE and CCHC results, thereby usable for likewise comparison. Linkage disequilibrium (LD) analyses identified independent variants (R2 < 0.1). For each independent locus, we highlighted the most likely functional or closest observed proxy to functional variant based on a literature review, and the lowest or close to the lowest PaSPU.
Bioinformatic annotation
Ensemble Variant Effect Predictor (VEP) (grch37.ensembl.org/) was used to determine the effect of each variant on genes, transcripts, protein sequences, and regulatory regions. We searched replicated variants and their close LD proxies (with LD cut-point of R2 > 0.8) in the publicly available database PhenoScanner [48] and GWAS Catalogue [49] for reported associations with any phenotype including adiposity and inflammation traits at P < 1 × 10–5 significance level. LD clumping was performed using the AMR ancestry panel. We used both Gene Expression Portal (GTEx Portal) [50] and PhenoScanner to examine variants and close proxies (R2 > 0.5) for functional significance and influence on tissue expression at GWAS significance level, which are genomic regions associated with expression levels of messenger RNA (mRNA). Finally, Haploreg was used to evaluate the regulatory potentials of variants on haplotype blocks, such as candidate regulatory SNPs at disease-associated loci [51]. This annotation tool was used to assess the effects of SNPs on regulatory motifs and expression Quantitative Trait Loci (eQTL).
Replication of shared genetic loci
All significant variants were taken forward to replication in the CCHC. GWASs were performed with SUGEN. Multi-trait analysis was completed following the same aSPU based approach used in the discovery stage. The criteria set for replication included, a) variant is nominally significant in multi-trait test (Paspu < 0.05), b) variant was nominally significant for CRP and at least one anthropometric trait, and c) direction of effect of each SNP was same in discovery vs replication cohort for the tested trait.
Causal pathway analyses
We performed a causal mediation pathway analyses of replicated variants to assess pleiotropic potential of identified variants, and whether an observed association is independently associated with several phenotypes and the genetic effect is transmitted through a common pathway that is upstream to the associated phenotypes (i.e., biologic pleiotropy, Additional file 2: Fig. S1); or induced due to relationships between the outcome phenotype and a “mediating” phenotype [53]. Elucidating these complexities could provide novel biological insights [54].
Mediation analyses allows for the assessment of whether a SNP has a direct effect on the phenotype of interest. The R package Mediation [55] was used for analysis, which conducts a three-step process. The total effect between a SNP and an outcome is assumed to be the sum of the average causal mediation effects (ACMEs) and the average causal direct effect (ADE). The first step includes estimating the distribution of the mediator phenotype as a function of the SNP, adjusted for genetic ancestry (e.g., mediator ~ genotype (variant) + PCs + covariates). The next step involves estimating the distribution of the outcome phenotype as a function of the mediator phenotype, genotype, and covariates (e.g., outcome ~ mediator + genotype + PCs + covariates). The final step combines the fitted models in the mediation equation, providing estimates and p-values for the direct (e.g., genotype → adiposity [outcome]) and indirect/mediating (e.g., genotype → mediator → adiposity [outcome]) associations. We did not allow for any mediator by genotype interaction [16]. We performed mediation analyses in the biggest subset of the discovery cohort that were genotyped by the same MEGA array, excluding samples with missingness since mediation method required complete observation across output and mediating phenotypes; the total available sample size stood at 15,600. We considered age and sex as covariates in the mediator and outcome models, together with first 10 PCs. We additionally performed sex stratified analyses but 95% confidence intervals and point estimates overlapped with sex adjusted results. With this method, quasi-Bayesian Monte Carlo simulations are used for the estimation of standard errors. The number of simulations was set to 1000, with robust standard errors using sandwich estimators [56]. We also used same mediation package to further perform sensitivity analysis on the mediated and direct effects for violation of the sequential ignorability assumption and assessing the effect of unmeasured confounders. For genotypes, we used allele dosages and models were estimated assuming additive genetic effects.
For each significantly replicated SNP, if a SNP was previously associated with inflammation, we performed mediation analyses assuming CRP as mediator and an adiposity trait as the outcome (Additional file 3: Fig. S2); conversely, where a SNP was primarily associated with adiposity in prior studies, then we used adiposity phenotype as the mediator and CRP as the outcome. Statistical significance of ADE in either scenario would suggest an independent association of the lead SNP with the outcome and a potentially biologic pleiotropy effect.
Results
Sample description
Most participants were women (N = 22,733 (63.4%) and the average age was 52.9 years (SD = 14; Table 1). Most study participants were overweight and centrally obese, with an average of BMI of 29.2 kg/m2 and WHR of 86.6 in the discovery group, and mean BMI of 30.8 kg/m2 and mean WHR of 91.2 in CCHC replication group. Inflammation was apparent with a mean value of CRP of 4.2 mg/dL in the discovery group. A small number of samples (N = 33 for BMI, N = 21 for WHR in females, N = 13 for CRP) were excluded as outliers.
GWAS and multi-trait association analyses in PAGE
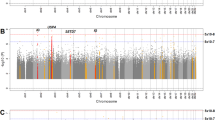
More than 16 million SNPs were available for analyses post QC and were evaluated in our combined phenotype analysis of 4 traits (BMI, WHR in men, WHR in women and CRP). No genomic inflation was observed in univariate GWAS (Additional file 5: Table S1). In univariate GWAS, we observed 3 independent loci (i.e., LD R2 < 0.1) exceeding genome-wide significance level (P < 5 × 10−8) for BMI, one locus for WHR in men and one for WHR in women, and 11 loci for CRP in meta-analyzed results (Additional file 5: Table S2). All observed loci were previously known. In subsequent multi-trait association assessment, SNPs representing 7 independent loci exceeded the multi-trait test threshold (PaSPU < 1.25 × 10−8) (Additional file 5: Table S3).
Phenotype decomposition of multi-trait significant variants in PAGE
All SNPs except for rs11642015 (FTO) were associated with CRP at GWAS significance level; the FTO SNP was associated with BMI (Additional file 5: Table S3). SNPs rs11208712 and rs9987289 (both intergenic) were also associated with BMI at nominal level. SNPs rs62158854 (IL1RN), rs1169288 (HNF1A) and rs439401 (APOE) were nominally associated with WHR-women. The remaining rs12064564 on chromosome 1 was nominally associated with WHR-men.
Functional assessments
Phenoscanner and GWAS-catalogue probe indicated that multi-trait significant SNP on chromosome 16 (rs11642015, overlapping FTO gene) and its close proxies (R2 > 0.8) is primarily associated with anthropometric and fat mass traits (Additional file 4: Fig. S3, Additional file 5: Table S4). rs9987289 on chromosome 8 (LOC157273) is associated with lipid traits, liver enzymes, and glucose (diabetes) measures. rs1169288 on chromosome 12 (HNF1A), is primarily reported in association with lipid traits and CRP. rs11208712 (intergenic) on chromosome 1 is reported for blood immunity cells and CRP. Variant rs62158854 on chromosome 2 (IL1RN gene) is mostly associated with blood immunity cells. We found only one study reported rs12064564 on chromosome 1 (intergenic) associated with CRP. The APOE variant (rs439401, on chromosome 19) is frequently reported in association with lipid traits, and in some studies in connection with Alzheimer’s disease.
Tissue gene expression
Probe of GTEx, and PhenoScanner for multi-trait significant variants suggested that except for rs12064564 and rs11208712 on chromosome 1, the remaining SNPs were associated with higher mRNA expression in several tissues including skin, thyroid, brain, adipose, artery, whole blood, esophagus, testis, and colon (Additional file 6: Fig. S4, Additional file 5: Table S5). SNP rs11208712 exhibited gene expression only with whole blood and pancreas, though not at GWAS significance level.
Replication with CCHC
We carried forward the 7 loci for replication in the CCHC population. 2 of the 7 variants met replication criteria in CCHC multi-trait association results (Table 2, Additional file 5: Table S3): variant rs439401 (APOE) on chromosome 19, and rs11208712 (intergenic) on chromosome 1.
Regulatory significance of replicated multi-trait SNPs
We searched Haploreg for regulatory features for overlap of the replicated variant with enhancers with enrichment of histone 3 lysine 4 monomethylated 1 [H3K4me1] and H3K27 acetylation [H3K27ac], and promoter epigenomic markers with enrichment of H3K4me3 and H3K9ac. Lead SNP rs439401 (APOE) exhibited higher frequencies of enhancer and promoter annotations in the epithelial and brain tissues (Additional file 5: Table S6), enhancer annotations with embryonic stem cells, muscles, heart, digestive, and various cells with hormonal functionality. The remaining rs11208712 on chromosome did not illustrate regulatory annotation in Haploreg database.
Causal mediation analyses
We performed mediation analyses for replicated SNPs. Primarily known for association with inflammation, we assumed CRP as mediator in association between rs11208712 and BMI (the phenotype at association with SNP at nominal level). Results suggest directly positive and indirectly negative (via CRP) associations of the SNP with BMI, though total effect (e.g., direct + indirect effect) was positive but marginally nonsignificant (beta = 0.12, 95% CI [− 0.01, 0.26]) (Table 3). In comparison, rs439401 (APOE) was previously associated with adiposity (WHR), and therefore was used as mediator in association between the SNP and CRP. Analyses suggested positive indirect (via WHR) and direct (independent of WHR) associations with CRP. These results were supportive of biologic pleiotropy effects for APOE. Results were estimated with robust standard errors and remained consistent after sensitivity tests.
Discussion
We performed pleiotropy analyses on univariate meta-analyzed GWAS of BMI, WHR and CRP, as measures of overall and central adiposity and inflammation respectively, using data from self-identified Hispanics in PAGE cohort. We identified 7 independent signals with potential for pleiotropy, 2 of which were replicated with CCHC population including rs11208712 and rs439401.
The lead variant on chromosome 1 (rs11208712) is intergenic and the closest gene is leptin receptor (LEPR), a gene linked to obesity [57, 58]. Although this SNP has not previously been associated with inflammation, another SNP (rs4655582) which is a close proxy of this variant (R2 > 0.9) was associated with acute-phase serum amyloid A proteins (A-SAA) [59]. This family of apolipoproteins is secreted during the acute phase of inflammation [60]. Increased expression of this proinflammation lipolytic adipokine is suggested to play a role in systemic inflammation, free fatty acid production and is proposed as a link between obesity and CVD risk factors including insulin resistance and atherosclerosis [61]. Mediation analyses did not suggest significant association with adiposity once adjusted for mediator (CRP).
The only replicated likely pleiotropic SNP, rs439401 overlapping APOE gene is reported in association with different lipid markers. Analyses suggested higher gene expression levels associated with this SNP in adipose, skin, liver, thyroid, and adrenal tissues. Overall, several genetic variants in APOE has been widely related to pro-inflammatory measure [62, 63], and obesity [64]. One recent meta-analysis also supported strong correlation between better known APOE variants and CVD, especially CHD [65]. Although rs439401 is less interrogated compared to other APOE SNPs, but we found a study linking the SNP with high blood pressure [66], and another linking SNP’s allelic variation to poor metabolic profile (obesity, insulin resistance and high triglyceride level [67]. SNP rs584007, a variant in the same region in tight LD with rs439401, is frequently associated with CRP level [68]. rs439401 is also associated with lower risk of Alzheimer’s disease [69], and appears to be functionally independent of another APOE SNP (rs429358) which is the most widely studied Alzheimer’s disease variant (R2 ~ 0.1).
Mediation analyses illustrated a positive indirect association with CRP independent of WHR, and indirect positive association via WHR, and robust total positive association, consistent with prior suggestions. Interestingly, the SNP was also proposed as potentially pleiotropic in another study due to association with inflammatory reactions in leprosy through regulatory effects on lipid metabolism [70]. Authors suggested that SNPs’ risk allele is associated with low APOE gene expression level, leading to increased plasma lipoprotein level that facilitates survival of mycobacterium leprae in the skin and hence sustained inflammatory lesions. Increased lipoprotein is a known risk factor for arterial inflammation [71], but results from our mediation analyses suggest a concurrent and independent association with inflammation as well.
Inadequate power with replication set was a notable limitation to our approach as the remaining 5 likely pleiotropic SNPs did not replicate in CCHC. However, review of the literature suggests strong candidates for pleiotropy and larger replication sample set may signify their potentials. For example, the SNP on chromosome 12 (rs1169288) that overlaps HNF1A had previously been reported as a pleiotropic variant affecting cardiometabolic traits and CRP levels [72]. The FTO variant on chromosome 16 (rs11642015) is reported as pleiotropic SNP for inflammation and lipid traits [73], and associated with BMI adjusted diabetes [74].
The precise characterization of pleiotropic signals requires insight from joint studies of genetics and gene expression, particularly in the tissues of interest for the traits interrogated. However, functional annotation evaluation was able to provide suggestive evidence for possible functional roles associated with likely causal variants. For instance, mesenchymal cell H3K4me3 promoter annotation for rs439401 was suggestive of potential pleiotropic effects at the protein level. It also should be noted that annotation tools used for this study were primarily derived using European populations and may harbors limitations when used for functional annotation of variants in an admixed group like Hispanics.
Another notable limitation was our consideration of only one self-reported Mexican American study population for replication. While all PAGE study participants self-reported Hispanic/Latino ethnicity, a large range of study participants reported backgrounds from South and Central American populations. For example, study participants self-reported Dominican, Puerto Rican, Cuban, and Chilean background, among many others. It is well known that self-reported Hispanic/Latino populations display extensive ancestral diversity from Europe, Asia, Africa, and Native America [75, 76]. Thus, our lack of replication of some discovery genetic signals may have been influenced by the limited ancestral diversity CCHC study participants, when compared to the total PAGE population. However, we do have great confidence in our replicated signals, which were common in both study populations. Finally, caution should be exercised with causal interpretation of observed pleiotropic association which require longitudinal and randomized assessments.
The present study also has major strengths. This study is the first comprehensive assessment of pleiotropic associations between adiposity and inflammation traits in a self-identified Hispanics population. The results provide suggestive evidence for the regulatory effects of identified genetic variants on metabolic pathways and highlight the complexity and interrelatedness of seemingly independent phenotypic traits. The intentional selection of a genetically tri-admixed Hispanics population [76] with shorter haplotypes compared with European ancestry (EA) populations [77] greatly narrowed the number of SNPs identified for causal evaluations [78, 79], and allowed isolation of pleiotropic loci potentially specific to Hispanic/East Asian populations.
In conclusion, our study characterized potential pleiotropy between inflammation and adiposity susceptibility variants in self-identified Hispanics. We found 2 variants with evidence for biologic pleiotropy. The lead variant on APOE (rs439401) and rs11208712 were found to be harbor pleiotropic association with adiposity and inflammation. Particularly, both loci illustrate significant and concurrent associations with lipoproteins levels and inflammatory markers’ variations which indicate a common functional pathway with overlapping molecular underpinning. Additional loci with similar regulatory patterns were also isolated, though study lacked power to replicate associations. Results from this study demonstrate the importance of conducting multi-trait analyses, for improving our understanding of the etiology of the various metabolic pathways that regulate cardiovascular disease development.
Availability of data and materials
PAGE accession numbers are: phs000356 PAGE collectively; phs000223 PAGE-ARIC and phs000280 ARIC Cohort; phs000555 PAGE-HCHS/SOL and phs000810 HCHS/SOL Cohort; phs000220 PAGE-MEC; phs000227 PAGE-WHI and phs000200 WHI Cohort; phs000925 PAGE-IPM-BioMe. Summary of study results of meta-analyses and aSPU multi-trait analyses will be made available on dbGap repository upon publication approval.
References
Ogden CL, Fryar CD, Martin CB, Freedman DS, Carroll MD, Gu Q, Hales CM. Trends in obesity prevalence by race and hispanic origin—1999-2000 to 2017–2018. JAMA. 2020;324(12):1208–10.
Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Investig. 2000;106(4):473–81.
Pausova Z. From big fat cells to high blood pressure: a pathway to obesity-associated hypertension. Curr Opin Nephrol Hypertens. 2006;15(2):173–8.
Franssen R, Monajemi H, Stroes ES, Kastelein JJ. Obesity and dyslipidemia. Med Clin North Am. 2011;95(5):893–902.
Chikunguwo S, Brethauer S, Nirujogi V, Pitt T, Udomsawaengsup S, Chand B, Schauer P. Influence of obesity and surgical weight loss on thyroid hormone levels. Surg Obesity Related Dis. 2007;3(6):631–5.
Schmidt MI, Watson RL, Duncan BB, Metcalf P, Brancati FL, Sharrett AR, Davis C, Heiss G. Investigators ARiCS: Clustering of dyslipidemia, hyperuricemia, diabetes, and hypertension and its association with fasting insulin and central and overall obesity in a general population. Metabolism. 1996;45(6):699–706.
Jafar TH, Chaturvedi N, Pappas G. Prevalence of overweight and obesity and their association with hypertension and diabetes mellitus in an Indo-Asian population. CMAJ. 2006;175(9):1071–7.
Koh EH, Kim A-R, Kim H, Kim JH, Park H-S, Ko MS, Kim M-O, Kim H-J, Kim BJ, Yoo HJ. 11b-HSD1 reduces metabolic efficacy and adiponectin synthesis in hypertrophic adipocytes. J Endocrinol. 2015;225:147–58.
Gustafson B. Adipose tissue, inflammation and atherosclerosis. J Atheroscler Thromb. 2010;17(4):332–41.
Blackburn P, Després JP, Lamarche B, Tremblay A, Bergeron J, Lemieux I, Couillard C. Postprandial variations of plasma inflammatory markers in abdominally obese men. Obesity. 2006;14(10):1747–54.
Abeywardena MY, Leifert WR, Warnes KE, Varghese JN, Head RJ. Cardiovascular biology of interleukin-6. Curr Pharm Des. 2009;15(15):1809–21.
Wang A, Liu J, Li C, Gao J, Li X, Chen S, Wu S, Ding H, Fan H, Hou S. Cumulative exposure to high-sensitivity C-reactive protein predicts the risk of cardiovascular disease. J Am Heart Assoc. 2017;6(10):e005610.
Tajfard M, Tavakoly Sany SB, Avan A, Latiff LA, Rahimi HR, Moohebati M, Hasanzadeh M, Ghazizadeh H, Esmaeily H, Doosti H. Relationship between serum high sensitivity C-reactive protein with angiographic severity of coronary artery disease and traditional cardiovascular risk factors. J Cell Physiol. 2019;234(7):10289–99.
Fu Y, Wu Y, Liu E. C-reactive protein and cardiovascular disease: From animal studies to the clinic. Exp Ther Med. 2020;20(2):1211–9.
Reilly SM, Saltiel AR. Adapting to obesity with adipose tissue inflammation. Nat Rev Endocrinol. 2017;13(11):633–43.
Barzilay J, Forsberg C, Heckbert S, Cushman M, Newman A. The association of markers of inflammation with weight change in older adults: the Cardiovascular Health Study. Int J Obes. 2006;30(9):1362–7.
Park EJ, Lee JH, Yu G-Y, He G, Ali SR, Holzer RG, Österreicher CH, Takahashi H, Karin M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140(2):197–208.
Soukas A, Cohen P, Socci ND, Friedman JM. Leptin-specific patterns of gene expression in white adipose tissue. Genes Dev. 2000;14(8):963–80.
Way JM, Harrington WW, Brown KK, Gottschalk WK, Sundseth SS, Mansfield TA, Ramachandran RK, Willson TM, Kliewer SA. Comprehensive messenger ribonucleic acid profiling reveals that peroxisome proliferator-activated receptor γ activation has coordinate effects on gene expression in multiple insulin-sensitive tissues. Endocrinology. 2001;142(3):1269–77.
Gratten J, Visscher PM. Genetic pleiotropy in complex traits and diseases: implications for genomic medicine. Genome Med. 2016;8(1):1–3.
Stearns FW. One hundred years of pleiotropy: a retrospective. Genetics. 2010;186(3):767–73.
Kraja AT, Chasman DI, North KE, Reiner AP, Yanek LR, Kilpeläinen TO, Smith JA, Dehghan A, Dupuis J, Johnson AD. Pleiotropic genes for metabolic syndrome and inflammation. Mol Genet Metab. 2014;112(4):317–38.
Swenson BR, Louie T, Lin HJ, Méndez-Giráldez R, Below JE, Laurie CC, Kerr KF, Highland H, Thornton TA, Ryckman KK. GWAS of QRS duration identifies new loci specific to Hispanic/Latino populations. PLoS ONE. 2019;14(6): e0217796.
Schick UM, Jain D, Hodonsky CJ, Morrison JV, Davis JP, Brown L, Sofer T, Conomos MP, Schurmann C, McHugh CP. Genome-wide association study of platelet count identifies ancestry-specific loci in Hispanic/Latino Americans. Am J Human Genet. 2016;98(2):229–42.
Khan UI, Wang D, Sowers MR, Mancuso P, Everson-Rose SA, Scherer PE, Wildman RP. Race–ethnic differences in adipokine levels: the Study of Women’s Health Across the Nation (SWAN). Metabolism. 2012;61(9):1261–9.
Morimoto Y, Conroy SM, Ollberding NJ, Kim Y, Lim U, Cooney RV, Franke AA, Wilkens LR, Hernandez BY, Goodman MT. Ethnic differences in serum adipokine and C-reactive protein levels: the multiethnic cohort. Int J Obes. 2014;38(11):1416–22.
Nazare J-A, Smith JD, Borel A-L, Haffner SM, Balkau B, Ross R, Massien C, Almeras N, Despres J-P. Ethnic influences on the relations between abdominal subcutaneous and visceral adiposity, liver fat, and cardiometabolic risk profile: the International Study of Prediction of Intra-Abdominal Adiposity and Its Relationship With Cardiometabolic Risk/Intra-Abdominal Adiposity. Am J Clin Nutr. 2012;96(4):714–26.
Lê K-A, Ventura EE, Fisher JQ, Davis JN, Weigensberg MJ, Punyanitya M, Hu HH, Nayak KS, Goran MI. Ethnic differences in pancreatic fat accumulation and its relationship with other fat depots and inflammatory markers. Diabetes Care. 2011;34(2):485–90.
Frank AT, Zhao B, Jose PO, Azar KM, Fortmann SP, Palaniappan LP. Racial/ethnic differences in dyslipidemia patterns. Circulation. 2014;129(5):570–9.
Kim J, Bai Y, Pan W. An adaptive association test for multiple phenotypes with GWAS summary statistics. Genet Epidemiol. 2015;39(8):651–63.
Manolio TA: Collaborative genome-wide association studies of diverse diseases: programs of the NHGRI’s office of population genomics. 2009.
Kenny EM. Population Architecture using Genomics and Epidemiology (PAGE).
Matise TC, Ambite JL, Buyske S, Carlson CS, Cole SA, Crawford DC, Haiman CA, Heiss G, Kooperberg C, Marchand LL. The Next PAGE in understanding complex traits: design for the analysis of Population Architecture Using Genetics and Epidemiology (PAGE) Study. Am J Epidemiol. 2011;174(7):849–59.
Fisher-Hoch SP, Rentfro AR, Wilson JG, Salinas JJ, Reininger BM, Restrepo BI, McCormick JB, Pérez A, Brown HS, Hossain MM: Peer Reviewed: Socioeconomic Status and Prevalence of Obesity and Diabetes in a Mexican American Community, Cameron County, Texas, 2004–2007. Preventing chronic disease 2010, 7(3).
Gorber SC, Tremblay MS. The bias in self-reported obesity from 1976 to 2005: a Canada–US comparison. Obesity. 2010;18(2):354–61.
Carty CL, Bhattacharjee S, Haessler J, Cheng I, Hindorff LA, Aroda V, Carlson CS, Hsu C-N, Wilkens L, Liu S. Analysis of metabolic syndrome components in > 15 000 african americans identifies pleiotropic variants: results from the population architecture using genomics and epidemiology study. Circ Cardiovasc Genet. 2014;7(4):505–13.
Brooks GC, Blaha MJ, Blumenthal RS. Relation of C-reactive protein to abdominal adiposity. Am J Cardiol. 2010;106(1):56–61.
Kocarnik JM, Pendergrass SA, Carty CL, Pankow JS, Schumacher FR, Cheng I, Durda P, Ambite JL, Deelman E, Cook NR. Multiancestral analysis of inflammation-related genetic variants and C-reactive protein in the population architecture using genomics and epidemiology study. Circ Cardiovasc Genet. 2014;7(2):178–88.
Bien SA, Wojcik GL, Zubair N, Gignoux CR, Martin AR, Kocarnik JM, Martin LW, Buyske S, Haessler J, Walker RW. Strategies for enriching variant coverage in candidate disease loci on a multiethnic genotyping array. PLoS ONE. 2016;11(12):e0167758.
Hu Y, Bien SA, Nishimura KK, Haessler J, Hodonsky CJ, Baldassari AR, Highland HM, Wang Z, Preuss M, Sitlani CM. Multi-ethnic genome-wide association analyses of white blood cell and platelet traits in the Population Architecture using Genomics and Epidemiology (PAGE) study. BMC Genomics. 2021;22(1):1–11.
Kowalski MH, Qian H, Hou Z, Rosen JD, Tapia AL, Shan Y, Jain D, Argos M, Arnett DK, Avery C. Use of> 100,000 NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium whole genome sequences improves imputation quality and detection of rare variant associations in admixed African and Hispanic/Latino populations. PLoS Genet. 2019;15(12):e1008500.
Lin D-Y, Tao R, Kalsbeek WD, Zeng D, Gonzalez F II, Fernández-Rhodes L, Graff M, Koch GG, North KE, Heiss G. Genetic association analysis under complex survey sampling: the Hispanic Community Health Study/Study of Latinos. Am J Human Genet. 2014;95(6):675–88.
Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26(17):2190–1.
Hackinger S, Zeggini E. Statistical methods to detect pleiotropy in human complex traits. Open Biol. 2017;7(11):170125.
Schaid DJ, Tong X, Larrabee B, Kennedy RB, Poland GA, Sinnwell JP. Statistical methods for testing genetic pleiotropy. Genetics. 2016;204(2):483–97.
Geyer CJ. Practical markov chain monte carlo. Stat Sci. 1992;7:473–83.
Fadista J, Manning AK, Florez JC, Groop L. The (in) famous GWAS P-value threshold revisited and updated for low-frequency variants. Eur J Hum Genet. 2016;24(8):1202–5.
Kamat MA, Blackshaw JA, Young R, Surendran P, Burgess S, Danesh J, Butterworth AS, Staley JR. PhenoScanner V2: an expanded tool for searching human genotype–phenotype associations. Bioinformatics. 2019;35(22):4851–3.
Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42(D1):D1001–6.
Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, Hasz R, Walters G, Garcia F, Young N. The genotype-tissue expression (GTEx) project. Nat Genet. 2013;45(6):580–5.
Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40(D1):D930–4.
Imai K, Keele L, Tingley D. A general approach to causal mediation analysis. Psychol Methods. 2010;15(4):309.
Solovieff N, Cotsapas C, Lee PH, Purcell SM, Smoller JW. Pleiotropy in complex traits: challenges and strategies. Nat Rev Genet. 2013;14(7):483–95.
Torday JS. Pleiotropy, the physiologic basis for biologic fields. Prog Biophys Mol Biol. 2018;136:37–9.
Imai K, Keele L, Tingley D, Yamamoto T. Causal mediation analysis using R. In: Advances in social science research using R. Springer; 2010: 129–154.
Zeileis A. Object-oriented computation of sandwich estimators. J Stat Softw. 2006;16:1–16.
Mammes O, Aubert R, Betoulle D, Pean F, Herbeth B, Visvikis S, Siest G, Fumeron F. LEPR gene polymorphisms: associations with overweight, fat mass and response to diet in women. Eur J Clin Invest. 2001;31(5):398–404.
Gregoor JG, van der Weide J, Mulder H, Cohen D, van Megen HJ, Egberts AC, Heerdink ER. Polymorphisms of the LEP-and LEPR gene and obesity in patients using antipsychotic medication. J Clin Psychopharmacol. 2009;29(1):21–5.
Marzi C. Is acute-phase serum amyloid a protein a risk factor for type 2 diabetes: epidemiologic perspective including a genetic approach. München, Ludwig-Maximilians-Universität, Diss., 2014; 2014.
Uhlar CM, Whitehead AS. Serum amyloid A, the major vertebrate acute-phase reactant. Eur J Biochem. 1999;265(2):501–23.
Yang R-Z, Lee M-J, Hu H, Pollin TI, Ryan AS, Nicklas BJ, Snitker S, Horenstein RB, Hull K, Goldberg NH. Acute-phase serum amyloid A: an inflammatory adipokine and potential link between obesity and its metabolic complications. PLoS Med. 2006;3(6):e287.
Kocarnik JM, Richard M, Graff M, Haessler J, Bien S, Carlson C, Carty CL, Reiner AP, Avery CL, Ballantyne CM. Discovery, fine-mapping, and conditional analyses of genetic variants associated with C-reactive protein in multiethnic populations using the Metabochip in the Population Architecture using Genomics and Epidemiology (PAGE) study. Hum Mol Genet. 2018;27(16):2940–53.
Hubacek JA, Peasey A, Pikhart H, Stavek P, Kubinova R, Marmot M, Bobak M. APOE polymorphism and its effect on plasma C-reactive protein levels in a large general population sample. Hum Immunol. 2010;71(3):304–8.
Long J, Liu P, Liu Y, Lu Y, Xiong D, Elze L, Recker R, Deng H. APOE and TGF-β1 genes are associated with obesity phenotypes. J Med Genet. 2003;40(12):918–24.
Zhao QR, Lei YY, Li J, Jiang N, Shi JP. Association between apolipoprotein E polymorphisms and premature coronary artery disease: a meta-analysis. Clin Chem Lab Med (CCLM). 2017;55(2):284–98.
Boulenouar H, Mediene BS, Ouhaibi DH, Larjam HS, Houti L, Hammani-Medjaoui I. Genetic variants on chromosome 19 (rs439401 and rs4420638) are associated with obesity and high blood pressure in the Algerian population. Baвилoвcкий жypнaл гeнeтики и ceлeкции. 2019;23(5):608–14.
Iqbal Kring SI, Barefoot J, Brummett BH, Boyle SH, Siegler IC, Toubro S, Hansen T, Astrup A, Pedersen O, Williams RB. Associations between APOE variants and metabolic traits and the impact of psychological stress. PLoS ONE. 2011;6(1): e15745.
Prins BP, Kuchenbaecker KB, Bao Y, Smart M, Zabaneh D, Fatemifar G, Luan Ja, Wareham NJ, Scott RA, Perry JR. Genome-wide analysis of health-related biomarkers in the UK Household Longitudinal Study reveals novel associations. Sci Rep. 2017;7(1):1–9.
Belloy ME, Eger SJ, Guen YL, Napolioni V, Greicius MD. Initiative AsDN: Two APOE splice sQTLs reduce Alzheimer’s disease risk in APOE 4/4 carriers: genetics/genetic factors of Alzheimer’s disease. Alzheimers Dement. 2020;16:e043539.
Wang D, Zhang DF, Li GD, Bi R, Fan Y, Wu Y, Yu XF, Long H, Li YY, Yao YG. A pleiotropic effect of the APOE gene: association of APOE polymorphisms with multibacillary leprosy in Han Chinese from Southwest China. Br J Dermatol. 2018;178(4):931–9.
Barter P. The inflammation: lipoprotein cycle. Atheroscler Suppl. 2005;6(2):15–20.
Ligthart S, De Vries PS, Uitterlinden AG, Hofman A, Group CIW, Franco OH, Chasman DI, Dehghan A. Pleiotropy among common genetic loci identified for cardiometabolic disorders and C-reactive protein. PLoS ONE. 2015;10(3):e0118859.
Ligthart S, Vaez A, Hsu Y-H, Stolk R, Uitterlinden AG, Hofman A, Alizadeh BZ, Franco OH, Dehghan A. Bivariate genome-wide association study identifies novel pleiotropic loci for lipids and inflammation. BMC Genomics. 2016;17(1):1–10.
Han L, Tang L, Wang C, Chen Z, Zhang T, Chen S, Liu S, Peng X, Mai Y, Duan S. Fat mass and obesity? associated gene rs11642015 polymorphism is significantly associated with prediabetes and type 2 diabetes subsequent to adjustment for body mass index. Biomed Rep. 2014;2(5):681–6.
Bryc K, Velez C, Karafet T, Moreno-Estrada A, Reynolds A, Auton A, Hammer M, Bustamante CD, Ostrer H. Genome-wide patterns of population structure and admixture among Hispanic/Latino populations. Proc Natl Acad Sci. 2010;107(supplement_2):8954–61.
Conomos MP, Laurie CA, Stilp AM, Gogarten SM, McHugh CP, Nelson SC, Sofer T, Fernández-Rhodes L, Justice AE, Graff M. Genetic diversity and association studies in US Hispanic/Latino populations: applications in the Hispanic Community Health Study/Study of Latinos. Am J Human Genet. 2016;98(1):165–84.
Consortium GP. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65.
van de Bunt M, Cortes A, Consortium I, Brown MA, Morris AP, McCarthy MI. Evaluating the performance of fine-mapping strategies at common variant GWAS loci. PLoS Genet. 2015;11(9):e1005535.
Kichaev G, Pasaniuc B. Leveraging functional-annotation data in trans-ethnic fine-mapping studies. Am J Human Genet. 2015;97(2):260–71.
Acknowledgements
Not applicable.
Funding
The PAGE Study is funded by the National Human Genome Research Institute with co-funding from the National Institute on Minority Health and Health Disparities. Assistance with data management, data integration, data dissemination, genotype imputation, ancestry deconvolution, population genetics, analysis pipelines and general study coordination was provided by the PAGE Coordinating Center (NI-HU01HG007419). Genotyping services were provided by the Center for Inherited Disease Research, which is fully funded through a federal contract from the National Institutes of Health (NIH) to The Johns Hopkins University, contract number HHSN268201200008I. Genotype data quality control and quality assurance services were provided by the Genetic Analysis Center in the Biostatistics Department of the University of Washington, through support provided by the Center for Inherited Disease Research contract. PAGE was also funded by grants R56HG010297 and R01HG010297. PAGE data and materials included in this report were funded through the following studies and organizations: The Mount Sinai BioMe Biobank is supported by The Andrea and Charles Bronfman Philanthropies. The MEC characterization of epidemiological architecture is funded through the NHGRI PAGE program (U01HG004802 and its NHGRI ARRA supplement). The MEC study is funded by the National Cancer Institute (R37CA54281, R01CA63, P01CA33619, U01CA136792 and U01CA98758). The WHI program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, and US Department of Health and Human Services through contracts 75N92021D00001, 75N92021D00002, 75N92021D00003, 75N92021D00004 and 75N92021D00005. The HCHS/SOL is a collaborative study supported by contracts from the National Heart, Lung and Blood Institute (NHLBI) to the University of North Carolina (HHSN268201300001I / N01-HC-65233), University of Miami (HHSN268201300004I / N01-HC-65234), Albert Einstein College of Medicine (HHSN268201300002I / N01-HC 65235), University of Illinois at Chicago (HHSN268201300003I / N01-HC-65236 Northwestern University), and San Diego State University (HHSN268201300005I / N01-HC-65237). The following Institutes/Centres/Offices have contributed to the HCHS/SOL through a transfer of funds to the NHLBI: National Institute on Minority Health and Health Disparities; National Institute on Deafness and Other Communication Disorders; National Institute of Dental and Craniofacial Research; National Institute of Diabetes and Digestive and Kidney Diseases; National Institute of Neurological Disorders and Stroke; and NIH Institution-Office of Dietary Supplements. The Genetic Analysis Center at the University of Washington was supported by NHLBI and NIDCR contracts (HHSN268201300005C AM03 and MOD03. The CCHC is supported by MD000170 P20 funded from the National Center on Minority Health and Health Disparities (NCMHD), the University of Texas Houston Health Sciences Center, Center for Clinical and Translational Science CCTS-CTSA award UL1 TR00371 from NCATS. MYA is funded by NIDDK grant # 3R01DK122503 – 02W1. HMH is funded by NHLBI training grant T32 HL129982, ADA Grant #1-19-PDF-045, and R01HL142825.
Author information
Authors and Affiliations
Contributions
MYA, KEN, JEB, KCT, JBM, and SPFH participated in the study conception. MYA, ARB, HGP, CMS, HMH, HHC, AGH, LEP, WZ, KEN, and JEB performed data analyses. MYA and KEN drafted the manuscript. All co-authors performed critical reviews. KEN, JEB, KCT, CLA, JBM and SPFH supervised the study.All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All study participants provided written informed consent and each study was approved by relevant institutional review board. All methods were performed in accordance with the relevant guidelines and regulations set by Declaration of Helsinki. HCHS/SOL- The institutional review board at the coordinating center (University of North Carolina Office of Human Research Ethics, 07-1003) and Board Office, 200601-0471; University of California-San Diego Human Research Protection Program, 3677; University of Miami Human Subject Research Office, FWA00002247) approved study protocols. All participants gave informed consent. BioMe- Program for the Protection of Human Subjects, Mount Sinai Health System, Icahn School of Medicine at Mount Sinai CARDIA- University of Texas Health Science Center at Houston; WHI- Fred Hutchison Cancer Research Center; ARIC- The Johns Hopkins Medicine Institutional Review Board; Cameron County- Committee for the Protection of Human Subjects at the University of Teas Health Sciences Center at Houston; Human Research Protections Program at Vanderbilt University; MEC- USC Institutional Review Board.
Consent for publication
Not applicable.
Competing interests
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Conceptual framework for adiposity and inflammation pleiotropy.
Additional file 2
. Analytical framework for mediation analysis. Mediating phenotype is the known associated trait with SNP. Genetic Variant is assume to have multi-trait effect if the direct, mediated and total effect all exceed statistical significance level.
Additional file 3
. Association of PAGE derived multi-trait significant SNPs with phenotypic domains.
Additional file 4
. Tissue specific messenger RNA (mRNA) expression levels associated with PAGE derived multi-trait associated SNPs.
Additional file 5
. Supplementary Tables.
Additional file 6
. Appendix
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Anwar, M.Y., Baldassari, A.R., Polikowsky, H.G. et al. Genetic pleiotropy underpinning adiposity and inflammation in self-identified Hispanic/Latino populations. BMC Med Genomics 15, 192 (2022). https://doi.org/10.1186/s12920-022-01352-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-022-01352-3