
Abstract
Background
Mutations in the MYO15A gene are a widely recognized cause of autosomal recessive non-syndromic sensorineural hearing loss (NSHL) globally. Here, we examined the role and the genotype–phenotype correlation of MYO15A variants in a cohort of Chinese NSHL cases.
Methods
Eighty-one cases with evidenced MYO15A variants from the 2263 Chinese NSHL cases, who underwent next-generation sequencing (NGS), were enrolled in the study. We investigated the association of MYO15A variants with the severity, progression and age of onset of hearing loss, as well as compared it to the previous reports in different nationalities. The cases were divided into groups according to the number of truncating variants: 2 truncating, 1 truncating and 1 non-truncating, 2 non-truncating variants, and compared the severity of HL among the groups.
Results
MYO15A accounted for 3.58% (81/2263) of all NSHL cases. We analyzed 81 MYO15A-related NSHL cases, 73 of whom were with congenital bilateral, symmetric or severe-to-profound hearing loss (HL), however, 2 of them had a postlingual, asymmetric, mild or moderate HL. There were 102 variants identified in all MYO15A structural domains, 76.47% (78/102) of whom were novel. The most common types of detected variants were missense (44/102, 43.14%), followed by frameshift (27/102, 26.47%), nonsense (14/102, 13.72%), splice site (10/102, 9.80%), in frame (4/102, 3.92%), non-coding (2/102, 1.96%) and synonymous (1/102, 0.98%). The most recurrent variant c.10245_10247delCTC was detected in 12 cases. We observed that the MYO15A variants, located in its N-terminal, motor and FERM domains, led to partial deafness with better residual hearing at low frequencies. There were 34 cases with biallelic truncating variants, 37 cases with monoallelic truncating variants, and 13 cases with biallelic non-truncating variants. The biallelic non-truncating variants group had the least number of cases (12/81), and most of them (10/12) were with profound NSHL.
Conclusions
MYO15A is a major gene responsible for NSHL in China. Cases with MYO15A variants mostly showed early-onset, symmetric, severe-to-profound hearing loss. This study is by far the largest focused on the evaluation of the genotype–phenotype correlations among the variants in the MYO15A gene and its implication in the outcome of NSHL. The biallelic non-truncating MYO15A variants commonly caused profound HL, and the cases with one or two truncating MYO15A variants tended to increase the risk of HL. Nevertheless, further investigations are needed to clarify the causes for the variable severities and progression rates of hearing loss and the detected MYO15A variants in these cases.
Similar content being viewed by others


Background
Hearing loss (HL) is one of the most common human pathologies that significantly affects the quality of life [1]. About 60% of congenital HL is caused by genetic factors [2, 3]. Non-syndromic sensorineural hearing loss (NSHL) is considered a major cause of HL. To date, mutations in 124 genes have been identified in individuals affected with NSHL, among which mutations in 78 genes were related to autosomal recessive non-syndromic sensorineural hearing loss (ARNSHL), mutations in 51 autosomal dominant genes and 5 X-linked genes were correlated with NSHL (Hereditary Hearing Loss Homepage, http://hereditaryhearingloss.org, updated on 30 August 2021). The most common variations that were found in ARNSHL were in the genes GJB2, SLC26A4, CDH23, MYO15A and OTOF [4, 5]. Genetic variations in MYO15A were considered the third most common cause of ARNSHL in Iran due to prevalent consanguineous marriages [5, 6]. Whereas in the cohort of Korean ARNSHL patients, MYO15A mutations were recognized as the fourth most important deafness gene variants after those detected in other genes like GJB2, SLC26A4 and CDH23 [7, 8].
MYO15A (OMIM #602666) is a 71 kb long gene that contains 66 exons. It is localized on chromosome 17p11.2 (chr17:18012020–18083116; hg19 assembly) and encodes the myosin-XV protein with 35,390 amino acids [9]. Myosin proteins are a large family of actin-based molecular motors that bind actin filaments to produce force and motion, thus contributing to the hydrolysis of ATP.
The MYO15A protein contains an N-terminal domain (amino acids (AA) 1–1223), a motor domain (AA 1224–1899), three light-chain binding IQ motifs (AA1909–1942), two myosin-tail homologies 4 domains (MyTH4, AA 2066–2174 and 3051–3161), two band F, ezrin, radixin, myosin domains (FERM, AA 2687–2867 and 3217–3497), an Src-homology-3 domain (SH3, AA 2865– 2959) and a C-terminal PDZ ligand motif [6, 10, 11].
It is reported that MYO15A mutations cause sensorineural HL in human autosomal recessive deafness 3 (DFNB3, OMIM #600316) [7]. The DFNB3 locus was discovered in patients from a remote village in Indonesia, where 2.2% (47/2185) of the population was affected by hearing loss [12, 13]. So far, more than 200 MYO15A variants have been reported in more than 20 countries and regions, such as Algeria, Arab, Brazil, China, France, Germany, India, Iran, Israel, Japan, Mexico, the Netherlands, Oman, Pakistan, Palestine, Qatar, South Korea, Spain, Tunisia, Turkey and the United States. However, due to the large size of the gene and its many exons, simple techniques for detecting variants are discordant with it. Therefore, the clinical characteristics of MYO15A related to NSHL hearing level, age of onset, the degree of progression, associated symptoms and hotspot mutations were not clearly identified. So far, MYO15A had been reported sporadically in China. In this study, 81 cases from 74 families identified with at least one MYO15A pathogenic or likely pathogenic variants, or uncertain significant variants, diagnosed by next-generation sequencing (NGS) from 2263 Chinese cases with NSHL, were enrolled to analyze the correlation between the MYO15A genomic variants and NSHL pathological phenotype. Co-segregation of variants was confirmed in probands and healthy parents, as well as more family members if available, via NGS and Sanger sequencing. This study is by far the largest focused on MYO15A variants and their implication in the outcome of NSHL. As well as we were able to detect the gene frequency and the recurrent variant of the MYO15A in Chinese patients with NSHL. The association of MYO15A variants with hereditary deafness patients, their severity, progression and age of onset was further conducted.
Methods
Purpose of test
The performed test aimed to examine the role and the genotype–phenotype correlation of MYO15A variants in a cohort of Chinese NSHL patients.
Subjects and clinical evaluation
There were 2263 participants from 1842 families with NSHL from the Genetic Testing Center for Deafness at the College of Otolaryngology Head and Neck Surgery, Chinese PLA General Hospital enrolled in the study, from June 2015 to September 2021. Trio WES was performed in 95 cases and their parents, trio/quadro NGS in 2009 cases and their family members, and singleton NGS in 159 cases.
And 81 cases from 74 families with detected MYO15A variants, related to NSHL, were analyzed for the assessment of the correlation between the MYO15A genotype and the NSHL phenotype. Detailed interviews were conducted with probands and their families to obtain their medical and familial histories.
All underwent testing that included physical examination, otoscopy, pure tone audiometry (PTA), tympanometry, assessment of auditory brainstem responses (ABR), distortion product otoacoustic emission (DPOAE), multiple auditory steady-state evoked responses (ASSR), temporal bone computerized tomography scans, and magnetic resonance of the brain. The definition for the severity of hearing impairment, according to pure-tone audiometry (PTA) of the better ear, was made based on the average hearing threshold level at four frequencies (500, 1000, 2000 and 4000 Hz) of air conduction. 26–40 dB HL were considered to be mild hearing loss; 41–55 dB HL, moderate hearing loss; 56–70 dB, moderately severe hearing loss; 71–90 dB HL, severe hearing loss; > 90 dB HL, profound hearing loss. The occurrence of hearing loss was categorized as prelingual (≤ 3 years) or post-lingual (> 3 years). Asymmetric hearing loss (AHL) was defined as greater than 15 dB between the ears at 0.5, 1, and 2 kHz or greater than 20 dB at 4 kHz on the audiogram (American Academy Otolaryngology-Head Neck Surgery 1997) [14] as reported previously [15].
Peripheral blood samples were collected from all cases, their parents and siblings (if any). All cases obtained informed consent for the performed molecular genetic analysis and their clinical data publication. The study was approved by the Ethics Committee of the Chinese PLA General Hospital (reference number S2016-120–02). Written informed consent was obtained from the participants and in the case of young cases from their parents.
Targeted deafness gene capture and NGS
Targeted deafness gene capture and NGS were performed as previously reported [16]. DNA samples of 64 cases from 58 families were subjected to targeted NGS, 35 cases of them conducted trio (proband and parents) targeted NGS and 29 cases conducted quarto (proband, parents and sibling) targeted NGS. The proband received the panel test containing 168 deafness-related genes (Additional files 1: Table S1). All coding exons, along with 100-bp flanking regions were sequenced on the Illumina HiSeq 2000 (Illumina, San Diego, CA, USA) using the MyGenostics gene enrichment system (MyGenostics, Boston, MA, USA).
Whole-exome sequencing (WES)
Illumina NovaSeq6000 sequencing platform was used to conduct the WES by MyGenostics (Beijing, China) (detailed procedures shown in Additional files 3). DNA samples from 17 MYO15A-related cases and their parents were subjected to trio WES and subsequently validated by Sanger sequencing. The nomenclature of the mutation described in Table 1 is based on MYO15A cDNA and protein accession numbers NM_016239.3 and NP_057323.3, respectively. We used the genomic coordinates from GRCH37/hg19 constructed from the human genome.
Bioinformatics
After sequencing the targeted region, quality control was performed to ensure the accuracy of the data. Low-quality data were filtered out to obtain clean sequencing data. Burrows-Wheeler alignment was used to align the clean sequence to the human reference genome hg19. Genome Analysis Toolkit (GATK) was used to detect single-nucleotide and insertion/deletion polymorphisms (indel). The NCBI ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/, last accessed date 16 December 2021), the Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk/ac/, last accessed date 16 December 2021), the Genome Aggregation Database (gnomAD, https://gnomad.broadinstitute.org, last accessed date 16 December 2021) and the Deafness Variation Database (DVD v8, https://deafnessvariationdatabase.org) were used to obtain the variants information, including gene information, variant consequence, minor allele frequency (MAF), altered protein function, and related disease information. The predictive score of pathogenicity of the variation was calculated, and the effect of amino acid substitution on protein structure and function was evaluated by Polyphen2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://provean.jcvi.org/). Pathogenicity was assessed according to the expert specification of the American Society for Medical Genetics and Genomics/ Association of Medical Pathology (ACMG/AMP) guidelines for genetic HL[17].
Sanger sequencing
Presumed pathogenic or likely pathogenic variants and variants of uncertain significance detected by WES and deafness gene panel in the probands were subsequently validated by a polymerase chain reaction (PCR) amplification and Sanger sequencing. Segregation analysis was performed on the probands and their family members. The primer sets are listed in Additional files 2: Table S2.
Results
Detected variants
Clinical features and genotypes of the pathogenic, likely pathogenic and uncertain MYO15A variants are summarized in Table 1. In particular, 4 cases were found to carry homozygous variants, 77 carried compound heterozygous variants.
In our study we have found 102 MYO15A variants, among which the most recurrent variants were c.10245_10247delCTC (0.27%, 12/4526), followed by c.10419_10423delCAGCT (0.24%, 11/4526), c.10251_10253delCTT (0.15%, 7/4526), c.4441 T > C (0.09%, 4/4526), c.4898 T > C (0.09%, 4/4526), c.3524dupA (0.07%, 3/4526), c.5964 + 3G > A (0.07%, 3/4526), c.6177 + 1G > T(0.07%, 3/4526), c.8827insT (0.07%, 3/4526) and c.9690 + 1G > A (0.07%, 3/4526). Other variants appeared only once or twice (Table 1).
Our analysis showed that the most common type of MYO15A variants was missense (44/102, 43.14%), followed by frameshift (27/102, 26.47%), nonsense (14/102, 13.72%), splice site (10/102, 9.80%), in frame (4/102, 3.92%), non-coding (2/102, 1.96%) and synonymous (1/102, 0.98%) (Fig. 1). The variants showed the various degree of HL, although the cases with the same variant type showed different phenotypes. In frame and splice variants showed more possibilities to cause profound HL, and frameshift and missense variants related to various degrees of HL (Fig. 1).

The degree of HL and the types of detected variants in the identified MYO15A variations. *The Multiple column represented the cases with the same variations showed different degrees of HL
The variants were located in 41 of the 66 protein-coding exons of the MYO15A gene (Table 1) and identified in all domains in this study. Seventy-eight novel and 24 reported variants were identified, and all of them were confirmed by Sanger sequencing. (Fig. 2).

The locations of the detected 102 MYO15A variants. The figure shows the locations of 102 MYO15A variants correlated with NSHL found in this study. The previously reported ones are shown at the bottom. Pathogenic variants were expressed in red words, likely pathogenic variants in green words, and VUS in black words
According to the guidelines of the ACMG/AMP on hereditary hearing loss, the variations in the MYO15A were manually classified [17, 18]. Based on the ACMG/AMP rating, ClinVar, HGVS and DVD database, respectively, the pathogenicity of the 102 MYO15A variants identified in this study included 40 pathogenic (P), 24 likely pathogenic (LP) and 38 variants with uncertain significance (VUS). (Table 2) We identified 36 cases with bi-allelic MYO15A pathogenic or likely pathogenic variants. The others with VUS in one of the alleles (LP/VUS, P/VUS and VUS/VUS) were also included in the study that classified as the best candidate of DFNB3. We also compared the severity of HL by the pathogenicity of variants. The results were inconclusive, and even the cases with the same variations showed various phenotypes (Table 3).
Variants with HIGH impact (e.g., frameshift variants, splice variants, stop gain variants, etc.) were counted as protein-truncating variants (PTVs) [19]. The 81 cases were divided into groups according to the number of PTVs: 2 truncating (34 cases); 1 truncating and 1 non-truncating (37 cases); 2 non-truncating variants (13 cases) (Table 4). We compared the severity of HL among the groups. The 2 non-truncating variants group had the least number of cases (12/81), and most of them (10/12) were with profound NSHL. Thus, we suggested that cases with the monoallelic or biallelic truncating MYO15A variant may increase the risk of HL.
Although synonymous variation is generally considered as non-pathogenic, the variant c.8340G > A(p.Thr2780Thr) identified in the case M488 (Fig. 3) was considered to be pathogenic (PVS1_Very Strong, PM2_Moderate, PP5_Supporting) based on the ACMG/AMP classification in our cohort. In the NCBI ClinVar database, it was shown that the c.8340G > A (p.Thr2780Thr) predicted loss of exon 45 and led to a stop codon. (National Center for Biotechnology Information. ClinVar; [VCV000236038.1], https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000236038.1 (accessed Sept. 20, 2021).)

The audiograms and the pedigree of case M488. a Pedigree of case M488 and her family members. b All case M488 and her parents had the same profound HL of total deafness type
Clinical findings
Among the 2263 cases from 1842 families with NSHL included in this study, including 1215 males and 1048 females. Age ranged from a few days after birth to 65 years with a mean age of 15.01 ± 13.67 years and the median age of 7.92 years. In our cohort, 1654 cases had prelingual HL and 609 had postlingual HL; 71 cases were mild, 238 were moderate, 179 were moderately severe, 512 were severe and 1263 were profound HL.
There were 81 (3.58%, 81/2263) cases from 74 families identified with at least one MYO15A pathogenic or likely pathogenic variant, or uncertain significant variant. Among them, 45 were males and 36 females, aged from 3 months to 43 years, with an average age of 10.41 ± 10.32 years. The ethnic distribution among the cases was as follows: one case was belonged to Korean ethnic group, one of Manchu, one of Tujia, while the others were all Han. None of the participants had a history of using aminoglycoside antibiotics.
Most of the audiological assessments and clinical history of the affected members showed a prelingual (92.59%, 75/81), symmetrical (97.53%, 79/81), bilateral (100%, 81/81), non-syndromic (100%, 81/81), sensorineural (100%, 81/81) HL (Fig. 4). Only a few showed a postlingual (7.41%, 6/81) and asymmetrical (2.47%, 2/81) HL. Analysis of the high-resolution CT scan of the temporal bone in the affected members showed a normal middle and inner ear structure.

Audiological phenotype of MYO15A-related HL
The cases showed large variations in the degree of HL. The degree of HL was profound in 61 cases (75.30%, 61/81), severe in 12 (14.81%, 12/81), moderately severe in 4 (4.94%, 4/81), moderate in 3 (3.70%, 3/81) and mild in 1 (1.22%, 1/81). The last had the right ear with a mild HL and the left ear with a profound HL. Audiogram forms showed 6 cases with a flat type, 50 cases with total deafness, 10 cases with a descending type, whereas 24 remained undefined.
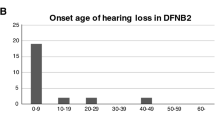
The age of onset among cases ranged from a few days after birth to 41 years. The hearing loss in 79.01% (64/81) of the cases appeared at birth, in 13.58% (11/81) was detected during the first 1–3 years, in 6.17% (5/81) HL arose around the age of 4–10 years, in 1.23% (1/81) was reported after 18 years (with severe deafness in the left ear and moderate deafness in the right ear, especially at the age of 41). (Fig. 5).

Age of onset of MYO15A-related HL
In our study, it was found that the genotype–phenotype correlation between the variants in the MYO15A gene and the HL in some cases was different from that of the others. For example, the cases M80 and M646 with an asymmetric unilateral severe deafness bore the compound heterozygous variants c.2957delC(p.Thr986fs)/c.9478C < T(p.Leu3160Phe) and c.1179insC(p.Glu396Argfs*36)/c.1261C > T (p.Pro421Ser), respectively. (Fig. 6).

The audiograms and the pedigree of case M80 (a, b) and M646 (c, d)
Affected subjects also showed progression with the different onset of HL. Case M623 with c.10251_10253delCTT homozygous variants was found in this study, who passed the hearing screening at birth, but was diagnosed with HL at the age of 3 and the HL demonstrated a progressive trait. His brother carrying the same c.10251_10253delCTT homozygous variant showed severe bilateral sensorineural HL at the age of 3 years.
Through the telephone follow-up of 56 MYO15A-related cases, the effect of using hearing aids and cochlea implants was satisfactory in most of the participants.
Discussion
Mutations in MYO15A were initially identified in HL individuals of consanguineous families from Bengkala, Bali in 1995 [12, 13]. Screening for the reported variants in the MYO15A gene with 66 exons was a very difficult and expensive task at that time. Therefore, the MYO15A gene was rarely sequenced in familial segregated deafness unless significant genetic linkage data implicated the presence of the DFNB3 locus. Instead, efforts were invested in screening for variations in smaller genes that have been identified as important contributors to inherited HL in humans, such as GJB2, which has only one protein-coding exon. The widespread contribution of MYO15A mutations on human HL was not recognized until the NGS became cost-effective and widely adopted around the world [20]. Now, mutations in MYO15A are a widely recognized cause of recessively inherited NSHL globally. More than 200 MYO15A variants have previously been reported ranging along with the domains and motifs of the encoded by MYO15A protein myosin XVA (Table 5) [8, 11, 13, 20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68].
Many studies analyzed the mutations in the GJB2 and SLC26A4 genes among cases with NSHL in different parts of the world. The obtained results demonstrated that the prevalence of the variants in GJB2 and SLC26A4 in HL accounted for about 15% to 25% and 2% to 12.6%, respectively, all dependent on the region localized [69]. The reported frequency of MYO15A variations in HL was 1.1% to 28% in respect to the different regions [70]. Besides Farjami et al. [70] reported that the MYO15A variant frequency in NSHL was 4.9% considering the variant rate of the GJB2 gene of 20%. In our study, the estimated prevalence of MYO15A variants in NSHL was 3.58%, which was similar to Farjami’s report. Moreover, Farjami et al. [70] proposed a total of 192 recessive MYO15A variants related to HL. The evaluated proportions of the various types of variants detected by him were similar to those noticed in our study. The composition of the detected variant types was similar in the different intensities of the HL (see Fig. 1). The c.10245_ 10247delCTC variant was identified as the most recurrent HL variant in our cohort. According to the MAF of 0.000016 in the Exome Aggregation Consortium (ExAC) database, 0.000389 in East Asian population and 0.000281 in total population by gnomAD, the c.10245_10247delCTC had been previously reported pathogenic, causing ARNSHL in the Japanese, Korean and Chinese individuals [7, 21, 71]. Therefore, we suggest that this variant is the hotspot of the MYO15A-related NSHL variant in East-Asian populations.
In the past two decades, scholars worldwide have gradually made a progress in the understanding of the correlation between the genotype and the resultant phenotype of MYO15A variants. During the first decade, it was thought that the hearing phenotype of ARSNHL was congenital, bilateral, full-frequency, severe to profound sensorineural hearing loss (SNHL). In 2007, Nal et al. [22] reported for the first time that an N-terminal variant (p.Glu1112fs*1124) in the exon 2 of the MYO15A gene resulted in a mild hearing loss with residual hearing at low frequency. At that time, it was considered that the phenotype of the hearing loss in cases with MYO15A variants was closely related to the region where this gene variant was located. However, subsequent studies showed that the correlation between the genotype and phenotype of MYO15A seemed to be more complex. Notably, the congenital non-progressive NSHL was investigated as the main consequence of the MYO15A variants. Interestingly, in families with ARNSHL with the same MYO15A pathogenic variant, the degree of the hearing phenotype was different [23, 24]. Different hearing phenotypes of non-congenital binaural severe SNHL were reported. Except for the residual hearing in the low-frequency region [25], it also included congenital moderate and severe SNHL with descending hearing curve [22, 23, 26, 27], all-frequency moderate and severe SNHL [28], progressive high-frequency descending severe SNHL [29], delayed and progressive moderate and severe SNHL [7, 30]. Allelic heterogeneity is common in hearing loss and is associated with clinical phenotype heterogeneity [72]. The variability of phenotypes makes clinical diagnosis and variant interpretation in genetic hearing loss diagnosis and maintenance [17]. And in our study, we found that the MYO15A variants-related hearing phenotype of SNHL in China was similar to the previous reports.
Nevertheless, some reports showed that MYO15A pathogenic variants cause moderate-to-severe HL, although they previously had been presented to cause profound HL [7, 31]. We found three cases in our cohort with MYO15A variants in the N-terminal, motor and MyTH domains that were diagnosed with a subtle HL. The hypothesis indicated that the predicted amino acid substitutions of the intrinsically disordered N-terminal domain were structurally less menacing, leading to a subtler HL. Based on these results, we believe that MYO15A variants may be the cause leading to the postlingual onset of partial deafness, the molecular mechanism of which requires further investigation. The occurrence of this non-severe hearing phenotype may be related to the following factors: the weak pathogenicity of MYO15A alleles, the existence of modified genes to reduce the degree of HL, and the influence of environmental factors. In addition, the progress of technologies for genetic diagnosis recently has further enriched the phenotypic spectrum of MYO15A. In the past, linkage analysis was often used in the study of inbreeding hereditary ear families. Those cases with severe hearing phenotypes caused by homozygous variants were always given priority to be included in the relevant genetic research. However, with the use of the WES technology and Molecular Genetics techniques, sporadic and medium-sized families around the world started to be increasingly diagnosed, and more cases with compound heterozygous variants with different phenotypes were identified, which allowed the MYO15A variants to show more diverse phenotypic characteristics.
We have detected a synonymous variant in MYO15A which was considered as a pathogenic variant. Generally, synonymous variants are considered to be non-pathogenic and are not expected to change the function of proteins. In recent years, this paradigm has been challenged with the evidence that the changes in the codon usage affected the efficiency and speed of translation, which in turn modified the folding and function of proteins [73]. Furthermore, the possible pathogenic mechanism of the abnormal splice site caused by a single nucleotide substitution at the codon wobble site and its implication in the phenotypes of HL was often ignored. Its pathogenicity was suggested by both NCBI ClinVar and DVD databases. NCBI ClinVar database, c.8340G > A (p.Thr2780Thr) predicted loss of exon 45 (116 bp), leading to a stop codon 2803 of 3531, and was the only synonymous variant considered as pathogenic. The other synonymous variants were classified as benign, likely benign, uncertainly significant, and to some extent conflicting interpretations of pathogenicity (National Center for Biotechnology Information. ClinVar; [VCV000236038.1], https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000236038.1 (accessed Sept. 20, 2021).) Danial-Farran N et al. [32] reported that c.8340G > A (p.Thr2780Thr), in the last nucleotide of exon 46 eliminated the full exon inclusion isoform, indicating that this variant impaired splicing of exon 46. Therefore, c.8340G > A (p.Thr2780Thr) was also classified as PTV.
There was a limited understanding about the impact of MYO15A PTV across multiple phenotypes. In this study, the cases with biallelic non-truncating MYO15A variants commonly related with profound HL, and the cases with one or two truncating variants tended to show more prone to HL. Therefore, it suggested a correlation between genotype and phenotype in MYO15A-related NSHL.
Consistent with previous genetic studies, MYO15A variants are considered to play an important role in the pathogenesis of HL in China. There were several limitations of this study. First, the approach yet could not detect variants in the promoter or enhancer region and copy number variants. In addition, the follow-up time varies, some cases lack long-term follow-up results and objective evaluation, particularly the cochlear implant cases.
Conclusion
In summary, we found that a total of 3.58% of the Chinese population with NSHL were related to MYO15A variants. MYO15A variants associated with NSHL were proven by NGS and validated by Sanger sequencing. Here, we report 78 novel and 24 reported MYO15A variants, which further enriched the MYO15A variant spectrum regarding the NSHL. Auditory features of the affected individuals were consistent with that previously reported for the recessive variants in the MYO15A gene. The hearing loss in most affected individuals was severe to profound, but in a few cases showed mild to moderate deafness. We suggest that the detected large variations in the phenotype of MYO15A-related NSHL might be correlated with the epigenetics and other factors that require further investigation. Noteworthy, screening for MYO15A variants in NSHL patients is of high necessity for efficient genetic diagnosis, patients’ counseling and clinical intervention.
Availability of data and materials
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The novel MYO15A variants were submitted to ClinVar under accession number: SUB10564921.
Abbreviations
- ABR:
-
Auditory brainstem response
- ACMG:
-
Americal college of medical genetics and genomics
- ARNSHL:
-
Autosomal recessive non-syndromic sensorineural hearing loss
- ASSR:
-
Auditory steady state response
- DPOAE:
-
Distortion product otoacoustic emission
- DVD:
-
Deafness variation database
- ExAC:
-
Exome aggregation consortium
- FERM:
-
Fezrin, radixin, myosin
- GATK:
-
Genome analysis toolkit
- HGMD:
-
Human gene mutation database
- HGVS:
-
Human genome variation society
- HL:
-
Hearing loss
- MAF:
-
Minor allele frequency
- MyTH4:
-
Myosin-tail homology 4
- NGS:
-
Next-generation sequencing
- NSHL:
-
Non-syndromic sensorineural hearing loss
- OAE:
-
Otoacoustic emission
- PCR:
-
Polymerase chain reaction
- PTA:
-
Pure-tone audiometry
- PTV:
-
Protein-truncating variants
- SNHL:
-
Sensorineural hearing loss
- VUS:
-
Variant of uncertain significance
- WES:
-
Whole-exome sequencing
References
Smith RJ, Bale JF Jr, White KR. Sensorineural hearing loss in children. The Lancet. 2005;365(9462):879–90.
Abou Tayoun AN, Al Turki SH, Oza AM, Bowser MJ, Hernandez AL, Funke BH, et al. Improving hearing loss gene testing: a systematic review of gene evidence toward more efficient next-generation sequencing–based diagnostic testing and interpretation. Genet Med. 2016;18:545–53.
Khalil A, Karroum SB, Barake R, Dunya G, Abou-Rizk S, Kamar A, et al. Post-lingual non-syndromic hearing loss phenotype: a polygenic case with 2 biallelic mutations in MYO15A and MITF. BMC Med Genet. 2020;21:1.
Duman D. Autosomal recessive nonsyndromic deafness genes: a review. Front Biosci. 2012;17:2213.
Hilgert N, Smith RJH, Van Camp G. Forty-six genes causing nonsyndromic hearing impairment: Which ones should be analyzed in DNA diagnostics? Mutat Res Mutat Res. 2009;681:189–96.
Nasrniya S, Miar P, Narrei S, Sepehrnejad M, Nilforoush MH, Abtahi H, et al. Whole-exome sequencing identifies a recurrent small in-frame deletion in MYO15A causing autosomal recessive nonsyndromic hearing loss in 3 Iranian pedigrees. Lab Med. 2021; XX:e0–12.
Chang MY, Lee C, Han JH, Kim MY, Park H-R, Kim N, et al. Expansion of phenotypic spectrum of MYO15A pathogenic variants to include postlingual onset of progressive partial deafness. BMC Med Genet. 2018;19:29.
Park JH, Kim NKD, Kim AR, Rhee J, Oh SH, Koo J-W, et al. Exploration of molecular genetic etiology for Korean cochlear implantees with severe to profound hearing loss and its implication. Orphanet J Rare Dis. 2014;9:167.
Fang Q, Indzhykulian AA, Mustapha M, Riordan GP, Dolan DF, Friedman TB, et al. The 133-kDa N-terminal domain enables myosin 15 to maintain mechanotransducing stereocilia and is essential for hearing. eLife. 2015;4:e08627.
Garcı́a-Alvarez B, de Pereda JM, Calderwood DA, Ulmer TS, Critchley D, Campbell ID, et al. Structural determinants of integrin recognition by Talin. Mol Cell. 2003;11:49–58.
Kalay E, Uzumcu A, Krieger E, Çaylan R, Uyguner O, Ulubil-Emiroglu M, et al. MYO15A (DFNB3) mutations in Turkish hearing loss families and functional modeling of a novel motor domain mutation. Am J Med Genet A. 2007;143A:2382–9.
Friedman TB, Liang Y, Weber JL, Hinnant JT, Barber TD, Winata S, et al. A gene for congenital, recessive deafness DFNB3 maps to the pericentromeric region of chromosome 17. Nat Genet. 1995;9:86–91.
Wang A. Association of unconventional myosin MYO15 mutations with human nonsyndromic deafness DFNB3. Science. 1998;280:1447–51.
Read AP, Newton VE. Waardenburg syndrome. J Med Genet. 1997;34:656–65.
Wang G, Li X, Gao X, Su Y, Han M, Gao B, et al. Analysis of genotype–phenotype relationships in 90 Chinese probands with Waardenburg syndrome. Hum Genet. 2021. https://doi.org/10.1007/s00439-021-02301-3.
Cui T-Y, Gao X, Huang S-S, Sun Y-Y, Zhang S-Q, Jiang X-X, et al. Four novel variants in POU4F3 cause autosomal dominant nonsyndromic hearing loss. Neural Plast. 2020;2020:1–12.
Oza AM, DiStefano MT, Hemphill SE, Cushman BJ, Grant AR, Siegert RK, et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum Mutat. 2018;39:1593–613.
The ACMG Laboratory Quality Assurance Committee, Richards S, Aziz N, Bale S, Bick D, Das S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–23.
Takayama J, Tadaka S, Yano K, Katsuoka F, Gocho C, Funayama T, et al. Construction and integration of three de novo Japanese human genome assemblies toward a population-specific reference. Nat Commun. 2021;12:226.
Rehman AU, Bird JE, Faridi R, Shahzad M, Shah S, Lee K, et al. Mutational spectrum of MYO15A and the molecular mechanisms of DFNB3 human deafness. Hum Mutat. 2016;37:991–1003.
Miyagawa M, Nishio S, Hattori M, Moteki H, Kobayashi Y, Sato H, et al. Mutations in the MYO15A Gene Are a significant cause of nonsyndromic hearing loss: massively parallel DNA sequencing–based analysis. Ann Otol Rhinol Laryngol. 2015;124 1_suppl:158S-168S.
Nal N, Ahmed ZM, Erkal E, Alper ÖM, Lüleci G, Dinç O, et al. Mutational spectrum of MYO15A : the large N-terminal extension of myosin XVA is required for hearing. Hum Mutat. 2007;28:1014–9.
Cengiz FB, Duman D, Sırmacı A, Tokgöz-Yilmaz S, Erbek S, Öztürkmen-Akay H, et al. Recurrent and private MYO15A mutations are associated with deafness in the Turkish population. Genet Test Mol Biomark. 2010;14:543–50.
Shearer AE, Hildebrand MS, Webster JA, Kahrizi K, Meyer NC, Jalalvand K, et al. Mutations in the first MyTH4 domain of MYO15A are a common cause of DFNB3 hearing loss. Laryngoscope. 2009;119:727–33.
Fattahi Z, Shearer AE, Babanejad M, Bazazzadegan N, Almadani SN, Nikzat N, et al. Screening for MYO15A gene mutations in autosomal recessive nonsyndromic, GJB2 negative Iranian deaf population. Am J Med Genet A. 2012;158A:1857–64.
Li W, Guo L, Li Y, Wu Q, Li Q, Li H, et al. A novel recessive truncating mutation in MYO15A causing prelingual sensorineural hearing loss. Int J Pediatr Otorhinolaryngol. 2016;81:92–5.
Mehregan H, Mohseni M, Jalalvand K, Arzhangi S, Nikzat N, Banihashemi S, et al. Novel mutations in MYTH4-FERM domains of myosin 15 are associated with autosomal recessive nonsyndromic hearing loss. Int J Pediatr Otorhinolaryngol. 2019;117:115–26.
Bashir R, Fatima A, Naz S. Prioritized sequencing of the second exon of MYO15A reveals a new mutation segregating in a Pakistani family with moderate to severe hearing loss. Eur J Med Genet. 2012;55:99–102.
Gu X, Guo L, Ji H, Sun S, Chai R, Wang L, et al. Genetic testing for sporadic hearing loss using targeted massively parallel sequencing identifies 10 novel mutations. :12.
Miyagawa M, Nishio S, Ikeda T, Fukushima K, Usami S. Massively parallel DNA sequencing successfully identifies new causative mutations in deafness genes in patients with cochlear implantation and EAS. PLoS ONE. 2013;8:8.
Naz S, Imtiaz A, Mujtaba G, Maqsood A, Bashir R, Bukhari I, et al. Genetic causes of moderate to severe hearing loss point to modifiers: Genetic causes of hearing loss point to modifiers. Clin Genet. 2017;91:589–98.
Danial-Farran N, Brownstein Z, Gulsuner S, Tammer L, Khayat M, Aleme O, et al. Genetics of hearing loss in the Arab population of Northern Israel. Eur J Hum Genet. 2018;26:1840–7.
Manzoli GN, Bademci G, Acosta AX, Félix TM, Cengiz FB, Foster J, et al. Targeted Resequencing of Deafness Genes Reveals a Founder MYO15A Variant in Northeastern Brazil: Deafness Genes Reveals a Founder MYO15A Variant Brazil. Ann Hum Genet. 2016;80(6):327–31. https://doi.org/10.1111/ahg.12177.
Sloan-Heggen CM, Bierer AO, Shearer AE, Kolbe DL, Nishimura CJ, Frees KL, et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Human Genet. 2016;135(4):441–50. https://doi.org/10.1007/s00439-016-1648-8.
Diaz-Horta O, Duman D, Foster J II, Sırmacı A, Gonzalez M, et al. Whole-Exome Sequencing Efficiently Detects Rare Mutations in Autosomal Recessive Nonsyndromic Hearing Loss. PLoS ONE. 2012;7(11): https://doi.org/10.1371/journal.pone.0050628.
Atik T, Onay H, Aykut A, Bademci G, Kirazli T, Tekin M, et al. Comprehensive Analysis of Deafness Genes in Families with Autosomal Recessive Nonsyndromic Hearing Loss. PLoS ONE. 2015;10(11): https://doi.org/10.1371/journal.pone.0142154.
Gao X, Zhu Q, Song YS, Wang GJ, Yuan YY, Xin F, et al. Novel compound heterozygous mutations in the MYO15A gene in autosomal recessive hearing loss identified by whole-exome sequencing. J Translat Med. 2013;11:284.
Yang T, Wei X, Chai Y, Li L, Wu H. Genetic etiology study of the non-syndromic deafness in Chinese Hans by targeted next-generation sequencing. Orphanet J Rare Dis. 2013;8:85.
Danial-Farran N, Brownstein Z, Gulsuner S, Tammer L, Khayat M, Aleme O, et al. Genetics of hearing loss in the Arab population of Northern Israel. Eur J Hum Genet. 2018;26:1840–7.
Chen Y, Wang Z, Wang Z, Chen D, Chai Y, Pang X, et al. Targeted Next-Generation Sequencing in Uyghur Families with Non-Syndromic Sensorineural Hearing Loss. PLOS ONE. 2015;10:e0127879.
Brownstein Z, Friedman LM, Shahin H, Oron-Karni V, Kol N, Rayyan A, et al. Targeted genomic capture and massively parallel sequencing to identify genes for hereditary hearing loss in middle eastern families. Genome Biol. 2011;12:R89.
Zhang J, Guan J, Wang H, Yin L, Wang D, Zhao L, et al. Genotype-phenotype correlation analysis of MYO15A variants in autosomal recessive non-syndromic hearing loss. BMC Med Genet. 2019;20:60.
Vozzi D, Morgan A, Vuckovic D, D’Eustacchio A, Abdulhadi K, Rubinato E, et al. Hereditary hearing loss: a 96 gene targeted sequencing protocol reveals novel alleles in a series of Italian and Qatari patients. Gene. 2014;542:209–16.
Imtiaz F, Taibah K, Ramzan K, Bin-Khamis G, Kennedy S, Al-Mubarak B, et al. A comprehensive introduction to the genetic basis of non-syndromic hearing loss in the Saudi Arabian population. BMC Med Genet. 2011;12:91.
Sloan-Heggen CM, Babanejad M, Beheshtian M, Simpson AC, Booth KT, Ardalani F, et al. Characterising the spectrum of autosomal recessive hereditary hearing loss in Iran. J Med Genet. 2015;52:823–9.
Vona B. Targeted next-generation sequencing of deafness genes in hearing-impaired individuals uncovers informative mutations. Genet Med. 2014;16:9.
Palombo F, Al-Wardy N, Ruscone GAG, Oppo M, Kindi MNA, Angius A, et al. A novel founder MYO15A frameshift duplication is the major cause of genetic hearing loss in Oman. J Hum Genet. 2017;62:259–64.
Brownstein Z, Abu-Rayyan A, Karfunkel-Doron D, Sirigu S, Davidov B, Shohat M, et al. Novel myosin mutations for hereditary hearing loss revealed by targeted genomic capture and massively parallel sequencing. Eur J Hum Genet. 2014;22:768–75.
Richard EM, Santos-Cortez RLP, Faridi R, Rehman AU, Lee K, Shahzad M, et al. Global genetic insight contributed by consanguineous Pakistani families segregating hearing loss. Hum Mutat. 2019;40:53–72.
Liburd N, Ghosh M, Riazuddin S, Naz S, Khan S, Ahmed Z, et al. Novel mutations of MYO15A associated with profound deafness in consanguineous families and moderately severe hearing loss in a patient with Smith-Magenis syndrome. Hum Genet. 2001;109:535–41.
Neveling K, Feenstra I, Gilissen C, Hoefsloot LH, Kamsteeg E-J, Mensenkamp AR, et al. A Post-Hoc Comparison of the Utility of Sanger Sequencing and Exome Sequencing for the Diagnosis of Heterogeneous Diseases. Hum Mutat. 2013;34:1721–6.
Bai X, Nian S, Feng L, Ruan Q, Luo X. Identification of novel variants in MYO15A, OTOF, and RDX with hearing loss by next-generation sequencing. Molec Genet Genom Med. 2019. https://doi.org/10.1002/mgg3.808.
Woo H-M, Park H-J, Baek J-I, Park M-H, Kim U-K, Sagong B, et al. Whole-exome sequencing identifies MYO15A mutations as a cause of autosomal recessive nonsyndromic hearing loss in Korean families. BMC Med Genet. 2013;14:72.
Moteki H, Azaiez H, Booth KT, Shearer AE, Sloan CM, Kolbe DL, et al. Comprehensive genetic testing with ethnic-specific filtering by allele frequency in a Japanese hearing-loss population: Comprehensive genetic testing with ethnic-specific filtering. Clin Genet. 2016;89:466–72.
Sarmadi A, Nasrniya S, Narrei S, Nouri Z, Abtahi H, Tabatabaiefar MA. Whole exome sequencing identifies novel compound heterozygous pathogenic variants in the MYO15A gene leading to autosomal recessive non-syndromic hearing loss. Mol Biol Rep. 2020;47:5355–64.
Chen J-R, Tang Z-H, Zheng J, Shi H-S, Ding J, Qian X-D, et al. Effects of genetic correction on the differentiation of hair cell-like cells from iPSCs with MYO15A mutation. Cell Death Differ. 2016;23:1347–57.
Belguith H, Aifa-Hmani M, Dhouib H, Said MB, Mosrati MA, Lahmar I, et al. Screening of the DFNB3 Locus: Identification of Three Novel Mutations of MYO15A Associated with Hearing Loss and Further Suggestion for Two Distinctive Genes on This Locus. Genet Test Mol Biomark. 2009;13:147–51.
Ammar-Khodja F, Bonnet C, Dahmani M, Ouhab S, Lefe GM, Ibrahim H, et al. Diversity of the causal genes in hearing impaired Algerian individuals identified by whole exome sequencing. Molec Genet Genom Med. 2015;3(3):189–96. https://doi.org/10.1002/mgg3.131.
Kalay E, Uzumcu A, Krieger E, Çaylan R, Uyguner O, Ulubil-Emiroglu M, et al. MYO15A (DFNB3) mutations in Turkish hearing loss families and functional modeling of a novel motor domain mutation. Am J Med Genet A. 2007;143A:2382–9. https://doi.org/10.1002/ajmg.a.31937.
Duman D, Sirmaci A, Cengiz FB, Ozdag H, Tekin M. Screening of 38 Genes Identifies Mutations in 62% of Families with Nonsyndromic Deafness in Turkey. Genet Test Mol Biomark. 2011;15:29–33. https://doi.org/10.1089/gtmb.2010.0120.
Riahi Z, Bonnet C, Zainine R, Louha M, Bouyacoub Y, Laroussi N, et al. Whole Exome Sequencing Identifies New Causative Mutations in Tunisian Families with Non-Syndromic Deafness. PLoS ONE. 2014;9:e99797.
Shahin H, Walsh T, Rayyan AA, Lee MK, Higgins J, Dickel D, et al. Five novel loci for inherited hearing loss mapped by SNP-based homozygosity profiles in Palestinian families. Eur J Hum Genet. 2010;18:407–13.
Bademci G, Foster J, Mahdieh N, Bonyadi M, Duman D, Cengiz FB, et al. Comprehensive analysis via exome sequencing uncovers genetic etiology in autosomal recessive nonsyndromic deafness in a large multiethnic cohort. Genet Med. 2016;18:364–71.
Budde BS, Aly MA, Mohamed MR, Breß A, Altmüller J, Motameny S, et al. Comprehensive molecular analysis of 61 Egyptian families with hereditary nonsyndromic hearing loss. Clin Genet. 2020;98:32–42.
Xia H, Huang X, Guo Y, Hu P, He G, Deng X, et al. Identification of a Novel MYO15A Mutation in a Chinese Family with Autosomal Recessive Nonsyndromic Hearing Loss. PLOS ONE. 2015;10:e0136306.
Zhou H, Kuermanhan A, Zhang Z, Wang W, Dong J, Zhou Z, et al. Identification of a novel homozygous mutation in the MYO15A gene in a Kazakh family with non-syndromic hearing loss. Int J Pediatr Otorhinolaryngol. 2019;125:128–32.
Akbariazar E, Vahabi A, Abdi Rad I. Report of a Novel Splicing Mutation in the MYO15A Gene in a Patient With Sensorineural Hearing Loss and Spectrum of the MYO15A Mutations. Clin Med Insights Case Rep. 2019;12:117954761987190. https://doi.org/10.1177/1179547619871907.
Lezirovitz K, Pardono E, de Mello Auricchio MTB, de Carvalho e Silva FL, Lopes JJ, Abreu-Silva RS, et al. Unexpected genetic heterogeneity in a large consanguineous Brazilian pedigree presenting deafness. Eur J Hum Genet. 2008;16:89–96.
Tsukada K, Nishio S, Hattori M, Usami S. Ethnic-Specific Spectrum of GJB2 and SLC26A4 Mutations: Their origin and a literature review. Ann Otol Rhinol Laryngol. 2015;124(1_suppl):61S-76S.
Farjami M, Asadi R, Afzal Javan F, Alimardani M, Eslami S, Mansoori Derakhshan S, et al. The worldwide frequency of MYO15A gene mutations in patients with autosomal recessive non-syndromic hearing loss: a meta‐analysis. Iran J Basic Med Sci. 2020;23.
Xu P, Xu J, Peng H, Yang T. Compound heterozygous mutations in TMC1 and MYO15A are associated with autosomal recessive nonsyndromic hearing loss in two Chinese han families. Neural Plast. 2020;2020:1–7.
Keats BJ, Berlin CI. Genomics and hearing impairment. Genome Res. 1999;9:7–16.
Sheikh TI, Mittal K, Willis MJ, Vincent JB. A synonymous change, p.Gly16Gly in MECP2 Exon 1, causes a cryptic splice event in a Rett syndrome patient. Orphanet J Rare Dis. 2013;8:108.
Acknowledgements
We would like to extend our gratitude to the patients and their families for their participation and cooperation.
Funding
This work was supported by grants from Beijing Natural Science Foundation (7191011, 7192234), the Project of the Reproductive Health and Serious Birth Defect Prevention Research Project (National Key Research Project, 2016YFC1000704 and 2016YFC1000706), National Natural Science Foundation of China (81730029, 81873704, 82171158, 82171155 and 81870731), and Fostering Funds of Chinese PLA General Hospital for National Distinguished Young Scholar (2017-JQPY-001). Scientific Research Foundation of Qilu Hospital of Shandong University (Qingdao) (No. QDKY2020QN05). Youth Science Fund Project of National Natural Science Foundation of China (81900953). Natural Science Foundation of Hainan Province (819MS110).
Author information
Authors and Affiliations
Contributions
YY and PD conceived the study, participated in its design and coordination. SH carried out molecular genetic studies in hearing loss pedigrees. YF analyzed the genotype–phenotype correlations in all patients and drafted the paper. YY and XG technically edited and revised the manuscript. MH, GW and DK participated in repairing data supplement and reorganizing the revised manuscript. All authors have read and approved the final paper.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of the Chinese PLA General Hospital (reference number S2016-120–02). We obtained fully informed written consent from all participants, and the Institutional Review Board (IRB) approval was obtained from the Research Ethics Committee of the Chinese PLA General Hospital (approval number S2016-103–01). Fully informed written consent for participation and publication of clinical data was obtained from each subject or the guardians of subjects < 16 years old (yo). The study protocol was carried out as per the relevant guideline.
Consent for publication
We obtained the written informed consents for publication from all the participants. Written informed consents for publication were obtained from the next of kin on the behalf of the minors/children participants involved in this study.
Competing interests
The authors declare that no competing interest exists.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1 Table S1 L
ist of the 168 deafness genes and miRNA.
Additional file 2. Table S2
Primer sets of Sanger sequencing used in this study.
Additional file 3.
Detailed WES procedures.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fu, Y., Huang, S., Gao, X. et al. Analysis of the genotype–phenotype correlation of MYO15A variants in Chinese non-syndromic hearing loss patients. BMC Med Genomics 15, 71 (2022). https://doi.org/10.1186/s12920-022-01201-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-022-01201-3